The LINK-A lncRNA activates normoxic HIF1α
signalling in triple-negative breast cancer
Aifu Lin
1,13, Chunlai Li
1,13, Zhen Xing
1,13, Qingsong Hu
1, Ke Liang
1, Leng Han
2, Cheng Wang
3,
David H. Hawke
4, Shouyu Wang
1, Yanyan Zhang
1, Yongkun Wei
1, Guolin Ma
5, Peter K. Park
1,
Jianwei Zhou
6,
Yan Zhou
7, Zhibin Hu
3, Yubin Zhou
5, Jefery R. Marks
8, Han Liang
4,9, Mien-Chie Hung
1,10,11,
Chunru Lin
1,10,14and Liuqing Yang
1,10,12,14Although long non-coding RNAs (lncRNAs) predominately reside in the nucleus and exert their functions in many biological
processes, their potential involvement in cytoplasmic signal transduction remains unexplored. Here, we identify a cytoplasmic
lncRNA, LINK-A (long intergenic non-coding RNA for kinase activation), which mediates HB-EGF-triggered, EGFR:GPNMB
heterodimer-dependent HIF1α phosphorylation at Tyr 565 and Ser 797 by BRK and LRRK2, respectively. These events cause
HIF1α stabilization, HIF1α–p300 interaction, and activation of HIF1α transcriptional programs under normoxic conditions.
Mechanistically, LINK-A facilitates the recruitment of BRK to the EGFR:GPNMB complex and BRK kinase activation. The
BRK-dependent HIF1α Tyr 565 phosphorylation interferes with Pro 564 hydroxylation, leading to normoxic HIF1α stabilization.
Both LINK-A expression and LINK-A-dependent signalling pathway activation correlate with triple-negative breast cancer (TNBC),
promoting breast cancer glycolysis reprogramming and tumorigenesis. Our findings illustrate the magnitude and diversity of
cytoplasmic lncRNAs in signal transduction and highlight the important roles of lncRNAs in cancer.
Triple-negative breast cancer (TNBC) continues to
be a severe health problem
1–3, demanding the
consideration of emerging long non-coding RNAs
(lncRNAs) as biomarkers and therapeutic targets in
combatting this disease
4–6. Accumulating evidence
demonstrates that lncRNAs have broad functional
roles in the nucleus: regulation of transcriptional
activation,
X
chromosome
inactivation,
heterochromatin formation, and maintenance of
telomeres
7–14. Alterations of these functions promote
tumour formation, progression and metastasis of many
cancer types
15–20. However, many known lncRNAs
reside either within the cytosol or shuttle between the
nucleus and cytoplasm
21, playing important roles in
modulating messenger RNA translation, decay and
cytoplasmic protein trafcking
22–24. Intriguingly, many
protein kinases and metabolic enzymes bind RNA
through their non-canonical RNA- binding domains
25– 27, raising an important question of whether
cytoplasmic lncRNAs are relevant in the regulation of
fundamental cellular processes.
The
hypoxia-inducible
factor
(HIF)
transcriptional program is involved in TNBC
progression,
recurrence
and
metabolic
reprogramming
28–30. Although it is well known that the
hydroxylation
of
HIF1α
mediated
by
proline
hydroxylase domain (PHD) proteins triggers
VHL-dependent HIF1α ubiquitylation and degradation under
normoxic conditions
31,32, under certain circumstances
in tumour, HIF1α can accumulate under normoxic
conditions, promoting angiogenesis and cancer
progression
33,34. However, the mechanism underlying
normoxic HIF1α stabilization in TNBC remains elusive.
Here, we identified a highly prognostic lncRNA
in TNBC, long intergenic non-coding RNA for kinase
activation (LINK-A) (also known as LOC339535 and
NR_015407), which is critical for the growth
factor-induced normoxic HIF1α signalling pathway.
1Department of Molecular and Cellular Oncology, The University of Texas M D Anderson Cancer Center, Houston, Texas 77030, USA. 2Department of Biochemistry and Molecular Biology, The University of Texas Health Science Center at Houston McGroven Medical School, Houston, Texas 77030, USA. 3Department of Epidemiology and Biostatistics and Ministry of Education (MOE), School of Public Health, Nanjing Medical University, 210029, China. 4Department of System Biology, The University of Texas M D Anderson Cancer Center, Houston, Texas 77030, USA. 5Center for Translational Cancer Research, Institute of Biosciences and Technology, Texas A&M University Health Science Center, Houston, Texas 77030, USA. 6Department of Molecular Cell Biology and Toxicology, School of Public Health, Nanjing Medical University, 140 Hanzhong Road, Nanjing 210029, China. 7Department of Oncology, Yixing People’s Hospital, 75 Zhenguan Road, Yixing 214200, China.
8Department of Surgery, Division of Surgical Science, Duke University, School of Medicine, Durham, North Carolina 27710, USA. 9Department of Bioinformatics and Computational Biology, Division of Quantitative Sciences, The University of Texas M D Anderson Cancer Center, Houston, Texas 77030, USA. 10The Graduate School of Biomedical Sciences, The University of Texas M D Anderson Cancer Center, Houston, Texas 77030, USA. 11Center for Molecular Medicine and Graduate Institute of
Cancer Biology, China Medical University, Taichung 404, Taiwan. 12Center for RNA Interference and Non-Coding RNAs, The University of Texas M D Anderson Cancer Center, Houston, Texas 77030, USA. 13These authors contributed equally to this work.
a P = 1.38 × 10–3 P = 5.43 × 10–5 P = 2.48 × 10–3 P = 0.06 P = 6.04 × 10–6 P = 2.57 × 10
–6
P = 0.95 P = 0.86 P = 0.82 P = 0.57 P = 1 Training set P = 0.50 P = 0.92 5 4 4 P = 0.88 P = 1 LINK-A P = 2.08 × 10–4 80 P = 3.05 × 10–4 3 3 2 2 40 P = 0.34 P = 0.76 1 1 20 P = 0.35 0 0 d Validation set P = 6.31 × 10–13 P = 6.88 × 10–6 P = 3.07 × 10–5 e 100 Low LINK-A f LINK-A pulldown Peptide number 60 P = 0.034 P = 0.27 P = 0.68 High LINK-A 40 P = 0.69 P = 0.11 P = 0.98 P = 0.10 50 20 0 0 P = 0.0189 g MDA-MB-231 LINK-A 0 100 200 Month h Streptavidin pulldown Biotinylated 300 i Streptavidin pulldown Biotinylated Input Beads LINK-A 0.30 0.25 0.20 0.15 0.10 0.05 0 RIP ∗ GAPDH FLAG–BRK IB: FLAG Streptavidin– HRPInput Beads LINK-A
Myc–LRRK2 IB: Myc-tag Streptavidin– HRP BRK-WT BRK-ΔSH3 SH3 SH2 Kinase domain CT SH2 Kinase LRRK2 WT WD LRRK2 /KD2 1 11 72 78 170 191 321 451 ΔWD40 1 983 1291 1879 2164 2515 Biotinylated j LINK-A (as.)
Biotinylated LINK-A (sen.) k Streptavidin pulldown
Bio-LINK-A 1 2 3 4 5 6 A B C D E 1 2 3 4 5 6 1 2 3 4 5 6 A A A B B B C C C D D D E E E 1 2 3 4 5 6 1 2 3 4 5 6 A B C D E IB: BRK GST GST–BRK Streptavidin–HRP GST–LRRK2 IB: LRRK2
A1 to E2, individual LINK-A probes (ant.) E3, blank E4, mixed LINK-A probes (ant.) IB: eIF4B
E5, mixed LacZ probes E6, synthesized biotin-scramble probe
1 1540 LINK-A (5
′–3′)
A1 A2 A3 E1 E2 Probes (3′–5′)
Streptavidin–
HRP MDA-MB-231
Figure 1 LINK-A is a TNBC-upregulated cytoplasmic lncRNA with prognostic value. (a,b) Scatter plots comparing LINK-A expression in breast tumour samples with different ER, PR and HER2 status including ER− /PR− /HER2−
(n =119), ER− /PR− /HER2+ (n = 30), ER+ /PR+ /HER2− (n =482), and ER+ /PR+ /HER2+ (n = 80) (a), or in breast tumour tissue samples with different subtypes including basal (n = 139), HER2 (n = 67), LumA (n = 417), LumB (n = 191) and normal-like (n = 23) (b). Statistical significance
was determined by two-way ANOVA. The boxes show the median and the interquartile range. The whiskers show the minimum and maximum. (c,d) RNAScope detection of LINK-A expression in human breast cancer and adjacent normal tissues (training (c) and validation (d) set, respectively). Left panel in c: representative images (scale bars, 100 µm); d and right
panel in c: statistical analysis. Training set: TNBC (n = 10), ER− /PR− /HER2+
(n = 7), ER+ /PR+ /HER2− (n = 18), and ER+ /PR+ /HER2+ (n = 2); validation
set: ER− /PR− /HER2− (n = 38), ER− /PR− /HER2+ (n = 2), ER+ /PR+ /HER2−
(n = 6), ER+ /PR+ /HER2+ (n =9) and normal tissue (n = 20) (median,
two-way ANOVA). Horizontal black lines represent median. Coloured error bars represent 95% quantile. (e) Recurrence-free survival analysis of LINK-A status in breast cancer patients detected by qRT–PCR (n = 123 patients,
Bead only
Transcription intermediary factor 1β Prelamin A/C Matrin-3 2 2 4 LINK-A (as.)
Heterogeneous nuclear ribonucleoprotein A1 Tubulin β3 Matrin-3 3 2 3 LINK-A (sen.) Hypoxia-induced factor 1α
Leucine-rich repeat serine/threonine kinase 2
Protein tyrosine kinase 6
Transmembrane glycoprotein NMB
Epidermal growth factor receptor
12
4
5
6
4
ARM ANK LRR ROC/COR Kinase ARM ANK LRR ROC/COR Kinase
E xp re ss io n le ve l (l o g2 ) L IN K -A s ta in in g r e la tiv e i n te ns ity (× 1 0 4) P e rc e n ta g e o f in p u t E R –/P R –/ H E R 2 – T N B C E R –/P R –/ H E R 2 + N o rm a l E xp re ss io n le ve l (l o g2 ) R e cu rr e n ce -f re e s u rv iv a l B a sa l B la n k H E R 2 L u m A L u m B N o rm a li ke P o si tiv e N e g a tiv e L IN K -A s ta in in g r e la tiv e i n te ns ity (× 1 0 4) B la n k W T Δ W D 4 0 In p u t B e a d s T N B C E R –/P R –/ H E R 2 + b c
Gehan–Breslow test). (f) A list of the top LINK-A-associated proteins identified by RNA pulldown and MS analysis in MDA-MB-231 cells. (g) RIP–qPCR detection of indicated RNAs retrieved by BRK-, LRRK2-or eIF4B-specific antibodies in MDA-MB-231 cells. ErrLRRK2-or bars, s.e.m.,
n = 3 independent experiments (∗ P < 0.05, two-tailed paired Student’s
t -test). (h,i) In vitro RNA–protein binding assay showing the interaction
of biotinylated LINK-A with wild-type (WT) FLAG-tagged BRK and a deletion mutant (h), or WT Myc-tagged LRRK2 and a deletion mutant (i). Dot-blot of RNA–protein binding samples indicates equal RNA transcript present in the assay. Bottom panel: graphic illustration of BRK or LRRK2 domain deletion mutants. IB, immunoblot. (j) Upper panel: In vitro RNA– protein binding followed by dot-blot assays using biotinylated LINK-A sense (sen.) or antisense (as.) transcripts and GST-tagged, bacterially expressed BRK or LRRK2 proteins. The hybridized RNA fragments were detected by streptavidin–HRP. Bottom panel: graphic illustration of LINK-A probes. (k) Immunoblot detection of proteins retrieved by in-vitro-transcribed biotinylated LINK-A full-length (FL) or deletion mutants expressed in MDA-MB-231 cells. Unprocessed original scans of blots are shown in Supplementary Fig. 7.
Mechanistically, LINK-A is required for
HB-EGF-triggered, EGFR:GPNMB
heterodimer-mediated
recruitment of BRK to GPNMB, and subsequent
enzymatic activation of BRK. The activated BRK,
together with LRRK2 that is also recruited by
LINK-A, phosphorylates HIF1α at Tyr 565 and Ser 797,
respectively. Whereas the phosphorylation at Tyr 565
inhibits hydroxylation at the adjacent Pro 564, which
prevents
HIF1α
degradation
under
normoxic
conditions, Ser 797 phosphorylation facilitates HIF1α–
p300 interaction, leading to activation of HIF1α
target genes on HB-EGF stimulation. Furthermore, we
demonstrated that LRRK2, a constitutively active
kinase in Parkinson’s disease, is a RNA-binding kinase
that phosphorylates HIF1α in human cancers.
Importantly, both LINK-A expression and activation of
the LINK-A-mediated normoxic HIF1α signalling
pathway correlated with TNBC. Therefore, targeting
LINK-A may serve as a favourable strategy to block a
normoxic HIF1α signalling pathway in TNBC with
promising therapeutic potential.
RESULTS
LINK-A is a cytoplasmic lncRNA with prognostic value
for TNBC
To identify TNBC-relevant lncRNAs, we examined the
lncRNA expression profile in two stage III TNBC tissues
and their paired adjacent non-cancerous tissues,
finding 21 diferentially expressed lncRNAs (ref. 16).
We further searched the expression pattern of these
21 lncRNAs in the TCGA database. Interestingly,
statistical analysis of a combined 711 RNA-seq
transcriptome profiles indicated that the expression of
LINK-A is frequently elevated in TNBC patient cohorts
in comparison with cohorts of ER
−/PR
−/HER2
+,
ER
+/PR
+/HER2
−and ER
+/PR
+/HER2
+patients. Diferential LINK-A
expression
between
ER
−/PR
−/HER2
+,
ER
+/PR
+/HER2
−and
ER
+/PR
+/HER2
+cohorts was not statistically significant (Fig. 1a).
Consistently,
basal-like breast cancer, which lacks or shows low
levels of ER, PR
and HER2 proteins
35,36, exhibited significantly
increased LINK-A
expression in comparison with HER2
+, LumA, LumB
and normal-like
subtypes (Fig.
1b).
LINK-A is a ∼1.5-kb-long intergenic
non-protein-coding RNA
(ref. 37), which was confirmed by our northern
blot and RACE
analyses in MDA-MB-231 cells (Supplementary Fig.
1a,b). Given that LINK-A has a predicted open reading
frame (ORF) of 139 amino acids, we performed in vitro
translation assays, showing that neither the sense nor
the antisense transcript of LINK-A encodes protein
(Supplementary Fig. 1c). We next examined LINK-A
expression in breast cancer tissue microarrays
(clinicopathological
parameters
listed
in
Supplementary Table 1) using the RNAScope 2.0 HD
assay. In both the training and validation sets of tissue
samples, the expression of LINK-A was significantly
increased in TNBC tissues compared
with normal breast tissues, and ER
−/PR
−/HER2
+,
ER
+/PR
+/HER2
−,
and ER
+/PR
+/HER2
+subtypes (Fig. 1c,d),
demonstrating the
strong correlation of LINK-A expression with TNBC.
Additionally,
we examined the LINK-A expression level in a
Duke breast
cancer cohort, finding that high levels of LINK-A
correlated with
unfavourable recurrence-free survival for breast
cancer patients (Fig. 1e). Consistently, LINK-A was
highly expressed in TNBC cell lines compared with
oestrogen receptor (ER)- or HER2-positive breast
cancer cell lines (Supplementary
Next, we examined the subcellular localization of
LINK-A, finding that LINK-A predominately resides in
the cytoplasm or close to the cellular membrane,
which was distinct from typical nuclear lncRNAs
including BCAR4 (ref. 16) and HOTAIR (ref. 20)
(Supplementary Fig. 1e–g). Cell fractionation analysis
showed that >90% of LINK-A is localized within the
cytosolic fraction compared with the nuclear
enrichment of BCAR4 (Supplementary Fig. 1h,i). We
reasoned that LINK-A has important roles in the
cytosol.
Identification and characterization of
LINK-A–protein interaction
We performed an RNA pulldown assay followed by
mass spectrometry
15,16(MS) to identify
LINK-A-associated proteins that might be involved in
cytoplasmic processes. Interestingly, the sense LINK-A,
but not the antisense or beads control, specifically
associated
with
two
transmembrane
proteins,
epidermal growth factor receptor (EGFR) and
transmembrane glycoprotein NMB (GPNMB), tyrosine
protein kinase 6 (also known as breast tumour kinase,
BRK; refs 38,39), leucine-rich repeat kinase 2 (LRRK2;
refs 40,41), and HIF1α in the breast cancer cell (Fig.
1f, Supplementary Fig. 1j and Supplementary Table
2). An RNA pulldown assay in cell lysate and an RNA–
protein binding assay using recombinant EGFR, BRK,
LRRK2, HIF1α and GPNMB confirmed that LINK-A
associated with all of the proteins mentioned above
in vivo, but only BRK and LRRK2 directly
interacted with LINK-A (Supplementary Fig. 1k–n).
The specific interaction between LINK-A and BRK or
LRRK2
was
also
confirmed
by
an
RNA
immunoprecipitation (RIP) assay (Fig. 1g).
To map the BRK domains required for LINK-A
binding, we generated BRK SH3 (amino acids 11–72),
SH2 (amino acids 78–170), kinase domains (amino
acids 191–445), and regulatory carboxy- terminal
(amino acids 446–451) deletion mutants (Fig. 1h,
bottom panel). Deletion of either the SH3 domain or
the C-terminal region of the kinase domain of BRK
alone impaired the interaction between LINK-A and
BRK, suggesting that LINK-A interacts with two
separate domains of BRK (Supplementary Fig. 1o).
Double deletion of these two domains abolished the
LINK-A–BRK interaction in vitro and in vivo (Fig. 1h
and Supplementary Fig. 1p). A similar strategy was
used to map the domain required for LRRK2–LINK-A
interaction, showing that deletion of the WD40
domain, an atypical RNA- binding domain
16,25,
abolished the direct interaction (Fig. 1i and
Supplementary Fig. 1q).
To map RNA motifs essential for the LINK-A–protein
interactions, we conducted an in vitro RNA–
protein binding coupled with dot-blot assay
15,16,
finding that BRK interacted with LINK-A at two
regions, nucleotides 481–540 (dot B3) and
nucleotides
781–840 (dot C2) (corresponding to the two
domains of BRK at the SH3 domain and the
C-terminal tail) (Fig. 1j). LINK-A nucleotides 1261–1320
(dot D4) interacted with LRRK2 (Fig. 1j). Consistently,
double deletion of LINK-A (nucleotides 471–550
and nucleotides 771–850) abolished the BRK–LINK-A
interaction without afecting the LRRK2–LINK-A
interaction, whereas deletion of LINK-A (nucleotides
1251–1330) specifically abolished LRRK2– LINK-A
association (Fig. 1k). The predicted secondary
structure of LINK-A indicates that the RNA motifs
required for BRK and LRRK2 interactions form
individual branching stem loops,
山E止L 一C 「
ILH
H
aIII-
,L
自 口一]鬥巳 Y怕NXN Protein汗 MmFMSummary of phosphorylation sites Site Peptide sequence
C Growth HB- HB-DTSSP - +
-
+ +「一一--,
r
一一--,
口川門口什川 nD GPNMB Y一
5的
2巨
5-
K EYNPIENSPGNWR S + phospho (Y)-三
一
Loz
300 250-
11--1.
EGFR GPNMB BRK Y351 --〈
KE一
-3宮一,
主 E.一
EDVYLSHDHNIP工KW + phospho (Y) 1801
••••••
• EGFR HIF1α Y5日 5 一--且E-2=一ggc KNP-
Fz
一
SE
山 E
Tz
QDTDLDLEI叫 LAPYIPMDDDFQ LR.S + phospho (Y) 130•
•
直•
‘
GPNMB '--E-C
B-一
3嘗
宮一
R.LLGQSMDESGLPQLTSYDCEVNAPIQ 100 HIF1α S797GSRN+ pho o (ST) IB EGFR(Ab-13) Stripped _".IB GPNMB
•
•一工
一
于
E
一
E
一
-SE
,
一←
PL一
U FLAG-GPNMBi
;;g
tB
5 : 山B
+ +E
HPEGFRSESES HB-EGF r了?亡?亡? 亡? 1 .. 1 HB-EGFr:-T T?T?TT
C E O 苟 二3 f 吉 芷 f..d} ,_ c P 三雪 LL :.= 歪 歪8
宋詩FRS
甘冒旦出音宮
室主
ZZhE
CL 山〈∞ l且已工工 t=-z
E
E||B HlM呵1
••
1
E 主
1|
主 IIB G川 BI - 1 |BGPI
出
二
|
二二二:二三|
MDA-MB-231 g FLAG-GPNMB -=
'"←
a: :Y:: -C
';←
正U
E::
E 的(') a: 的。 正 ξIIB F山 G-tag g _Q 0> 回 (')3
LL hJIlB
叭MBazzzgττ
=
τd
ffi
L
IB P句rl
-
1
1 IB BRK 1... . . . 酬 可I
IB川|
|
:j1
IB HIF10t1-
1 生1 IB p-Tyr 1|
-L
IB p-Ser1
'
_
1
MDA-MB-231 CBB ∞ C 山且∞0 山且 IB FLAG IB p-GPNMB 6U 一 L4A 」L nD凶仁尸
口他||L
u ←F 』llF G川 VJ…何也叩叫仰自引FF聞 RHR (Tyr 52司一
•
馴=
的•
M問一
問知哪一
一
三
•=E•
心
•一M一
=一
叫
•
[
九
一凹
二二二
之 c0叫 C\J BRK-\f\IT g ←- 芸 芸,
一但
•
岫
=
三
一
一
一
•
九
一 M [
BRK-. SH3 FLAG-BRK co 豈有這三語言屯 HB-EGFr:::-?
古?亡?T?T,一?去 亡
-=?一
.
TT
一一叫 一=
I BRK-. SH2司一
叮
]
-
叮
叫
」
三
一﹒
]]叮
BRK-. KD1 BRK-. KD sph fpP + + LC山,也工一 LL一
一
?三
8
8
IB 酬 ---
-
-1
LL I l 一 1 IB GPNMB 1﹒
-
- -
-1 L Input:; BRK-. KD2•
IB p-GPNMB 1 (Tyr 525) - - _ _ 色 "'1 1 11 72 78 170 191 321 451 Input 1也 -
--
-
!P
--
---
I
IB GPNMB 1.. 7 . .. .-1 MDA-MB-231 Figu陀 2 LfNK-A is involved in an HB-EGF-triggered, EG FR:GPNMB
mediated signalling pathway,(a) Summary 01 the phosphorylation sites
01 the indicated proteins identilied Irom RNA pulldown lollowed by MS analysis, (b) Immunoprecipitation (IP) lollowed by immunoblot
(IB) detection 01 the indicated proteins in MDA-MB-231 cells treated with the indicated growth lactors lor 30 min, (c) Immunoblot detection using the indicated antibodies in MDA-MB-231 cells
stimulated with vehicle, EGF or HB-EGFlollowed by DTSSP chemical
crosslinking ( 1mM,
30min), (d,e) His tag (d) or FLAG tag (e) pulldown lollowed
by
immunoblot detectlon using the indicated antibodies 川 MDA-MB-231 cells
translected with the indicated expression vecto內的Ilowed by HB
-EGF
stimulation ,ECD, extracellul司r domain; TM, transmembrane domain;
ICD, intracellular domain , (f) Immunoprecipitation followed by
immunoblot detection 01 GPNMB, B RK and HIFla phosphorylation in MDA-MB-231
cells treated with the indicated growth lactors,(g) fn vitro kinase assay
using the indicated recombinant proteins, lollowed by Coomassie blue
staining (CBB), and immunoblot detection using the indicated antibodies ,(h,il Immunoprecipitationlollowed by immunblot
detection using the
indicated antibodies in cells trans ected with the indicated expression vecto
內的Ilowed by HB-EGF stimulation,Lelt panel (自 ): graphic illustration 01
BRK domain delellon mutants,Unprocessed original scans 01 blots are
shown in Supplementary Fi日
7
,丘 I
suggesting that they contribute to specific RNA–
protein interactions
(Supplementary
Fig. 1r).
Characterization of a HB-EGF-triggered, EGFR:GPNMB-
dependent and LINK-A-mediated signalling pathway in TNBC
Our MS data revealed a series of phosphorylation
sites of GPNMB
(Tyr 525), BRK (Tyr 351), and HIF1α (Tyr 565 and Ser
797) (Fig. 2a
and Supplementary Fig. 2a–d and Supplementary
Table 2), leading us
to generate phosphorylation site-specific antibodies
(Supplementary Fig. 2e–i) to investigate whether
LINK-A modulates a previously unknown signalling pathway.
Given that LINK-A associated with the orphan
receptor GPNMB
and EGFR, which are involved in metastatic TNBC
(refs 42–44),
we reasoned that EGFR and GPNMB may interact
with each other
in TNBC cells on EGF family ligands. Although all
EGFR ligands
efectively activated EGFR (Supplementary Fig. 2j),
HB-EGF robustly
induced the specific interaction between EGFR and
GPNMB (Fig. 2b),
indicating that EGF ligands could diferentially trigger
the formation
of the EGFR homodimer or the heterodimer between
EGFR and other receptors
45. To test this, we
performed a crosslinking assay, finding that EGF
predominately triggered EGFR homodimerization with
a
lesser
degree
of
EGFR:GPNMB
heterodimerization but HB-EGF stimulated
EGFR:GPNMB heterodimerization with less EGFR
homodimerization (Fig. 2c). Knockdown of LINK-A
exhibited minimal efects on the HB-EGF-induced
EGFR:GPNMB interaction
as
well
as
GPNMB
phosphorylation on ligand stimulation (Supplementary
Fig. 2k,l), suggesting that HB-EGF preferentially
triggered EGFR:GPNMB heterodimer formation. We
further mapped the domains mediating EGFR–GPNMB
binding, and found that the kinase domain (KD) in
EGFR intracellular domains (ICD) interacts
with GPNMB ICD (Fig. 2d,e and Supplementary Fig.
2m).
HB-EGF
robustly
induced
site-specific
phosphorylation of EGFR, GPNMB, BRK and HIF1α (Fig.
2f ) and pretreatment of TNBC cell lines with
cetuximab impaired EGFR–GPNMB interaction
(Supplementary Fig. 2n,o). These observations led
us to fully characterize this HB-EGF-triggered,
EGFR:GPNMB-dependent signalling pathway in TNBC.
First, an in vitro kinase assay indicated that EGFR,
but not BRK,
phosphorylated GPNMB at Tyr 525 (Fig. 2g) and the
exogenously
expressed wild-type GPNMB but not the Y525F mutant
was phosphorylated in vivo on HB-EGF stimulation
(Fig. 2h). Next, we observed the interaction between
GPNMB and BRK following
ligand stimulation, which was abolished in the
presence of the GPNMB Y525F mutant (Fig. 2h).
Furthermore, the ligand-triggered BRK Tyr 351
phosphorylation was abolished in GPNMB
Y525F-overexpressing cells
(Fig.
2h).
Biochemical
experiments showed that BRK SH2 domain deletion
(amino acids 78–170) eliminated the ligand-dependent
interaction with Tyr-525-phosphorylated GPNMB (Fig.
2i). These data suggest that the EGFR-dependent
GPNMB Tyr 525 phosphorylation is required for further
recruitment of BRK through its SH2 domain and
subsequent phosphorylation at Tyr 351.
LINK-A facilitates the recruitment and activation of BRK
We then conducted an immuno-RNA fluorescence in
situ hybridization (FISH) assay to examine the
proximity of LINK-A to the ligand-bound receptors
on ligand treatment, finding the
overlap between LINK-A and EGFR on HB-EGF
stimulation (Supplementary Fig. 3a), which was further
validated by in vivo RIP assay (Supplementary Fig.
3b). We examined the co-localization of BRK and
the EGFR:GPNMB receptor complex in the presence
or absence of LINK-A. Our data indicate that
both BRK and phospho-BRK (Tyr 351) faithfully
co-localized with EGFR on HB- EGF stimulation (Fig. 3a
and Supplementary Fig. 3c). In contrast, depletion of
LINK-A abolished the recruitment of BRK to EGFR
and subsequent phosphorylation of BRK without
afecting the internalization of EGFR (Fig. 3a and
Supplementary Fig. 3c). We then performed rescue
experiments in which LINK-A was knocked down by
locked
nucleic
acids
(LNAs)
followed
by
reintroduction of LNA-resistant full-length LINK-A
or one of the following deletion mutants: ßBRK
(ß471–550 and ß771–850) or ßLRRK2 (ß1251–1330)
(Fig. 3b, lower panel, and Supplementary Fig. 3d,e),
finding that knockdown of LINK-A abolished the
HB-EGF-induced BRK–GPNMB interaction, as well as BRK
Tyr 351 phosphorylation (Fig. 3c,d); reintroduction of
full-length LINK-A or ßLRRK2 but not the ßBRK mutant
rescued these phenotypes (Fig. 3c,d). These data
suggest that LINK-A–BRK interaction facilitates the
recruitment of BRK to the tyrosine-phosphorylated
membrane receptor GPNMB, as well as subsequent
autophosphorylation of BRK.
LINK-A elicits the conformational change of BRK for
kinase activation
It has been reported that the activity of BRK is
auto-inhibited by interaction between the SH2 domain and
the Tyr-447-phosphorylated C-terminal domain
46–48.
Our data indicate that LINK-A interacts with BRK at two
regions, the SH3 domain and the C-terminal domain
(see Fig. 1h and Supplementary Fig. 1p), raising a
possible role for LINK-A in eliciting a BRK
conformational change that mitigates the
confor-mation required for self-inhibition. Indeed, we found
that full-length LINK-A and ßLRRK2 LINK-A markedly
enhanced the autophospho- rylation and kinase
activity of BRK, whereas both the control lncRNA and
ßBRK LINK-A showed minimal efects (Fig. 3e,f ).
We next conducted a protease digestion assay by
incubating BRK with caspase-1 in the presence of
full-length LINK-A or ßBRK LINK-A, finding that
caspase-1 barely cleaved BRK at amino acid
397 in the presence of an unrelated lncRNA
RP11-383G10.5, but robustly cleaved BRK only in the
presence of full-length LINK-A (Fig. 3g), suggesting
that a potential conformational change occurred in
BRK to expose the digestion site on LINK-A binding.
Notably, deletion of either of the two regions of LINK-A
involved in BRK interaction failed to promote the
caspase-1-dependent
BRK
cleavage (Fig.
3g),
suggesting that simultaneous binding of LINK-A to
the two BRK domains is required to elicit the
conformational change in BRK. Our data suggest that
the binding of LINK-A to BRK promotes a
conformational
change,
leading
to
increased
accessibility
of
the
SH2
domain
and
the
autophosphorylation sites in the kinase domain. On
ligand stimulation, these events lead to the
recruitment of BRK to Tyr-525-phosphorylated GPNMB
and activation of BRK on Tyr 351 phosphorylation.
LINK-A-interacting BRK and LRRK2 phosphorylate HIF1α
We next performed in vitro phosphorylation assays,
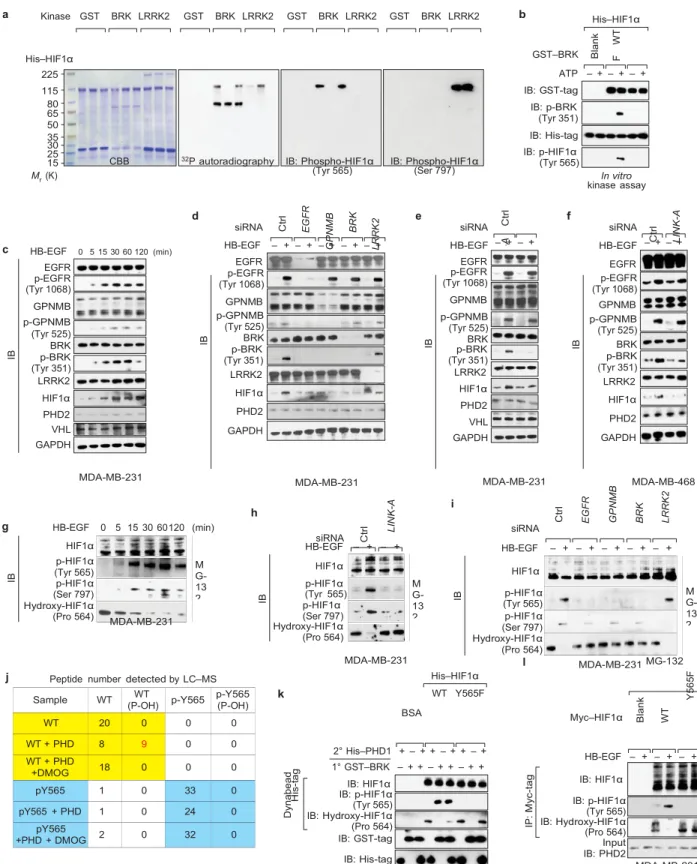
finding that activated BRK phosphorylated HIF1α at
Tyr 565 (Fig. 4a) and
a的〈
u
可」ZZωD-bEO」〈〈t
Z」正呵口的, 呵,法z
的之Z~斗b FIll--Ill-IIII-L
3H山的山的 L 工 σ 工 lHU 呵 La+σ 的 UH 工 a 且 lLH 工+LOO
Uit〉叫E斗〉由L-江一正臼臼」4
d LNA Scr L
爪
IK-A no. 5「一--, (\J
o
0<<
o 志在 ZI ..I
.
.
.
.
.
.
L1NK-A 主主可明才 .-可 r---r---r---r--1 HB-EGF 一+一+一+一+一+_
_
-
-_:r- 'W
"
-IB GPNMB量
IB p-GPNMB I _ I (Tyr 525) I| IB BRK 1-L仰K-A deletion mutantsFL53'' IB p-BRK
1
_
_
:
.
_
1
.
1
時間汀,一一3' t.BRK2 53'' t.BRK 5' 11噩噩耳- 3' t.LRRK25二' 二- 3' 咱1 5&1 781 8咱 1261 1320c
(Tyr 351) I| IB GAP叫
卜一一-一
MDA-MB-231 f。
o 0.0125 0α25 005 0.1 0.2 BRK (μg):=--。每
2,000 tβ 三斗 由F 冥 1 500三 ε
g;; 志 1.000Ei
;;;è 500_
c'
:T
>I
日 回 φ回缸一 FC3ot」由一丘。〉-F(豆G白 OO白P ('t_1;.",J0' L 一一一一一一」-"
,
V L/NK-Ae
Kinase GST-BRK g 盯 L扒IK-A 口(\ JVLJZ
豈宜 'T-;:::::_ U_ íí Protein AυAυAυAυAVAυ 6543241 口 ++++心心心
PNNN 仆件仟件 3AAA 即且GLBUFA RNA 是皆可司司 ATP可
?f?????
RNA
_ Caspase-1 們km 5闊1
---
1
---
可"'
-_
-.
_
--_
--
_
3叫KI
-
-1
Dto. 1'\ 25日 J---..r.: J且 150 l J LJ 1日 D 買E•
τE IB GST-tag 空空凹的仿制
.
I
1
/
5日﹒
|
...
.
/n vitro kinase assay
-
q
p
語M
,
(K)
IB GST-tag Stripped _. IB BRK (c-terminal)
Figu他 3 LfNK-A mediates recruitment 01 BRK to GPNMBlor kinase activation. (a) Immunolluorescence detection using the indicated antibodies
in MDA-MB-231 cells harbouring control (upper panel) or LfNK 可A
shRNA, lollowed by HB-EGF stimulation (upper panel). Scale A,這「之
字)主
....-
:
----1
I
R~vwv
l-bars, 20 [.lfl1. (b) Graphic illustration 01 the BR K- and LRRK2-Lf NK-A interactions (upper panel) and the corresponding deletion abolishing these interactions
in vitro-transcribed RNA transcripts as indicated in the presence or absence
01 [且 PlATP. The dot-blot indicates equal RNA transcript present in the assay. (f) Quantilication 01 BRK kinase activity in the presence 01 the indicated in vtiro-transcribed RNA transcripts using HIFl 0:
peptide (amino acids 557-566) as the substrate. Upper panel: release 01 Iree phosphate
ion (P;) amount measured at
o
D.,20 om ; lower p咽nel: calculation 01 BR K(Iower panel)峙,d) Immunolluorescence imaging 仗, scale ba月, 20
iJ.m) kinase activi句 (pmolmi
n-1 1
人
g-l). Error bars, s.e.m,.
月= 3 independent
。
r immunoblot (1 B) detection (d) was perlormed using the indicatedantibodies 川 MDA-MB-231 cells translected with LNA against LfNK-A lollowed by overexpr皂白 ion 01 the indicated rescue plasmids with
HB EGF stimulation. The dotted line on the blots 01 d indicates the position where the images 01 single blots were vertlcally cropped to luxtapose non-adjacent lanes,(e) In vitro kinase assay using recombinant BRK and
experiments ('P<O.05, two-tailed paired Student's t-test ). (g) Immunblot detection 01 BRK using the indicated antibodi的 in the presence 01 the indicated IncRNA transcripts with or without caspase-ldigestion. Lelt panel 皂raphic illustration 01 caspase-l-mediated BRK cleavage in the absence or presence 01 IncR NA. Unprocessed original scans 01 blots are shown In Suppl自mentary F皂i .7.
a Kinase GST BRK LRRK2 GST BRK LRRK2 GST BRK LRRK2 GST BRK LRRK2 b His–HIF1α His–HIF1α 225 115 80 65 50 35 30 25 15 Mr (K)
CBB 32P autoradiography IB: Phospho-HIF1α
(Tyr 565) IB: Phospho-HIF1α(Ser 797)
GST–BRK ATP – + – + – + IB: GST-tag IB: p-BRK (Tyr 351) IB: His-tag IB: p-HIF1α (Tyr 565) In vitro kinase assay d e f c HB-EGF 0 5 15 30 60 120 (min) EGFR p-EGFR (Tyr 1068) GPNMB p-GPNMB (Tyr 525) BRK p-BRK (Tyr 351) LRRK2 HIF1α PHD2 VHL GAPDH siRNA HB-EGF – + – + – + – + – + EGFR p-EGFR (Tyr 1068) GPNMB p-GPNMB (Tyr 525) BRK p-BRK (Tyr 351) LRRK2 HIF1α PHD2 GAPDH siRNA HB-EGF EGFR p-EGFR (Tyr 1068) GPNMB p-GPNMB (Tyr 525) BRK p-BRK (Tyr 351) LRRK2 HIF1α PHD2 VHL GAPDH – + – + siRNA HB-EGF EGFR p-EGFR (Tyr 1068) GPNMB p-GPNMB (Tyr 525) BRK p-BRK (Tyr 351) LRRK2 HIF1α PHD2 GAPDH – + – + MDA-MB-231 h g HB-EGF 0 5 15 30 60120 (min) MDA-MB-231 siRNA MDA-MB-231 i siRNA MDA-MB-468 HIF1α p-HIF1α (Tyr 565) p-HIF1α (Ser 797) Hydroxy-HIF1α (Pro 564) MDA-MB-231 HB-EGF HIF1α p-HIF1α (Tyr 565) p-HIF1α (Ser 797) Hydroxy-HIF1α (Pro 564) – + – + HB-EGF – + – + – + – + – + HIF1α p-HIF1α (Tyr 565) p-HIF1α (Ser 797) Hydroxy-HIF1α (Pro 564) j Peptide number detected by LC–MS
k MDA-MB-231 BSA l His–HIF1α WT Y565F MDA-MB-231 Myc–HIF1α MG-132 2° His–PHD1 + – + + – + + – + 1° GST–BRK – + + – + + – + + IB: HIF1α IB: p-HIF1α (Tyr 565) IB: Hydroxy-HIF1α (Pro 564) IB: GST-tag IB: His-tag HB-EGF – + – + – + IB: HIF1α IB: p-HIF1α (Tyr 565) IB: Hydroxy-HIF1α (Pro 564) Input IB: PHD2 MDA-MB-231
Figure 4 LINK-A-dependent BRK phosphorylation of HIF1α at Tyr 565 antagonizes HIF1α Pro 564 hydroxylation. (a) In vitro phosphorylation assay using recombinant proteins (WT or mutants as indicated). IB, immunblot. (b) In vitro kinase assay using bacterially expressed GST-tagged BRK WT or mutant and His-GST-tagged HIF1α. (c–f) Immunoblot detection using the indicated antibodies in MB-231 (c–e) or MDA-MB-468 (f) cells treated with HB-EGF at the indicated time point (c) or transfected with the indicated siRNAs followed by HB-EGF treatment (d–f). (g–i) Immunoblot detection using the indicated antibodies in MDA-MB-231 cells treated with MG-132 followed by HB-EGF treatment at
the indicated time (g) or in cells transfected with the indicated siRNAs followed by MG-132 and HB-EGF treatment (h,i). (j) LC–MS sequencing Sample WT (P-OH)WT p-Y565 p-Y565 (P-OH)
WT 20 0 0 0 WT + PHD 8 9 0 0 WT + PHD +DMOG 18 0 0 0 pY565 1 0 33 0 pY565 + PHD 1 0 24 0 pY565 +PHD + DMOG 2 0 32 0 M G-13 2 M G-13 2 M G-13 2 IB IB W T Y 5 6 5 F S 7 9 7 A W T Y 5 6 5 F S 7 9 7 A W T Y 5 6 5 F S 7 9 7 A IB IB C tr l H is -t a g D yn a b e a d E G F R G P N M B B R K C tr l L R R K 2 L IN K -A IB IB C tr l L IN K -A IP : M yc -t a g C tr l IB E G F R B la n k G P N M B W T Y 3 5 1 F B R K B la n k C tr l W T L R R K 2 L IN K -A Y 5 6 5F
of the HIF1α peptide (557–566) in an in vitro hydroxylation assay. The total peptide numbers of HIF1α proline non-hydroxylated versus proline hydroxylated (P-OH) under the indicated conditions are shown. The peptide number of hydroxylated WT peptide is indicated in red. (k) His-tag pulldown followed by immunoblot detection of HIF1α phosphorylation and hydroxylation (WT versus Y565F) in an in vitro kinase assay (1◦ ) followed by in vitro hydroxylation assay (2◦ ). (l) Immunoprecipitation (IP) followed by immunoblot detection of HIF1α phosphorylation and hydroxylation (WT versus Y565F) in MDA-MB-231 cells transfected with the indicated plasmids and treated with MG-132 followed by HB-EGF treatment. Unprocessed original scans of blots are shown in Supplementary Fig. 7.
LRRK2, another LINK-A-interacting protein kinase,
phosphorylated HIF1α at Ser 797, which was further
demonstrated by the marked inhibition of HIF1α
phosphorylation in the presence of a S797A point
mutant (Fig. 4a). The BRK kinase activity-deficient
mutant, Y351F, diminished the phosphorylation of
HIF1α in vivo (Fig. 4b). Both Tyr 565 and Ser 797 of
HIF1α are conserved (Supplementary Fig. 4a).
HB-EGF induced phosphorylation of GPNMB (Tyr
525) and BRK (Tyr 351), as well as HIF1α
protein stabilization under normoxic conditions (Fig.
4c and Supplementary Fig. 4b). Interestingly,
knockdown of EGFR abolished the ligand-dependent
phosphorylation of GPNMB (Tyr 525) and BRK (Tyr
351), as well as the stabilization of HIF1α; knockdown
of
GPNMB
abolished
HB- EGF-induced
BRK
phosphorylation and HIF1α protein stabilization, but
did not afect EGFR phosphorylation (Tyr 1068) (Fig.
4d). Knockdown of LINK-A in both MDA-MB-231 and
MDA-MB-468 cells eliminated HB-EGF-induced BRK
phosphorylation and HIF1α stabilization, but not
phosphorylation of EGFR or GPNMB (Fig. 4e,f ). In
contrast, LINK-A knockdown exhibited minimal efects
on hypoxia-dependent HIF1α stabilization, and hypoxia
failed to trigger phosphorylation of GPNMB and BRK
(Fig. 4c–f and Supplementary Fig. 4c). Finally,
depletion of BRK decreased ligand-triggered HIF1α
protein accumulation but did not afect the
phosphorylation status of EGFR or GPNMB (Fig. 4d).
Taken together, these data suggest a
linear
EGFR:GPNMB→LINK-A→BRK/LRRK2→HIF1α
signalling
cascade on HB-EGF stimulation under normoxic
conditions.
On HB-EGF stimulation, HIF1α underwent Tyr 565
and Ser 797
phosphorylation but the hydroxylation at Pro 564 was
inhibited, which led to HIF1α stabilization (Fig. 4g).
Knockdown of LINK- A abolished HB-EGF-induced
HIF1α Tyr 565 phosphorylation and enhanced the Pro
564 hydroxylation (Fig. 4h and Supplementary Fig.
4d). A similar pattern was observed with EGFR,
GPNMB and
BRK knockdown (Fig. 4i). These data suggest that
HB-EGF triggers an lncRNA-dependent signalling pathway
to stabilize HIF1α at the protein level.
Tyr 565 phosphorylation antagonizes Pro 564 hydroxylation to
stabilize HIF1α under normoxia
An in vitro hydroxylation assay demonstrated that the
HIF1α peptides (amino acids 557–566) but not
Tyr-565-phosphorylated peptides can be hydroxylated by PHD1
(Fig.
4j
and
Supplementary Fig.
4e–j
and
Supplementary Table 3). An in vitro kinase assay
followed by an in vitro hydroxylation assay further
showed that phosphorylation of wild- type HIF1α but
not the Y565F mutant by BRK prevented subsequent
hydroxylation at Pro 564 (Fig. 4k). Consistently,
HB-EGF-triggered Tyr 565 phosphorylation of HIF1α and
inhibition of hydroxylation at Pro 564, which was
abolished by overexpression of the Y565F mutant of
HIF1α (Fig. 4l).
A cycloheximide treatment experiment revealed that
on HB-EGF
stimulation, the HIF1α protein exhibited ≥4 h half-life
but knocking
down LINK-A reduced it to 1.5 h (Supplementary
Fig. 4k,l). In
TNBC cells exogenously expressing wild-type HIF1α or
the Y565D
mutant, the Y565D mutant exhibited a constitutively
prolonged half- life (Supplementary Fig. 4m–o). These
data
indicate
that
LINK-A- associated
BRK
phosphorylated HIF1α at Tyr 565, which prevents
HIF1α hydroxylation at adjacent Pro 564 and
stabilizes HIF1α
under
normoxia.
LINK-A-recruited LRRK2 phosphorylates Ser 797 of HIF1α to
potentiate its transcriptional activity
Knockdown of LINK-A or LRRK2, or overexpression of
the HIF1α
S797A mutant abolished Ser 797 phosphorylation of
HIF1α as well
as its association with p300, which was concurrent
with the release
of FIH (ref. 49), a protein that binds to HIF1α and
inhibits its
trans-activation function (Fig. 5a,b). We also examined the
kinase activity of LRRK2 in the presence of LINK-A,
finding that full-length LINK- A, ßBRK LINK-A or
ßLRRK2 LINK-A exhibited minimal efect on the kinase
activity of LRRK2 (Supplementary Fig. 5a). The rescue
experiments indicated that full-length LINK-A fully
rescued
HIF1α
phosphorylation and
protein
stabilization; ßBRK LINK-A rescued only HIF1α Ser 797
phosphorylation and ßLRRK2 LINK-A restored HIF1α Tyr
565 phosphorylation and protein stabilization, but
failed to rescue the phosphorylation of HIF1α at Ser
797 (Fig. 5c). Recent studies have shown that certain
lncRNAs encode small protein pep- tides
50–52. Whereas
our data have demonstrated that a predicted ORF of
LINK-A has no protein-coding products in vitro (see
Supplemen- tary Fig. 1a–c), we further mutated the
predicted translational start codon ATG (nucleotides
318–321), or the potential stop codon TGA
(nucleotides 732–735), of this ORF in a functional
rescue experi- ment, finding that the phosphorylation
of BRK (Tyr 351) and HIF1α (Tyr 565), two major
cellular efects mediated by LINK-A, was fully
rescued by wild-type LINK-A as well as ATG→TAG or
TGA→TGT
mutants of LINK-A (Supplementary Fig. 5b–d). These
observations
suggested that the cellular efect of LINK-A is mainly
dependent on its
RNA function instead of the potential translational
products. Taken together, we demonstrated that
LINK-A, in coordination with two protein kinases BRK and
LRRK2, mediated a growth factor-triggered
signalling cascade to synergistically regulate the
phosphorylation and protein stabilization of HIF1α
under normoxia.
LINK-A-dependent normoxic HIF1α signalling promotes tumour
growth and correlates with TNBC
Next, we examined the transcriptional activity of
HIF1α on HB- EGF stimulation by ChIP-seq, finding that
under normoxia, HB-EGF triggered the recruitment of
HIF1α to the promoters of HIF1α target genes and
regulated
the
HIF1α-dependent
transcriptional
program (Fig. 5d,e and Supplementary Table 4).
Knockdown of LINK-A in TNBC cells impaired
HIF1α-target gene expression on HB-EGF stimulation (Fig.
5f,g and Supplementary Fig. 5e). Consistently, in vitro
glucose uptake and lactate production assays
confirmed that LINK-A deficiency impaired glycolysis
(Supplementary Fig. 5f–l). Consistent with the in vitro
colony formation assays (Fig. 5h), mice with
xenografts of LINK-A-depleted tumour cells rarely
developed tumour mass in vivo (Fig. 5i,j and
Supplementary Fig. 5m).
The LINK-A-mediated signalling pathway was also
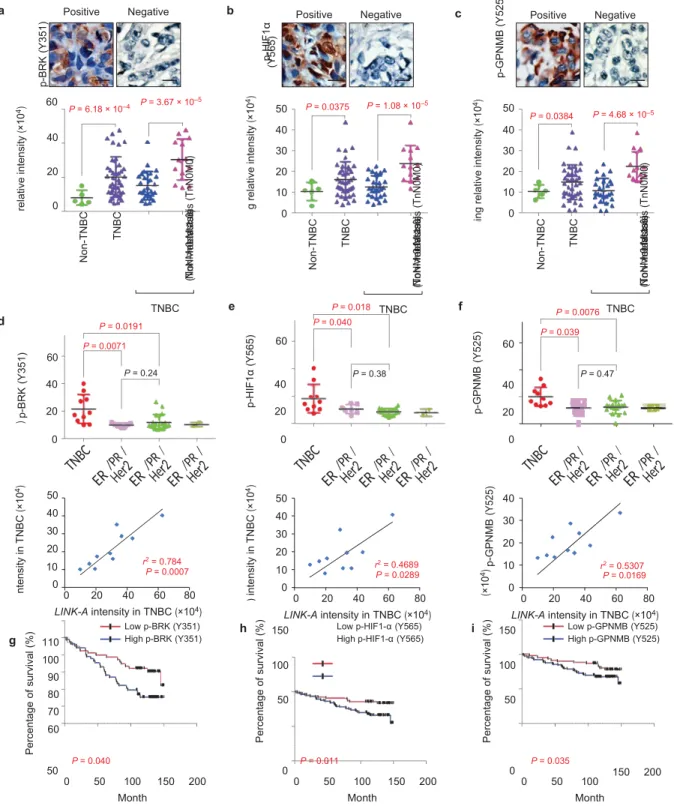
activated in TNBC tissues, as evidenced by a
significantly higher staining density of
GPNMB (Tyr 525), BRK (Tyr 351),
phospho-HIF1α (Tyr 565) and phospho-phospho-HIF1α (Ser 797) in TNBC
samples compared with non-TNBC samples (Fig. 6a–c
and Supplementary Fig. 6a). Furthermore, within the
TNBC category, breast cancer with advanced
lymph-node metastasis showed increased phospho-BRK (Tyr
351), phospho-HIF1α (Tyr 565) and phospho-GPNMB
(Tyr 525) levels compared with tissue samples with no
lymph-node metastasis (Fig. 6a–c,d–f, upper panel).
Importantly, there is a strong correlation
a siRNA HB-EGF MG-132 – + – + – + b c MG-132 Myc–HIF1α Blank WT S797A
HB-EGF – + – + – + LNA Scr LINK-A LINK-A no. 5 d P = 0.000001 2 IB: HIF1α IB: p-HIF1α (Ser 797) IB: p300 IB: FIH IB: p300 IB: FIH MDA-MB-231 IB: HIF1α IB: p-HIF1α (Ser 797) IB: p300 IB: FIH IB: FIH IB: p300 MDA-MB-231 HB-EGF IB: HIF1α IB: p-HIF1α (Tyr 565) IB: p-HIF1α (Ser 797) IB: GAPDH – + – + – + – + – + MDA-MB-231 1 0 1 2 3 4 5 6 2 1 0 1 2 3 4 5 6
e Signalling pathway of DE gene
HIF1α signalling pathway Fructose and mannose metabolism Glycolysis/gluconeogenesis Biosynthesis of amino acids Carbon metabolism Oocyte meiosis Progesterone-mediated oocyte maturation
f 0.40 ∗ 0.35 0.30 0.25 0.20 0.15 0.10 0.05 0 ChIP: Anti-HIF1α ∗∗ ∗ Anti-HIF1α IgG ∗ Long-term depression Protein expor HB-EGF – + – + – + – + – + – + – + – + g 25 ∗ 20 15
Enrichment score (–log10(P value))
∗ MDA-MB-231 –HB-EGF +HB-EGF 10 ∗ 5 ∗ 0 siRNA ∗ ∗ ∗ ∗ ∗ ∗ NS
ANGPTL4 ALDOA ANKRD37 BHLHE40 EGR1 IGFBP3 LDHA MAPK1 SLC16A3 PKM2 RPLP0
h 200 ∗ 150 i shRNA Ctrl j 0.5 0.4 0.3 ∗∗ ∗∗ 100 50 LINK-A no. 2 Ctrl LINK-A no. 3 0.2 0.1 0 –0.1 0 shRNA
Figure 5 LINK-A-recruited LRRK2 phosphorylates HIF1α at Ser 797, enhances HIF1α transcriptional activity and promotes tumour growth. (a,b) Immunoprecipitation (IP) followed by immunoblot (IB) detection using the indicated antibodies in MDA-MB-231 cells transfected with the indicated siRNAs (a) or plasmids (b), and treated with MG-132 followed by HB-EGF treatment. (c) Immunoblot detection using the indicated antibodies in MDA-MB-231 cells transfected with LNA against LINK-A followed by overexpression of the indicated rescue plasmids and HB-EGF stimulation. The dotted line indicates the position where the images of single blots were vertically cropped to juxtapose non-adjacent lanes. (d) HIF1α ChIP-seq analysis showing the top enriched HIF-binding consensus motifs. (e) HIF1α ChIP-seq analysis showing signalling pathways in MDA-MB-231 cells treated
with HB-EGF. (f,g) ChIP–qPCR detection of HIF1α occupancy on indicated target gene promoters (f) and qRT–PCR analysis of HIF1α target genes expression (g) in MDA-MB-231 cells transfected with control or
LINK-A siRNLINK-A followed by HB-EGF treatment. (h) Colony formation assay in
MDA-MB-231 cells transduced with control and LINK-A shRNAs. Scale bars, 200 µm. For f–h, error bars, s.e.m.; n = 3 independent experiments
(∗ P < 0.05 and ∗∗ P < 0.01, two-tailed paired Student’s t -test). (i,j) In
vivo
analyses of tumour growth (i) or weight (j) in mice that were subcutaneously injected with MDA-MB-231 cells harbouring control or LINK-A shRNA. Data are mean ± s.e.m., n = 5 mice per group (∗∗ P < 0.01, two-tailed
t siRNA Ctrl LINK-A Ctrl LINK-A Ctrl LINK-A Ctrl LINK-A
0 1 2 3 4 5 6 Promoter ANKRD37 ARRDC3 EGLN3 ERRFI1
In p u t IP : H IF 1α sh R N A 3 sh R N A 2 C tr l R el at iv e ex pr es si on l ev el N um be r of c o lo n ie s In p u t IP : M yc -t a g P er ce nt ag e o f in p u t V ec to r V ec to r F L ΔB R K Δ LR R K 2 T um ou r w ei gh t (m g ) B its B its
paired Student’s t -test). Unprocessed original scans of blots are shown in
a Positive Negative b Positive Negative c Positive Negative 60 P = 6.18 × 10–4 40 20 0 P = 3.67 × 10–5 50 P = 0.0375 40 30 20 10 0 P = 1.08 × 10–5 50 P = 0.0384 P = 4.68 × 10–5 40 30 20 10 0 d P = 0.0191 P = 0.0071 60 TNBC e P = 0.018 P = 0.040 60 TNBC f P = 0.0076 P = 0.039 60 TNBC P = 0.24 P = 0.38 P = 0.47 40 40 40 20 20 20 0 0 0 50 40 30 20 10 r2 P = 0.0007= 0.784 0 0 20 40 60 80 LINK-A intensity in TNBC (×104) 50 40 30 20 10 rP = 0.02892 = 0.4689 0 0 20 40 60 80 LINK-A intensity in TNBC (×104) 40 30 20 10 r2 = 0.5307 P = 0.0169 0 0 20 40 60 80 LINK-A intensity in TNBC (×104) g 110 100 90 80 70 60 Low p-BRK (Y351) High p-BRK (Y351) h 150 100 50
Low p-HIF1-α (Y565)
High p-HIF1-α (Y565) i 150
100 50 Low p-GPNMB (Y525) High p-GPNMB (Y525) P = 0.040 P = 0.011 P = 0.035 50 0 50 100 Month 0 150 200 0 50 100 Month 0 150 200 0 50 100 Month 150 200
Figure 6 The LINK-A-dependent normoxic HIF1α signalling pathway correlates with TNBC. (a–c) Immunohistochemical staining using antibodies against phospho-BRK (Tyr 351) (a), phospho-HIF1α (Tyr 565) (b) or phospho-GPNMB (Tyr 525) (c) in human breast cancer tissues. Upper panel: representative images (scale bars, 100 µm; lower panel: statistics analysis based on non-TNBC tissues (n = 5) versus TNBC tissues (n = 40) and non-metastasis (TnN0M0) TNBC (n = 27) versus metastasis
(TnN > 0 M ≥ 0) breast tissues (n = 13) (median, two-way ANOVA). (d–f) Upper panel: statistical analysis of immunohistochemical staining using antibodies against phospho-BRK (Tyr 351) (d), phospho-HIF1α
(Tyr 565) (e) or phospho-GPNMB (Tyr 525) (f) in human breast cancer tissues including TNBC (n = 10), ER− /PR− /HER2+ (n = 7), ER+ /PR+ /HER2− (n = 18), and ER+ /PR+ /HER2+ (n = 2) (median, two-way ANOVA). Lower
panel: Pearson’s correlation analysis comparing staining density between
LINK-A expression and phospho-BRK (Tyr 351) (d), phospho-HIF1α (Tyr 565)
(e) or phospho-GPNMB (Tyr 525) (f) within the TNBC group (n = 10 tissue samples, Fisher’s exact test). (g–i) Kaplan–Meier survival analysis of phosphor-BRK (Tyr 351) (g), phosphor-HIF1α (Tyr 565) (h) and phospho-GPNMB (Tyr 525) (i) status in breast cancer patients (n = 160, Gehan–Breslow test). p-B R K ( Y 35 1) in te ns ity in T N B C (× 10 4) p-B R K ( Y 35 1) in te ns ity (× 10 4) p-B R K ( Y 35 1) s ta in in g re la tiv e in te ns ity (× 10 4) P er ce nt ag e o f su rv iv al ( % ) p-B R K ( Y 35 1) N on -T N B C T N B C N on - m et as ta si s (T nN 0M 0) M et as ta si s (T nN > 0 M ≥ 0 ) p-H IF 1α ( Y 56 5) in te ns ity in T N B C (× 10 4) p-H IF 1α ( Y 56 5) p-H IF 1α ( Y 56 5) s ta in in g re la tiv e in te ns ity (× 10 4) P er ce nt ag e of s ur vi va l ( % ) p-H IF 1α (Y 56 5) N on -T N B C T N B C N o m et as ta si s (T nN 0M 0) M et as ta si s (T nN > 0 M ≥ 0 ) p-G P N M B ( Y 52 5) in te ns ity in T N B C (× 10 4) p-G P N M B ( Y 52 5) p-G P N M B ( Y 52 5) s ta in in g re la tiv e in te ns ity (× 10 4) P er ce nt ag e of s ur vi va l ( % ) p-G P N M B ( Y 52 5) N on -T N B C T N B C N on - m et as ta si s (T nN 0M 0) M et as ta si s (T nN > 0 M ≥ 0 )
between LINK-A expression and the phosphorylation
status of BRK, HIF1α and GPNMB in these TNBC tissues
(Fig. 6d–f, lower panel), and breast cancer patients
with higher levels of these phosphoproteins
exhibited a shorter survival time (Fig. 6g–i).
Furthermore, the TCGA database revealed that both
BRK and LRRK2 are highly expressed in invasive breast
carcinoma (Supplementary Fig. 6b). Our data implicate
LINK-A and its associated signalling pathway as
potential biomarkers and therapeutic targets for TNBC.
DISCUSSION
Our study reveals that lncRNA directly interacts with
non-receptor tyrosine kinase and facilitates its
recruitment to the membrane-bound receptor complex
and subsequent activation on ligand stimulation,
broadening the known mechanisms of lncRNA action
(Fig. 6j). The regulatory mechanism of non-receptor
tyrosine kinase activation is largely unknown. We
propose a model in which LINK-A interacts with
non-receptor tyrosine kinases to facilitate their activation.
At the basal level, BRK, a prototype RNA-binding
non-receptor tyrosine kinase, is in a ‘closed’ conformation
and its kinase activity is auto-inhibited, mediated by
the self-inhibitory interaction between the SH2 domain
and the phospho-C-terminus (Tyr 447; ref. 46). The
binding of LINK- A to both the SH3 domain and the
C-terminal region of BRK leads to a more accessible
structure of BRK, which may contribute to higher
accessibility by other regulatory proteins and kinases
for its activation.
Most common cancer types show increased HIF1α
protein levels although hypoxic areas are missing
53,54.
Our study delineates an lncRNA–protein kinase module
that regulates normoxic HIF1α stabilization with
respect to functional implications in glycolytic
reprogramming and tumorigenesis. The
LINK-A-dependent
HIF1α signalling cascade and the
consequent efects on cancer cell glycolysis implicate
LINK-A and LINK-A-interacting kinases/receptors as
promising therapeutic targets for TNBC. Analyses of
the LINK-A expression status in the TCGA database and
breast cancer tissues both indicated that LINK-A
significantly correlates with TNBC, revealing an
lncRNA that can serve as a biomarker for further
classification of TNBC.
Our study identifies four previously unknown
phosphorylation sites of GPNMB, BRK and HIF1α in
a LINK-A-regulated signalling pathway for glycolysis
reprogramming in TNBC. These phosphorylation
events predict a worse outcome in TNBC patients,
suggesting that the LINK-A-dependent signalling
pathway plays a critical role in TNBC and may
provide wide-ranging therapeutic
targets
for
treating
TNBC.
口
METHODS
Methods and any associated references are available
in the
n
o
li n
e
v
e
r si
o
n
o
f t h
e
p
a
p
er
.
ACKNOWLEDGEMENT S
We thank S. Kopetz for providing cetuximab and J. Chen for providing SFB-tagged
expression vector. We thank D. Aten for assistance with figure presentation. This
work was supported by the NIH R00 award (R00DK094981), UT Startup and UT
STARS grants to C.Lin, and the NIH R00 award (R00CA166527), CPRIT award
(R1218), UT Startup and UT STARS grants to L.Y.
AUTHOR CONTRIBUTIONS
C.Lin, L.Y. and A.L. designed the research, and A.L., C.Li and Z.X. performed
most of the experiments, with participation of K.L., S.W., Q.H., Y.Zhang, G.M.
and Yubin Z. D.H.H. executed mass spectrometry analysis. Clinical specimens were
ascertained and processed by S.W., J.Z., Yan Z. and J.R.M. The histological staining
and corresponding analysis were performed by K.L. and Y.W. P.K.P. helped with
manuscript preparation. TCGA data and microarray data analysis was performed
by C.W., Z.H., L.H. and H.L. M.-C.H. provided reagents and conceptual advice L.Y.,
C.Lin and A.L. wrote the manuscript.