行政院國家科學委員會專題研究計畫成果報告
活性氧族群及一氧化氮於心臟細胞中調控內皮素所誘發乙型肌凝
重鍊蛋白基因所扮演的角色
Role of r eactive oxygen species and nitr ic oxide in modulation of
endothelin-1-induced beta-myosin heavy chain gene expr ession in
car diomyocytes.
計畫編號:NSC
90-2320-B-002-144-執行期限:90 年 8 月 1 日至 91 年 7 月 31 日
主持人:陳錦澤 台大醫學院內科
一、中文摘要 大多數心血管方面的疾病,都會造成 心臟的肥大,初期的心臟肥大可以代償心 臟功能,然而持續性的心臟肥大,便會導 致心臟的衰竭。在心臟細胞,由於機械應 力或內生性生長因子的刺激作用下,會造 成心臟細胞的肥大,心臟細胞的過度肥大 有可能導致心臟機能受損,進一步引起心 臟衰竭。內皮素(endothelin-1; ET-1)有 造成細胞肥大的作用,但對於此作用其細 胞內的機轉目前還不是很清楚,已知內皮 素可增加許多迅即基因如 c-fos、c-jun 及 egr-1 等的基因表現;另外,對一些與心 臟 細 胞 的 增 大 有 關 的 基 因 如 心 房 利 鈉 (atrial natriuretic peptide)、肌凝蛋 白重鏈(myosin heavy chain)、骨骼肌肉 肌動蛋白(actin)等,也有增加它們基因表 現的作用。近 來 有許多研究報告指出活性 氧族群(reactive oxygen species ; ROS)及 一氧化氮(nitric oxide; NO)可於細胞內 扮演一訊號傳遞者的角色,所以在本研究 中,我們進一步觀察活性氧族群於內皮素 所誘發細胞肥大現象所扮演的角色,並進 而闡釋其可能的細胞內機轉。由本報告抗 氧化劑可抑制內皮素所誘發心臟細胞肥 大,可推測抗氧化劑,在臨床上相關心臟 肥大或心臟衰竭疾病的預防治療上具有運 用潛力。 關鍵詞:內皮素、活性氧族群、乙型重鏈 基因、心臟細胞、細胞肥大 AbstractOBJ ECTIVES: We dissected the molecular
regulatory mechanism of reactive oxygen species (ROS) on endothelin-1-(ET-1)-induced β-myosin heavy chain (β-MyHC) gene expression and hypertrophic signaling in neonatal rat cardiomyocytes.
BACKGROUND: ET-1 causes hypertrophy
in the cardiomyocytes. Expression of cardiac
β-MyHC gene is increased in response to ET-1. Our previous study has demonstrated that ET-1 increases intracellular reactive oxygen species (ROS) in cardiomyocytes. However, the intracellular regulatory mechanism of ROS on ET-1-induced
β-MyHC gene expression and cardiac hypertrophy still remains unclear.
METHODS: Cultured neonatal rat cardiomyocytes were stimulated with ET-1,
3
H-leucine incorporation and the β-MyHC gene promoter activities were examined. We also examined the effects of antioxidant pretreatment on ET-1-induced cardiac hypertrophy and MAPK activties to elucidate the redox-sensitive pathway in cardiomyocyte hypertrophy and β-MyHC gene expression.
RESULTS: 3H-Leucine incorporation and
β-MyHC promoter activities were increased by ET-1. These ET-1 effects were blocked by the specific ETA receptor antagonist BQ-485.
Cardiomyocytes treated with antioxidants significantly reduced ET-1-induced
3
H-leucine incorporation and β-MyHC gene promoter activities. ET-1 activated mitogen-activated protein kinases (MAPKs; extracellular signal-regulated kinase [ERK], p38, and JNK), which were significantly inhibited by antioxidants. Either PD98059 or
SB203580 inhibited ET-1-increased
3
H-leucine incorporation, but only PD98059 decreased ET-1-induced β-MyHC promoter activities. Co-transfection of dominant negative mutant of Ras, Raf, and MEK1 decreased the ET-1-increased β-MyHC promoter activities, suggesting that the Ras-Raf-ERK pathway is required for ET-1 action. Deletion mapping revealed that the deletion construct containing 273 base pairs (bp) of β-MyHC 5’-flanking sequences upstream from the transcription initiation site is necessary and sufficient for ET-1-induced increase of transcription, whereas the construct containing only 188 bp of 5’-flanking region had no effect, indicating the ET-1-responsive element(s) is located between position -273 and –188 bp upstream from the transcription start site. This minimal construct of 273 bp upstream of transcription initiation site in the β-MyHC promoter region is also required for ROS-induced β-MyHC expression.
CONCLUSIONS: These data demonstrate
that ROS involved in ET-1-induced hypertrophic responses, β-MyHC expression and mediated ET-1-induced activation of MAPK pathways, which culminated in hypertrophic responses and β-MyHC expression. The ROS-MAPK (ERK)-meditated pathway plays an essential role in ET-1-induced hypertrophic responses and β-MyHC expression in cardiomyocytes.
Keywords: endothelin-1 • reactive oxygen
species • beta-myosin heavy chain gene • cardiomyocyte • hypertrophy
二、緣由與目的
Endothelin-1 (ET-1), a 21-amino acid peptide, is the most potent and long-lasting vasoconstrictor (1). It is also a hypertrophy-promoting factor for various cells including cardiomyocytes (2). There is a prototypical final molecular response of cardiomyocytes to hypertrophic signals that involves an increase in cell size and protein synthesis, enhanced sarcomere organization, and re-expression of embryonic cardiac genes,
β-myosin heavy chain (β-MyHC) (3). The
cause and effect of cardiac hypertrophy have been extensively documented, but the underlying molecular mechanisms that couple hypertrophic signals initiated at the cell membranes to the reprogramming of cardiomyocyte gene expression remains poorly understood. Previously, we reported that ET-1 induced hypertrophic responses by stimulating β-MyHC gene expression in cardiomyocytes (4). However, the molecular mechanism involved in ET-1-induced cardiac hypertrophy and β-MyHC transcriptional control are not well defined.
Recent evidence indicates that reactive oxygen species (ROS) function as intracellular messengers to modulate signaling pathways (5). ROS encompass many oxygen species including singlet oxygen, superoxide, hydrogen peroxide (H2O2) and hydroxyl radicals that are
produced by virtually every type of cell (5). Cells possess several antioxidant systems, including cellular reductants, such as glutathione (GSH), and cellular antioxidant enzymes, such as catalase, which act against ROS. Recent studies have revealed that regulation of the cellular redox state plays an important role in cellular signaling systems linked to cell growth and cell differentiation (5). Excess ROS have been reported to be involved in pathogenesis of cardiac hypertrophy (6,7). The involvement of ROS is implicated in the transition of compensated hypertrophy to heart failure (6). In the late stage of cardiac hypertrophy, ROS appear to be toxic for myocardium and induce myocardial dysfunction or injury (7). Cardiac hypertrophy is an adaptive response of the heart to virtually all forms of cardiac diseases. Conversely, very few studies have evaluated the molecular mechanism of ROS in the early stage of cardiac hypertrophy. Of interest, we (8) and others (9,10) have shown that ET-1 increases intracellular ROS in cardiomyocytes. Studies have demonstrated that angiotensin II (Ang II), tumor necrosis factor-α and noradrenalin cause hypertrophy in part via the generation of ROS in cardiomyocytes (10-11). We also found that ROS are involved in the Ang II-induced
β-MyHC gene expression in cardiomyocytes (12). These studies suggest that ROS are
involved in the hypertrophic signaling. However, the molecular mechanism of how ROS are involved in the signaling pathways of cardiac hypertrophy is not well defined. Furthermore, the role of redox-sensitive pathways in ET-1-induced cardiomyocyte hypertrophy and β-MyHC gene expression remains to be elucidated.
In this study, we investigated the role of redox-sensitive mechanisms on ET-1-induced cardiomyocyte hypertrophy and β-MyHC gene expression. We further determined which redox-sensitive signal transduction pathways were involved in this process. Cardiomyocyte hypertrophy was monitored by the increase in protein synthesis and the enhanced expression of embryonic gene β-MyHC. We show that ROS involved in ET-1-induced cardiomyocyte hypertrophy and β-MyHC gene expression. The ROS- extracellular signal-regulated kinase (ERK)-meditated pathway plays an essential role in ET-1-induced hypertrophic responses and
β-MyHC expression in cardiomyocytes.
三、結果與討論、
RESULTS
ET-1-increased 3H-leucine incor por ation
via ETA receptor s
Rat neonatal cardiomyocytes cultured in serum-free condition were treated with different concentrations of ET-1 (0.1-100 nM) for 48 h. The effect of ET-1 on protein synthesis was analyzed by measurement of
3
H-leucine incorporation into the myocytes. ET-1 increased 3H-leucine incorporation in myocytes (Figure 1A). The effects of ETA or
ETB receptor antagonist on the
ET-1-increased protein synthesis were investigated (Figure 1B). Pretreatment with ETA receptor antagonist BQ485 (100 nM) for
30 min significantly inhibited the ET-1-increased 3H-leucine incorporation, whereas ETB receptor antagonist BQ788 (100
nM) had no obvious effect, indicating that ET-1-induced increase of protein synthesis in cardiomyocytes was via ETA receptors.
ET-1-induced β-MyHC gene expression via ETA r eceptor s
ET-1 has been demonstrated to
stimulate β-MyHC gene expression in cardiomyocytes (4). As shown in Figure 2A, incubation with ET-1 (1, 10, 100 nM) dose-dependently increases β-MyHC gene promoter activities in the cultured cardiomyocytes. We further determine which ET-1 receptor subtype is responsible for the ET-1-induced β-MyHC gene expression. The effects of ETA or ETB receptor antagonist on
the ET-1-induced β-MyHC gene expression were investigated (Figure 2B). Pretreatment of myocytes with BQ485 (100 nM) but not BQ788 (100 nM) abolished ET-1-induced
β-MyHC gene expression (Figure 2B). These data indicate that ET-1 increases β-MyHC gene expressionvia ETA receptors.
Redox regulation of ET-1-induced hypertrophic responses and β-MyHC gene promoter activities in cardiomyocytes
To unravel the role of ROS in modulation of ET-1-induced hypertrophic responses, we examined the effect of antioxidants on cardiomyocyte hypertrophy induced by ET-1. As shown in Figure 3A, cells exposed to ET-1 (10 nM) or H2O2 (25
µM) for 48 h, 3H-leucine uptake was increased by 2.0 ± 0.5- and 1.6 ± 0.5-fold (n = 5) over the basal, respectively, and inhibited by antioxidants NAC (10 mM), catalase (350 U/mL), or the NADH/NADPH oxidase inhibitor diphenyleneiodonium (DPI; 10 µM), respectively. To further determine whether the antioxidants affect the ET-1-induced
β-MyHC gene expression, either catalase (350 U/ml) or NAC (10 mM) was pretreated before ET-1 stimulation. As demonstrated in Figure 3B, neither catalase nor NAC had any effect on the basal β-MyHC promoter activity. However, cardiomyocytes treated with ET-1 (10 nM) or H2O2 (25 µM) for 48 h led to a
2.3- and 1.9-folds increase in CAT activity respectively, as compared to control unstimulated cells. In the presence of catalase or NAC, ET-1-induced β-MyHC promoter activities were significantly inhibited. These data clearly indicate that ROS involved in ET-1-induced hypertrophic responses and
ET-1–induced protein synthesis and β-MyHC expression are mediated via the Ras/Raf/ERK pathway
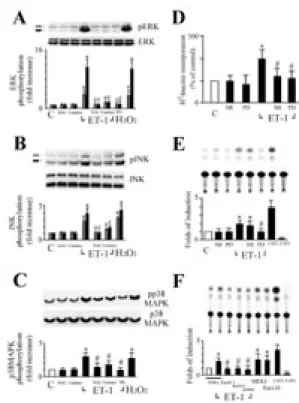
To define the mechanism of how ROS modulate hypertrophic signaling, we examined the effects of antioxidants on ET-1-induced MAPK activity. ERK was rapidly activated in cardiomyocytes exposed to ET-1 (10 nM) or H2O2 (25 µM), consistent
with previous reports (9). The ERK phosphorylation was observed at 30 min after exposure, and the -fold increase over the basal was 11.4 ± 2.2 (ET-1) or 10.7 ± 1.9 (H2O2), respectively (Figure 4A, n = 4). Two
subfamily members of the MAPK family, p38 and JNK were also activated by ET-1 or H2O2
but to a lesser degree than that of ERK activation. In the presence of 10 nM ET-1, the increase (-fold) in p38 and JNK activity was 2.9 ± 0.6 and 3.7 ± 0.6, respectively (Figure 4B and 4C, n = 4). Similar increased effects were also observed in the cells treated with 25 µM H2O2. All the increased activities
of ERK, p38, or JNK induced by ET-1were significantly suppressed by catalase or NAC treatment (Figure 4A, 4B and 4C). These findings suggest that ERK, p38, and JNK are critical components of the redox-sensitive signaling pathways activated by ET-1 in cardiomyocytes. We next determined the role of redox-sensitive activation of MAPK in ET-1-induced protein synthesis. Both PD98059, a specific inhibitor of MKK-1 (MEK), and SB203580, a specific inhibitor of p38, inhibited augmentation of 3H-leucine incorporation into the myocytes after ET-1 stimulation. (Figure 4D). Surprisingly, coincubated with PD98059 but not SB203580 abolished ET-1-increased β-MyHC promoter activity (Figure 5E). These findings suggest that activation of ERK is a necessary step for cardiomyocyte hypertrophy and β-MyHC gene expression induced by ET-1. In order to identify the signaling pathway involved in the ET-1-induced β-MyHC expression, we transient transfected cardiomyocytes with various dominant negative mutant, Ras (RasN17) or Raf-1 (Raf301) or a catalytically inactive mutant of ERK2 (mERK), all of which are associated with the Ras/Raf/ERK pathway. When cardiomyocytes cotransfected with the empty vector control PSRα revealed
no effect on ET-1-induced β-MyHC promoter activity (Figure 4F). However, cells cotransfected with RasN17, Raf301 or mERK resulted in a significant inhibition in ET-1-induced β-MyHC promoter activity. In contrast, cardiomyocytes cotransfected with a dominant positive mutant of Ras (RasL61) or MEK1 greatly increased their β-MyHC promoter activity. These results suggest that the Ras/Raf/ERK signaling pathway is involved in ET-1-induced β-MyHC gene expression in cardiomyocytes.
Localization of ET-1- or ROS-responsive regulator y elements in the β-MyHC promoter
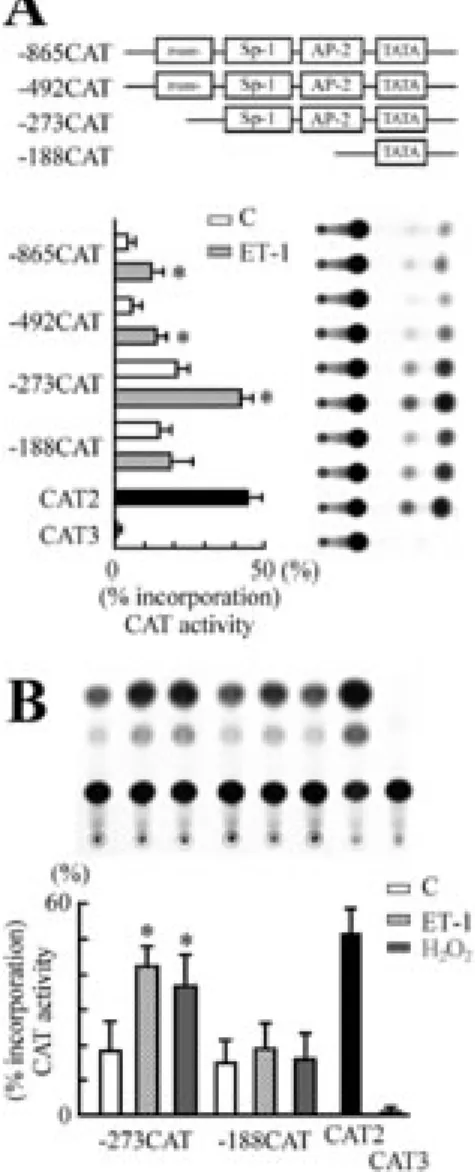
To localize the 5’ cis-acting elements in regulating the induction of the β-MyHC gene by ET-1, a series of recombinant plasmids containing upstream promoter deletion mutants linked to the CAT reporter gene were transfected into cardiomyocytes, and subsequently analyzed for CAT activities in the presence or absence of ET-1. At basal transcription, deletion mapping revealed the presence of a strong negative element in the region (-492/-273) of the β-MyHC promoter (Figure 5A). All the fusion plasmids containing -865/+77, –492/+77, or –273/+77 of the β-MyHC 5‘-flanking sequences significantly enhanced CAT activity in response to ET-1 (1.9-, 2.1-, and 2.2-fold induction respectively), whereas deletion up to –188 bp from the start site failed to respond to ET-1 stimulation (Figure 5A). These results suggest the presence of ET-1-responsive cis-acting regulatory element(s) is located between positions –273 and –188 bp upstream from the transcription start site. To further confirm whether ROS mediated ET-1-induced β-MyHC gene expression in the transcription regulation level, a plasmid containing a 273-base pair region 5’ of the start site in a CAT reporter construct was cotransfected with pSV- β ‘-galactosidase into cardiomyocytes.
β-MyHC promoter activities were increased in cardiomyocytes exposed to ET-1 (10 nM) or H2O2 (25 µM). On the contrary, ET-1 (10
nM) or H2O2 (25 µM) failed to increase the
promoter activities of β-MyHC with the minimal construct (-188/+77) (Figure 5B).
These results suggested the presence of an ET-1- or ROS-responsive regulatory element within the 273 bp.
DISCUSSION
In the present study, we demonstrated the roles of ROS-activated MAPK in ET-1-induced cardiomyocyte hypertrophy and β-MyHC gene expression. We showed that ROS mediate ET-1-induced β-MyHC gene expression and cardiomyocyte hypertrophy via the activation of ERK phosphorylation in cultured cardiomyocytes. Generation of ROS by ET-1 leads to the activation of ERK, which results in cardiac hypertrophy and β-MyHC gene expression.
In the present work, we provide evidence for a role of myocardial ETA
receptors in ET-1-induced β-MyHC gene expression and cardiomyocyte hypertrophy (Figure 1 & 2). These findings are consistent with previous study that ET-1-induced hypertrophic responses were completely blocked by pretreatment with ETA antagonist,
but not by ETB antagonist (2). Recently, it has
become apparent that ROS play a role in the stress-induced signal transduction pathway (15). We and others have shown that ET-1 generates ROS in cardiomyocytes (8-10). Our previous work found that ET-1 increased intracellular ROS via ETA receptor in
cardiomyocytes (8). In the present study, we also observed that antioxidants abolished the increases in protein synthesis and the induction of β-MyHC gene by ET-1. These results suggested that ET-1 causes hypertrophy via generation of ROS in cardiomyocytes. Thus we can propose that ROS generation modulate ET-1-induced
β-MyHC gene expression and cardiomyocyte hypertrophy via ETA receptors.
Several intracellular signaling pathways have been implicated in the induction of cardiac hypertrophy including MAPKs signaling cascade. By using western blotting, ET-1-stimulated ERK1/2, JNK and p38 phosphorylation were suppressed by antioxidants (Figure 4A, 4B, and 4C). These data support the hypothesis that ROS regulate hypertrophic signaling at least in part by modulating the activities of ET-1-dependent MAPK pathways in cardiac myocytes. Yue et
al. (16) reported that the ERK signal pathway, which is redox-sensitive (9,12), plays an essential role in ET-1-induced cardiomyocyte hypertrophy. Recently, Hirotani et al (10) found that ROS-activated apoptosis signal-regulated kinase 1 functions as a stress-adaptation signaling intermediate to activate MKK7-JNK and MKK3/6-p38 pathways (17), upstream activators for p38 (18) and JNK (19), which culminantly elicit characteristic hypertrophic responses in cardiomyocytes. In this study, inhibition of endogenous ERK or p38 by PD98059 or SB203580 suppressed ET-1-increased protein synthesis in cardiac myocytes. PD98059 or SB203580 at 20 µmol/L, which fully inhibits the endogenous ERK or p38 activity (Figure 4A and 4C). However, only PD98059 but not SB203580 could inhibit ET-1-induced
β-MyHC gene expression. These data agree with a previous study that blockade of ERK activation inhibits ET-1-induced expression of β-MyHC in myocytes (16). A number of transcription factors have been identified as effectors of hypertrophic stimuli in driving fetal cardiac genes. Given the complexity and heterogeneity of transcriptional activation in response to hypertrophic stimuli, both integrated and parallel hypertrophic transcriptional response pathways operate to control hypertrophic gene expression. The potential interaction of the cardiotrophic transcription factors in the regulation of
β-MyHC gene expression remains to be elucidated. Raf and ERK cascades are reported to mediate ET-1-induced cardiomyocyte hypertrophy (2, 20). Ras, the small GTP-binding protein, is known to be involved in mediating the effects of ET-1 in several cell types including cardiomyocytes (8). In order to identify the signaling pathway involved in the ET-1-induced β-MyHC expression in cardiomyocytes, cotransfection with various dominant positive or negative mutants of Ras, Raf or ERK confirm that the Ras/Raf/ERK signaling pathway is involved in ET-1-stimulated β-MyHC expression in cardiomyocytes (Figure 4E). The data are in agreement with that obtained with selective Raf or MEK inhibitors, indicating that the Raf/MEK/ERK pathway plays a critical role in myocyte hypertrophy (16). Taken together,
we conclude that ROS modulation of MAPKs activation exert a regulatory effect in ET-1-induced cardiomyocyte hypertrophy. This finding not only confirms a heretofore appreciated function of ROS in MAPKs activation and modulation the subsequent development of cardiac hypertrophy but also unravels a unique mechanism for the regulation of β-MyHC gene by ERK, which may have significant implications in the pathophysiology of heart failure.
The α- and β-MyHC genes are tandemly linked and regulated independently. Previous study indicates that putative regulatory sequences in the 5’-flanking region situated within 600 nucleotides upstream of the putative TATA box (21). The recombinant construct –1306/+77 contains the entire necessary promoter region that are sufficient for regulation of the β-MyHC gene expression by ET-1. As shown in Figure 5A, deletion series demonstrate that the strongest basal activity for the β-MyHC gene promoter was associated with the –273/+77 construct. A sharp increase in basal activity is observed upon removal of –492 to –273 region. This suggests the existence of a negative cis-acting regulatory element within this region that appears to be involved in determining the development-specific expression of the
β-MyHC gene. However, regardless of the presence of a repressor, deletion mapping revealed constructs containing –1306, -865, -492 or –273 bp allowed ET-1-induced transcription, whereas deletion containing –188 bp from the start site failed to respond to ET-1 induction, indicating the presence of ET-1-responsive cis-acting regulatory element(s), located between position –273 and –188 bp upstream from the transcription initiation site. This specific upstream sequence between –273 and –188 bp required for induction of β-MyHC gene by ET-1 corresponds to the region previously identified which contains a GATA motif at –269 to –264 bp (22) and a PKC response element at –215 to –196 bp (23). Previous studies have shown that inhibition of the ERK cascade with PD98059 blocked the phenylephrine-induced increase in the phosphorylation and the DNA binding ability of GATA protein (24). As shown in Figure
3B, both catalase and NAC inhibit ET-1- and H2O2-induced β-MyHC promoter activity,
raising the possibility that ROS via influencing ERK cascade could affect GATA protein to stimulate the gene expression. The molecular mechanism by which ET-1 through ROS in regulating β-MyHC gene expression remains to be investigated.
In our studies, antioxidants inhibit the effects of ET-1 on the activation of MAPKs signaling system. It is plausible that the ET-1 signaling pathway consists of redox-sensitive steps and that pretreatment with antioxidants could modulate the redox state of the cell. In summary, our data showed that antioxidants inhibit ET-1-induced β-MyHC gene expression and cardiomyocyte hypertrophy at least in part via the suppression of ET-1-activated ERK phosphorylation in a transcription-independent manner. These findings implicate the application of antioxidants in prevention of pathologic hypertrophy of the heart and protection of cardiac function. 四、計畫成果自評 由本報告抗氧化劑可抑制內皮素所 誘發心臟細胞肥大,可推測抗氧化劑在臨 床上相關心臟肥大或心臟衰竭疾病的預防 治療上具有運用潛力。 五、參考文獻
1. Yanagisawa M, Kurihara H, Kimura S, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988; 332: 411-415.
2. Yamazaki T, Komuro I, Kudoh S, et al. Endothelin-1 is involved in mechanical stress-induced cardiomyocyte hypertrophy. J Biol Chem 1996; 271: 3221-3228.
3. Chien KR, Knowlton KU, Zhu H, Chien S. Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiologic response. FASEB J 1991; 5: 3037-3046. 4. Wang DL, Chen JJ, Shih NL, et al. Endothelin stimulates cardiacá- and â-myosin heavy chain gene expression. Biochem Biophys Res Commun 1992; 183:
1260-1265.
5. Lander HM. An essential role for free radicals and derived species in signal transduction. FASEB J 1997; 11: 118-124. 6. Dhalla AK, Hill MF, Signal PK. Role of oxidative stress in transition of hypertrophy to heart failure. J Am Coll Cardiol 1996; 28:506-514.
7. Keith M, Geranmayegan A, Sole MJ, et al. Increased oxidative stress in patients with congestive heart failure. J Am Coll Cardiol 1998;31:1352-1356.
8. Cheng TH, Shih NL, Chen SY, Wang DL, Chen JJ. Reactive oxygen species modulate endothelin-1-induced c-fos gene expression in cardiomyocytes. Cardiovas Res 1999; 41: 654-662.
9. Tanaka K, Honda M, Takabatake T. Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. J Am Coll Cardiol 2001; 37: 676-685.
10. Hirotani S, Otsu K, Nishida K, et al. Involvement of nuclear factor-B and apoptosis signal-regulating kinase 1 in G-protein–coupled receptor agonist–induced cardiomyocyte hypertrophy. Circulation 2002;105:509-515.
11. Nakamura K, Kazuo F, Kouchi H, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-α and angiotensin II. Circulation 1998; 98: 794-799.
12. Shih NL, Cheng TH, Loh SH, et al. Reactive oxygen species modulate angiotensin II-induced β-myosin heavy chain gene expression via Ras/Raf/extracellular signal-regulated kinase pathway in neonatal rat cardiomyocytes. Biochem Biophys Res Commun 2001; 283: 143-148.
13. Lucknow B, Schutz G. CAT constructions with multiple unique restriction sites for the functional analysis of eukaryotic promoters and regulatory elements. Nucleic Acids Res 1987; 10: 5490.
14. Chen JJ, Shih NL, Hsu KH, Liew CC. Serial deletion constructs of human cardiac myosin heavy chain genes generated by PCR amplification. Mol Cellular Biochem 1993; 124: 81-84.
15. Adler V, Yin Z, Tew KD, et al. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999; 18:
6104–6111.
16. Yue TL, Gu JL, Wang C, et al. Extracellular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy. J Biol Chem 2000; 275: 37895-37901.
17. Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997; 275: 90–94.
18. Nemoto S, Sheng Z, Lin A. Opposing effects of Jun kinase and p38 mitogen-activated protein kinases on cardiomyocyte hypertrophy. Mol Cell Biol 1998; 18: 3518–3526.
19. Wang Y, Su B, Sah VP, et al. Cardiac hypertrophy induced by mitogen-activated protein kinase kinase 7, a specific activator for c-Jun NH2-terminal kinase in ventricular muscle cells. J Biol Chem. 1998; 273: 5423–5426.
20. Fuller SJ, Gillespie-Brown J, Sugden PH. Oncogenic src, raf, and ras stimulate a hypertrophic pattern of gene expression and increase cell size in neonatal rat ventricular myocytes. J Biol Chem 1998; 273: 18146-18152.
21. Yamauchi-Takihara K, Sole MJ, Liew J, Ing D, Liew CC. Characterization of human cardiac myosin heavy chain genes. Proc Natl Acad Sci USA 1989; 86: 3504-3508.
22. Morimoto T, Hasegawa K, Kaburagi S, et al. GATA-5 is involved in leukemia inhibitory factor-responsive transcription of the β-myosin heavy chain gene in cardiac myocytes. J Biol Chem 1999; 274: 12811-112818.
23. Kariya K, Karns LR, Simpson PC. An enhancer core element mediates stimulation of the rat beta-myosin heavy chain promoter by an alpha 1-adrenergic agonist and activated beta-protein kinase C in hypertrophy of cardiac myocytes. J Biol Chem 1994; 269: 3775-3782.
24. Morimoto T, Hasegawa K, Kaburagi S, et al. Phosphorylation of GATA-4 is involved in alpha 1-adrenergic agonist-responsive transcription of the endothelin-1 gene in cardiac myocytes. J Biol Chem 2000; 275: 13721-13726.
Figur e 1. ET-1 increased protein synthesis
via ETA in cardiomyocytes. (A)
ET-1-induced 3H-leucine incorporation into cardiomyocytes. Cardiomyocytes were cultured in 6-well plates and in serum-free medium for 24 h prior to the stimulation with different concentrations of ET-1 (0.1-100 nM) for 48 h. (B) ET-1-induced 3H-leucine incorporation into cardiomyocytes via ETA in
cardiomyocytes. Cardiomyocytes were cultured in 6-well plates and in serum-free medium for 24 h prior to the stimulation with ET-1 (10 nM) for 48 h in the presence of vehicle (control) or antagonists as indicated.
3
H-Leucine was added at 1 µCi per well. The cells were washed, fixed with cold 5% trichloroacetic acid, and the radioactivity incorporated into the trichloroacetic acid-precipitable material was determined. The data are mean ± SEM of five independent studies. *P<0.05 vs. control; #P<0.05 vs. ET-1 alone.
Figur e 2. Effect of ET-1 on β-MyHC promoter activity. (A) Induction of β-MyHC promoter activity by different concentration of ET-1 (1, 10, 100 nM). Cardiomyocytes were transfected with 15 µg of β-MyHCCAT chimeric gene for 24 h. After transfection, cardiomyocytes were then treated with ET-1 for 48 h. (B) Effect of ET-1 receptor antagonists on ET-1-induced β-MyHC gene expression. Cardiomyocytes were pretreated with either BQ485 (100 nM) or BQ788 (100 nM) for 30 min and then stimulated with ET-1. Cells were harvested and CAT activities were measured as described in
Methods. C (control), no drugs; CAT2 and
CAT3 are shown as positive and negative control. CAT activities represent the folds of induction in the treated cells compared with those in untreated controls after normalizing to that of β-galactosidase activities. The results are show as mean + SEM from three independent experiments. *P<0.05 vs. control; #P<0.05 vs ET-1 alone.
Figur e 3. ROS mediated ET-1-induced
hypertrophic responses. (A) Inhibition by antioxidants of ET-1-stimulated protein synthesis in cardiomyocytes. Cardiac myocytes plated in 6-well plates and maintained in serum-free medium for 24 h were treated with vehicle (basal) or ET-1 (10 nM) in the presence or absence of antioxidants for 48 h. The data are mean ± SEM of five independent studies. (B) Antioxidants inhibit ET-1-induced β-MyHC promoter activity. Cardiomyocytes were transfected with 15 µg of β-MyHCCAT chimeric gene for 24 h. Some cells were pretreated with either NAC or catalase for 30 min. Cardiomycytes were then treated with ET-1 (10 nM) for 48 h. H2O2 (25 µM) treated
cells (column 7) are shown as positive controls. Results are shown as mean + SEM from three independent experiments. *P<0.05 vs. control; #P<0.05 vs ET-1 alone.
Figur e 4. ET-1-induced protein synthesis and
β-MyHC gene expression via ERK by a redox-sensitive manner. (A) Through (C) ET-1-induced activation of ERK, JNK, and p38 was mediated by ROS-sensitive pathway. Cells were preincubated with either the catalase (350 U/ml), or NAC (10 mM) for 30 min and then stimulated with ET-1 (10 nM) or H2O2 (25 µ M) for 30 min.
Phosphorylation of ERK, JNK, or p38 was detected by Western blotting using phospho-ERK, phospho-JNK, and phospho-p38 antibodies as described in
Methods. Both catalase and NAC inhibited
ET-1-induced activation of ERK, JNK, or p38. Phosphorylations of ERK, JNK, or p38 were detected, and densitometric analyses were performed. Data are represented as fold increase relative to control groups. Results are shown as mean + SEM from four independent experiments. *P<0.05 vs control; #P<0.05 vs ET-1 (or H2O2) alone. (D) Either
PD98059 or SB203580 inhibited ET-1-stimulated protein synthesis in cardiomyocytes. Cardiac myocytes plated in 6-well plates and maintained in serum-free medium for 24 h were treated with vehicle (basal) or ET-1 (10 nM) in the presence or absence of PD98059 (PD; 20 µM) or SB203580 (SB; 20 µM) for 48 h. The data are mean ± SEM of five independent studies. (E)
ET-1-increased β-MyHC promoter activity was inhibited by PD98059 in cardiomyocytes. Cardiomyocytes were stimulated with ET-1 (10 nM) in the presence of PD98059 (PD; 20 µM) or SB203580 (SB; 20 µM), and CAT activity was assayed after 48 h. (F) ET-1-increased β-MyHC promoter activity via Ras/Raf/ERK pathway in cardiomyocytes. Cells were transfected with either pSRα-empty vector (5 µg), or an expression plasmid encoding the dominant negative mutant mERK, Raf301, or RasN17 (5 µg), was cotransfected with 15 µg of β-MyHC CAT plasmid. Cells cotransfected with an expression plasmid encoding MEK1 (5 µg) or RasL61 (5 µg) were used as positive controls. The above experiments were repeated three times with reproducible results. The results are show as mean + SEM. *P<0.05 vs. control; #P<0.05 vs. ET-1 alone.
Figur e 5. Localization of ET-1- or ROS-responsive elements in β-MyHC promoter. (A) A series of deletion mutants of
β-MyHC promoter gene plasmids were cotransfected into cardiomyocytes. Transfected cells were stimulated with ET-1 (10 nM) for 48 h, and CAT activities were measured. Results are shown as means ± SEM from three separate experiments. (B) Localization of the β-MyHC promoter region responses to ET-1 or H2O2 in cardiomyocytes.
Cardiomyocytes were transiently transfected with the β-MyHC CAT constructs (-273CAT or -188CAT) and treated with 10 nM ET-1 or 25 µM H2O2 for 48 h as described before.
Results are shown as means ± SEM from four separate experiments. *P<0.05 vs. control.
11

![HPSH [ 氧化數平衡反應式係數 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)