行政院國家科學委員會專題研究計畫 成果報告
以熱休克蛋白質 60 鍵結不同的腫瘤抗原的去氧核糖核酸疫
苗其抗腫瘤效用和作用機轉(2/2)
研究成果報告(完整版)
計 畫 類 別 : 個別型 計 畫 編 號 : NSC 95-2314-B-002-036- 執 行 期 間 : 95 年 08 月 01 日至 96 年 07 月 31 日 執 行 單 位 : 國立臺灣大學醫學院婦產科 計 畫 主 持 人 : 鄭文芳 共 同 主 持 人 : 陳祈安 計畫參與人員: 學士級-專任助理:林漢威、林怡君 處 理 方 式 : 本計畫可公開查詢中 華 民 國 96 年 10 月 31 日
計畫英文摘要
Objective
Vaccination based on tumor antigen is an attractive strategy for cancer prevention and therapy. Cervical cancer is highly associated with human papillomavirus, especially type 16. We developed DNA vaccines encoding heat shock protein 60 (HSP60) linked to HPV16 E6 or E7 to test if HSP60 chimeric DNA vaccines may generate strong E6 and/or E7-specific immune response and anti-tumor effects in vaccinated mice.
Methods
In vivo antitumor effects such as preventive, therapeutic, and antibody depletion experiments were performed. In vitro assays such as intracellular cytokine stainings, ELISA for Ab responses, and direct and cross-priming effects, were also performed.
Results
HSP60 chimeric DNA vaccines generated strong E6- or E7-specific immune responses and anti-tumor effects in vaccinated mice via direct and cross-priming effects. HSP60 was also linked with both E6 and E7 antigens and the HSP60/E6/E7 chimeric DNA vaccine generated more potent immunotherapeutic effects on E6- and E7-expressing tumors than HSP60/E6 or HSP60/E7 chimeric DNA vaccine alone.
Conclusion
Utilization of both E6 and E7 tumor antigens can advance the therapy of tumors associated with HPV-infections. The DNA vaccine encoding heat shock protein 60 co-linked to HPV16 E6 and E7 tumor antigens can generate more potent immunotherapeutic effects than E6 or E7 tumor antigens alone.
研究計畫之原由及目的
DNA vaccines have become an attractive approach for generating antigen-specific cancer vaccine and immunotherapy. Naked plasmid DNA can be repeatedly administered and easily prepared in large scale with high purity, and are highly stable relative to proteins and other biological polymers [1]. In addition, intra-dermal administration of DNA vaccines using a gene gun represents an efficient means of targeting dendritic cells, the most potent professional antigen-presenting cells that are specialized to prime T helper and cytotoxic cells in vivo [2] and [3].
One of the concerns about DNA vaccines is their limited potency, because they do not have the intrinsic ability to amplify in vivo as viral vaccines do [1]. Several strategies have been applied to increase their potency. For example, targeting antigens for rapid intracellular degradation [4], directing antigens to APCs by fusion to ligands for APC receptors [5], or fusing antigens to a pathogen sequence, such as fragment C of tetanus toxin [6] have been made. Linkage of antigens to HSP may be another potential approach for increasing the potency of DNA vaccines. HSP-based protein vaccine can also be administered by fusing antigens to HSP [7], [8] and [9]. Furthermore, immune response can be induced in mice with MHC that is either identical or different to the MHC of donor HSPs [10], [11] and [12]. These investigations have made HSPs more attractive to use in cancer vaccine and immunotherapy.
More than 99% of cervical cancers contain HPV, particularly the high-risk HPV type such as 16 and 18 [11]. Two HPV
oncoproteins, E6 and E7, are consistently expressed in HPV-associated cancer cells and are responsible for their malignant transformation. These two oncogenic proteins therefore represent ideal target antigens for developing vaccines and immunotherapeutic strategies against HPV-associated neoplasia. Numerous pre-clinical studies and some clinical studies have targeted the HPV16 oncogenic proteins E6 or E7 for vaccine development to control HPV-associated lesions [13], [14] and [15].
In the past, most HPV researchers focused on E7, and thus an E7 immuno-dominant epitope and its associated immune responses have been well characterized [6]. The gene gun approach was previously used to test several strategies that are able to rout the human papilloma virus type 16 E7 model antigen and result in enhanced E7-specific CD8+ T cell-mediated immune response and anti-tumor effects [16], [17], [18] and [19]. Since E6 represents another important target for potential vaccines to control HPV-associated lesions, it is also crucial to develop vaccines targeting E6.
We therefore developed a DNA vaccine encoding HSP60 linked to E6 and/or E7 to elucidate if DNA vaccines encoding HSP60 with two tumor antigens (E6 and E7) have the potential to prevent HPV infection and provide therapeutic effects on HPV-related cancers. 研究方法、進行步驟及執行進度
Plasmid DNA constructs and preparation
For the generation of pcDNA3-HSP60, HSP60 was first amplified with PCR using human placenta cDNA as template and a set of
primers,
5′-CCGGTCTAGAAGAAATGCTTCGGTTA
CCCACAG-3′ and 5′-CGCGGATCCACACTGCCTTGGGCTTC
CTGTCA-3′. The amplified product was then cloned into the XbaI/BamHI sites of the pcDNA3 vector (Invitrogen Corp., Carlsbad, California, USA). For the generation of pcDNA3-HSP60/E6, E6 was first amplified with PCR using DNA of the CaSki cell line as template and a set of primers, 5′-CGCGGATCCATGCACCAAAAGAGAAC
TGCAATGT-3′ and 5′-CCCAAGCTTTTACAGCTGGGTTTCTCT
ACGTGTTCT-3′. E6 was then cloned into the BamHI/HindIII sites of pcDNA3-HSP60 to generate pcDNA3-HSP60/E6.
To generate pcDNA3-HSP60/E7, E7 was first amplified using DNA of the CaSki cell line as template and with a set of primers, 5′-CCGGGATCCATGGAGATACACCTA -3′ and 5′-CCCAAGCTTTTGAGAACAGATGG -3′, and cloned into the BamHI/HindIII sites of
pcDNA3-HSP60 to generate pcDNA3-HSP60/E7. To generate pcDNA3-HSP60/E6/E7, E7 was cloned into the HindIII/HindIII sites of pcDNA3-HSP60/E6. Plasmid constructs were confirmed by DNA sequencing.
Cell line TC-1
The TC-1 tumor cell line was generated as described previously [20]. On the day of tumor challenge, tumor cells were harvested by trypsinization, washed twice with 1× Hanks buffered salt solution (HBSS), and finally re-suspended in 1× HBSS to the designated
concentration for injection.
293 DbKb cells
293 DbKb cells (a human embryonic kidney 293 cell line expressing the Db and Kb (293 DbKb), two C57BL/6 mouse MHC class I molecules) [21] were kindly provided by Dr. TC Wu, Johns Hopkins Medical Institutes, Baltimore, MD.
Transfection of 293 DbKb cells
293 DbKb were tansfected with various DNA, such as E6, E7, E6/E7, HSP60/E6, HSP60/E7, or HSP60/E6/E7 using LipofectAMINE reagent (Life Technologies) according to the manufacturer's instructions. The 293 DbKb cells were collected 48 h after transfection for further experiments.
Immunoblotting
Transfected 293 DbKb cells with various DNA constructs were lysed in immuno-precipitation assay buffer containing 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 1% NP40, 10% glycerol, 1 mg/ml BSA, 20 mM Tris, pH 8.0, and 2 mM orthovanadate and analyzed as previously described [22]. Briefly, 50 μg of cell lysates was resolved on a sodium dodecyl sulfate (SDS)-containing 12% polyacrylamide gel, transferred to polyvinylidene difluoride nylon membranes (Millipore, Bedford, Mass.), and probed with antibodies specific to E6 (Abcam, Cambridge, UK) or E7 (Zymed, San Francisco, CA) or β-actin (Chemicon International, Temecula, CA). The membrane was then probed with either horseradish peroxidase-conjugated goat anti-mouse or
anti-rabbit antibody.
The specific bands were visualized by an ECL (enhanced chemiluminescence) Western blot system (Amersham, Buckinghamshire, England). As shown in Fig. 1, the E6 (MW 18 kDa), E7 (MW 18 kDa), HSP60/E6 (MW 78 kDa), HPS60/E7 (MW 78 kDa), and HSP60/E6/E7 (MW 96 kDa) proteins showed similar expression levels.
Mice
Six to 8-week-old female C57BL/6J mice were purchased and kept in the animal facility of the School of Medicine, National Taiwan University. All animal procedures were performed according to approved protocols and in accordance with recommendations for the proper use and care of laboratory animals.
DNA vaccination
Preparation of DNA-coated gold particles and gene gun particle-mediated DNA vaccination were performed using a helium-driven gene gun according to a protocol described previously with some modifications [17] and [23]. Control plasmid (no insert), E6, E7, E6/E7, E6 mixed with E7, HSP60, HSP60 mixed with E6, HSP60 mixed with E7, HSP60/E6, HSP60/E7, HSP60/E6 mixed HSP60/E7, and HSP60/E6/E7-coated gold particles were delivered to the shaved abdominal region of mice using a low pressure-accelerated gene gun (BioWare Technologies Co. Ltd, Taipei, Taiwan) with a 50 psi discharge pressure of helium [23].
Intracellular cytokine staining and flow cytometry analysis
Mice were immunized with 2 μg of the various DNA vaccines and received a booster with the same regimen 1 week later. Splenocytes were harvested 1 week after the last vaccination. Before intracellular cytokine staining, pooled splenocytes from each vaccination group were incubated for 16 h with either 1 μg/ml of E6 peptide (aa 50–57) [13] or E7 peptide (aa 49–57) [24], containing an MHC class I epitope for detecting E6- or E7-specific CD8+ T cell precursors, or 50 μg/ml of E6 or E7 protein (kindly provided by Dr. CW Liao, Animal Technology Institute Taiwan, Miaoli, Taiwan) for detecting E6- or E7-specific CD4+ T cell precursors. Cell surface marker staining of CD4 or CD8 and intracellular cytokine staining for IFN-γ, as well as flow cytometric analysis, were performed using conditions described previously [17] and [25].
Cytotoxic T lymphocyte assay using transfected 293 DbKb cells as target cells
Cytotoxic T lymphocyte (CTL) assays were performed by quantitative measurements of lactate dehydrogenase (LDH) using CytoTox96 non-radioactive cytotoxicity assay kits (Promega Corp., Madison, Wisconsin, USA) according to the manufacturer's protocol. Various DNA-transfected 293 DbKb cells served as target cells while a Db-restricted E6- or E7-specific CD8+ T cell line (provided by Dr. TC Wu, Johns Hopkins Medical Institutes, Baltimore, MD) was used as effector cells. Untransfected 293 DbKb cells were used as a negative control. The 293 DbKb cells were collected 40–44 h after transfection.
CTL assays were performed with
effector cells and target cells mixed together at various effector/target (E/T) ratios, 1:1, 5:1, 15:1, and 45:1, in a final volume of 200 μl as described previously [16]. After a 5-h incubation at 37 °C, 50 μl of the cultured media was collected to assess the amount of LDH. The percentage of lysis was calculated from the following equation: 100 × (A − B) / (C − D), where A is the reading of experimental-effector signal value, B is the effector spontaneous background signal value, C is maximum signal value from target cells, and D is the target spontaneous background signal value.
CTL assay using dendritic cells pulsed with lysates of transfected 293 DbKb cells as target cells
CTL assays were performed using bone marrow-derived dendritic cells (DCs) pulsed with cell lysates as target cells and Db-restricted E6- or E7-specific CD8+ T cells as effector cells using the protocol described previously [26]. DCs were generated by culture of bone marrow cells in the presence of GM-CSF as described previously [27]. 293 DbKb cells were first transfected with various DNA constructs via lipofectamine. The protein concentration of lysates was determined using the BioRad protein assay (BioRad Laboratories Inc.) according to the vendor's protocol [16]. DCs were pulsed with different concentrations of cell lysates of various DNA-transfected 293 DbKb cells (50 μg/ml, 10 μg/ml, 2 μg/ml, and 0.4 μg/ml) in a final volume of 2 ml for 16–20 h. CTL assays were performed at a fixed E/T ratio of 9:1 using E7-specific T cells mixed with
prepared DCs in a final volume of 200 μl. Cytolysis was determined by quantitative measurements of LDH as described earlier.
In vivo tumor protection experiments
For the tumor protection experiments, mice (five per group) either received no vaccination or were immunized with 2 μg/mouse of various DNA vaccines with a gene gun. They were boosted with the same regimen as the first vaccination one week later. One week after the last vaccination, the mice were subcutaneously challenged with 5 × l04 of TC-1 cells/mouse in the right leg. They were monitored for evidence of tumor growth by palpation and inspection twice a week until they were sacrificed on day 60.
In vivo antibody depletion experiments
In vivo Ab depletions were performed as described previously [27] and [28]. Briefly, mice (five per group) were vaccinated with 2 μg/mouse of HSP60/E6 or HSP60/E7 DNA with a gene gun, boosted 1 week later, and challenged with 5 × 104 cells/mouse TC-1 tumor cells. Depletion was started 1 week before tumor challenge.
mAb GK1.5 was used for CD4 depletion [29], mAb 2.43 was used for CD8 depletion [30], and mAb PK136 was used for NK1.1 depletion [31]. Depletion was terminated on day 40 after tumor challenge. Mice were monitored for evidence of tumor growth by palpation and inspection twice a week until they were sacrificed on day 60.
In vivo tumor treatment experiments
The therapeutic potential of each
vaccine was assessed by performing an in vivo tumor treatment experiment using a previously described lung hematogenous spread model [16]. Two days after tumor challenge, mice received 16 μg/mouse of no insert DNA, E6 DNA, E7 DNA, E6 mixed with E7 DNA, HSP60 DNA, HSP60/E6 DNA, HSP60/E7 DNA, HSP60/E6 mixed with HSP60/E7 DNA, or HSP60/E6/E7 DNA by a gene gun, followed by a booster with the same regimen every 7 days for 4 weeks (a total of 64 μg DNA). Mice receiving no vaccination were used as negative control. The mice were sacrificed and the lungs were explanted on day 28. Pulmonary tumor nodules in each mouse were evaluated and counted by experimenters who were blinded to sample identity.
For the second in vivo tumor treatment experiment, mice were injected with 5 × 104 cells/mouse TC-1 tumor cells via the tail vein as described earlier. Seven days after tumor challenge, mice received various DNA vaccines such as HSP60/E6, HSP60/E7, HSP60/E6 mixed with HSP60/E7, and HSP60/E6/E7 as described earlier. The mice were sacrificed, and the lungs were explanted on day 28. The pulmonary tumors in each mouse were evaluated and counted as described earlier.
Statistical analysis
All data expressed as mean ± SEM were representative of at least two different experiments. Data for intracellular cytokine staining with flow cytometry analysis and tumor treatment experiments were evaluated by analysis of variance (ANOVA). Comparisons between individual data points
were made using Student's t-test. In the tumor protection experiment, the principal outcome of interest was the time to tumor development. The event time distributions for different mice were compared by the Kaplan and Meier and by log-rank analyses. A p value < 0.05 was considered statistically significant.
五、結果
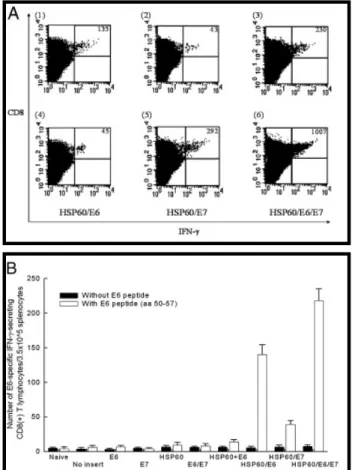
Vaccination with DNA encoding HSP60 linked to E6 alone, E7 alone, or both E6 and E7 significantly enhanced E6 and/or E7-specific CD8+ T cell response
The representative figures of flow cytometric analysis are shown in Fig. 2A. As shown in Fig. 2B, vaccination with HSP60/E6 (140.0 ± 14.1) or HSP60/E6/E7 (218.0 ± 17.0) DNA generated higher frequencies of E6-specific IFN-γ-secreting CD8+ T cell precursors when compared to mice vaccinated with E6 (6.5 ± 2.1), E7 (4.0 ± 1.4), E6/E7 (8.0 ± 3.5), HSP60 mixed with E6 (12.5 ± 2.8), or HSP60/E7 (39.0 ± 5.7) DNA (p < 0.01, one-way ANOVA). In addition, vaccination with HSP60/E7 (275.0 ± 24.0), or HSP60/E6/E7 (987.5 ± 27.6) DNA generated higher frequencies of E7-specific IFN-γ-secreting CD8+ T cell precursors when compared to mice vaccinated with E6 (7.5 ± 1.4), E7 (7.0 ± 2.8), E6/E7 (12.0 ± 2.8), HSP60 mixed with E7 (14.5 ± 2.1), or HSP60/E6 DNA (40.5 ± 7.0) (p < 0.01, one-way ANOVA) (Fig. 2C). We further evaluated whether HSP60 could enhance the E6, or E7-specific CD4+ T cell response when linked to E6, or E7 in a DNA vaccine, respectively. As shown in Figs. 2D and E, no increase in the number of E6- or E7-specific
IFN-γ-secreting CD4+ T cells in mice vaccinated with various DNA vaccines was observed.
Our data showed that physical linkage of HSP60 to E6 or E7 was required for enhancement of E6- or E7-specific CD8+ T cell activity, since DNA encoding HSP mixed with E6 or E7 DNA did not generate enhancement of E6- or E7-specific CD8+ T cell activity.
Enhanced presentation of E6, or E7, or E6 and E7 antigen through the MHC class I pathway in cells transfected with HSP60 linked with E6, or E7, or E6 and E7 DNA and in dendritic cells pulsed with various chimeric HSP60 proteins
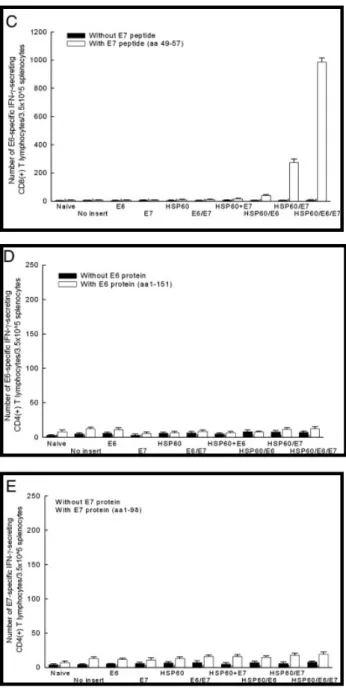
We further explored potential explanations for the observed increase in E6 and/or E7-specific CD8+ T cell precursors in mice vaccinated with HSP60/E6, HSP60/E7, or HSP60/E6/E7. One was that there was direct enhancement of MHC class I presentation of E6 or E7 in cells expressing HSP60/E6, HSP60/E7, or HSP60/E6/E7. As shown in Fig.
3A, 293 DbKb cells transfected with HSP60/E6
(57.6 ± 5.4%) and HSP60/E6/E7 (63.1 ± 5.2%) DNA generated significantly higher percentages of specific lysis at 45:1 E/T ratios compared with cells transfected with E6 and HSP60 (10.3 ± 1.4%), HSP60 (4.0 ± 0.4%), E6/E7 (4.4 ± 0.2%), or wild-type E6 (3.1 ± 0.1%) DNA when the effector cells were the E6-specific CD8+ T cell line (p < 0.001, one-way ANOVA).
Similar phenomena were also observed in 293 DbKb cells transfected with HSP60/E7 (44.2 ± 3.0%), and the HSP60/E6/E7
(50.9 ± 3.6%) DNA also generated significantly higher percentages of specific lysis at 45:1 E/T ratios compared with cells transfected with E7 and HSP60 (8.7 ± 1.4%), HSP60 (9.1 ± 0.5%), E6/E7 (9.0 ± 1.7%), or wild-type E7 (8.2 ± 1.4%) DNA when the effector cells were changed to the E7-specific CD8+ T cell line (p < 0.001, one-way ANOVA) (Fig. 3B).
Another potential mechanism for the observed enhancement of E7-specific CD8+ T cell immune responses in vivo was the so-called “cross-priming” effect [32], where the HSP70/E7 protein released from cells were taken up and processed by other antigen-presenting cells (APCs) via the MHC class I-restricted pathway [27]. As shown in
Fig. 3C, DCs pulsed with lysates of 293 DbKb
cells transfected with HSP60/E6 (57.2 ± 4.0%) or HSP60/E6/E7 (68.7 ± 4.1%) DNA generated a significantly higher percentage of specific lysis than DCs pulsed with lysates of 293 DbKb cells transfected with HSP60/E7 (20.2 ± 1.8%) or the wide-type E6 (10.7 ± 0.6%) DNA construct (p < 0.001, one-way ANOVA), when the effector cells were the E6-specific CD8+ T cell line.
In addition, DCs pulsed with 50 μg/ml lysates of 293 DbKb cells transfected with HSP60/E7 (52.6 ± 5.2%) or HSP60/E6/E7 (56.4 ± 6.3%) DNA generated a significantly higher percentage of specific lysis than DCs pulsed with lysates of 293 DbKb cells transfected with HSP60/E6 (19.0 ± 1.8%) or the wide-type E7 (8.8 ± 1.3%) DNA construct, when the effector cells were the E7-specific CD8+ T cell line (p < 0.001, one-way ANOVA) (Fig. 3D).
Our in vitro experiments revealed that HSP60/E6, HSP60/E7, and HSP60/E6/E7 chimeric molecules may enhance antigen-specific immunity via direct and/or cross-priming effects.
Vaccination with HSP60/E6, HSP60/E7, or HSP60/E6/E7 DNA enhanced tumor protection in mice challenged with an E6 and E7-expressing tumor cell line
To determine if the observed enhancement of E6- or E7-specific CD8+ T cell response translated into a significant E6- or E7-specific protective anti-tumor effect, we performed an in vivo tumor protection experiment using a previously characterized E6- and E7-expressing tumor model, TC-1 [20]. As shown in Fig. 4A, 100% of mice receiving HSP60/E6, HSP60/E7, or HSP60/E6/E7 DNA vaccination, when challenged with TC-1 tumor cells, remained tumor-free 60 days after TC-1 challenge. In comparison, all mice vaccinated with the wild-type E6, E7, E6/E7, or HSP60 mixed with E6 or E7 DNA developed tumors within 14 days of challenge.
These results indicated that fusion of HSP60 to E6 and/or E7 antigens is required for anti-tumor immunity of E6 and E7-expressing TC-1 tumor cells.
CD8+ T cells but not CD4+ T cells or natural killer cells were essential for anti-tumor effect generated by HSP60/E6 or HSP60/E7 DNA
To determine the subset of lymphocytes important for the anti-tumor effect, we performed in vivo Ab depletion experiments [16]. As shown in Figs. 4B and C, depleted of
CD8+ T cells grew tumors within 15 days after tumor challenge in all naive mice and those vaccinated with HSP60/E6 or HSP60/E6 DNA vaccine. In contrast, all of the non-depleted mice and those depleted of CD4+ T cells or NK1.1 cells remained tumor-free 60 days after umor challenge.
These results suggested that CD8+ T cells are required for anti-tumor immunity generated by HSP60/E6 and HSP60/E7 DNA vaccines.
Treatment with HSP60/E6/E7 led to significant reduction of pulmonary tumor nodules
As shown in Fig. 5, mice treated with HSP60/E6 DNA (3.7 + 1.7), HSP60/E7 (1.8 ± 1.6), or HSP60/E6/E7 (0.0 ± 0.0) all exhibited significantly fewer pulmonary tumor nodules than mice treated with the wild-type E6 (70.0 ± 9.0), wild-type E7 (73.6 ± 6.7), or E6/E7 (70.4 ± 3.1) DNA vaccine (p < 0.001, one-way ANOVA), when started on treatment 2 days after tumor injection.
The representative figures of pulmonary tumor nodules treated with various DNA vaccines after 7 days of TC-1 tumor injection are shown in Fig. 6A. As shown in
Fig. 6B, mice treated with HSP60/E6/E7 DNA
led to significantly lower lung weights (185.0 + 7.1 mg) than those with HSP60/E6 mixed with HSP60/E7 (330.0 ± 20.3 mg), or HSP60/E7 (362.0 ± 13.8 mg), or HSP60/E6 DNA (665.2 ± 14.8 mg) (one-way ANOVA, p < 0.05), when started on treatment 7 days after tumor injection.
These data indicated that HSP60, when linked with either E6 or E7 tumor antigens,
could generate more potent anti-tumor effects than wild-type E6 or E7 DNA vaccine in a lung hematogeous spread therapeutic model. Moreover, HSP60, when linked with E6 and E7 antigens together, could generate more potent anti-tumor effects than HSP60 linked with either E6 or E7 tumor antigen, or HSP60/E7 mixed with HSP60/E7.
六、討論
Mice vaccinated with HSP60/E6 or HSP60/E7 DNA enhanced E6- or E7-specific CD8+ T cell responses. One possible mechanism for the enhancement of CD8+ T cell responses was the effect of HSP60 in inducing direct-priming mechanism. Our in vitro data also revealed that HSP60 chimeric DNA vaccine might enhance CD8+ T cell responses via this mechanism. Ballistic DNA delivery might introduce chimeric HSP60/E6 or HSP60/E7 DNA directly into the dermal precursors or professional APCs to enhance the antigen processing and presenting processes [33]. It was also shown that direct-priming of CD8+ T cells by gene-transfected dendritic cells was the key event in gene gun-mediated DNA immunization [34]. We posit that HSP60/E6 or HSP60/E7 DNA vaccine was transfected into the DCs via the gene gun delivery system to directly enhance E6- or E7-specific T cell immunity.
Another possible mechanism for the enhancement of CD8+ T cell responses by the chimeric HSP60 DNA vaccine was the “cross-priming” of HSP/peptide complexes, where the HSP led exogenous proteins to the MHC-I restricted antigen presentation pathway. We demonstrated that cross-priming might be
one of possible mechanisms for the chimeric HSP60 DNA vaccine. HSP60/E6 and HSP60/E7 might be released from other cell types, such as keratinocytes (which were also transfected by gene gun vaccination) and then these chimeric HSP60/E6 or HSP60/E7 proteins were released exogenously to be taken up and processed by other APCs via the MHC class I-restricted pathway [35] and [36].
HSP complexes taken up by professional APCs are supposed to play an important role in introducing HSP-associated peptides into the MHC-I antigen presentation pathway [37]. These mechanisms may provide an explanation for the observed enhancement of E6- or E7-specific T cell immunity. CD14 [38] and Toll-like receptors (TLR) 2 and 4 [39] and [40] are involved in HSP60-mediated cell activation. As regards HSP60, studies have shown that CD14, TLR2, and TLR4 are involved in signal transduction in macrophages [38] and [41]. It is interesting to evaluate the in vivo mechanism of HSP60/E7 and HSP60/E6 DNA vaccine using TLR2 or TLR4 deficient mice in the future.
Heat-shock proteins of the HSP60 family are molecular chaperones that enhance immune responses. HSP60 can transport antigens and after internalization, mediates antigen-specific cytotoxic T cell response [36] and [42] as observed in this study. HSP60 protein has also been identified in many infectious agents as an immuno-dominant antigen with a protective effect. Immunization of laboratory animals by selected HSP60, HSP70, or HSP90 isolated from several pathogens induces protective host immunity and significantly reduce the clinical
manifestation of infection [43] and [44].
Self HSP60 protein and its derived peptide as carriers in a conjugated vaccine have been shown to protect against lethal Streptococcus pneumoniae [45] and [46]. Milan et al. reported that naked HSP60 DNA provides better protective effects than recombinant HSP60 protein [43]. Recently, naked HSP60 DNA has also been utilized to control adjuvant arthritis [47]. Naked human HSP60 DNA vaccine can also be used to inhibit insulitis and diabetes in the NOD mice by vaccination with a DNA construct encoding human HSP60 [48]. Mice, either unprimed or primed with M. Tuberculosis var. bovis (Bacillus Calmette-Guerin, BCG), produce a high and long-lasting titer of anti-peptide antibodies when immunized with repetitive malaria synthetic peptide (NANP) conjugated to mycobacterial HSP60 [49]. Although this carrier effect is associated with the risk of immune responses against self HSPs, it still provides a novel approach for the development of vaccines by a fusion of peptides or antigens to HSP60.
The 293 DbKb cells are always used as the antigen-presenting cells in various immunologic assays. Ideally, immunologic assays that evaluate HPV-related vaccines are better used in actual HPV-generated tumor cells. However, it is impossible to create HPV-generated tumor cells in the animal system. Hence, we tried to use 293 DbKb cells to test the immunogenicity of various HSP60 chimeric DNA vaccines. Although it is not relevant to actual HPV-generated tumor cells, 293 cells previously transfected and selected to stably express murine class I MHC molecules
H2- Kb and H2- Db (designated 293 KbDb) are good alternative target cells in evaluating the efficacy of DNA vaccine in the animal model. DNA vaccine encoding heat shock protein 60 co-linked to HPV16 E6 and E7 tumor antigens generates more potent immunotherapeutic effects than E6 or E7 tumor antigens alone. Both E6 and E7 can be utilized as target antigens of cancer vaccine and immunotherapy. The fusion of HSP60 family to E6 or E7 enhances E6- or E7-specific CD8+ T cell-mediated immune responses. In the past, most HPV researchers focused on E7 [50]. Since E6 represents another important target for potential vaccines to control HPV-associated lesions, it is crucial to develop vaccines targeting E6. To combine E6 and E7 as target tumor antigens is important in aiding the future development of HPV vaccines. In addition, the ability of HSP60 to enhance immune responses when linked to two different antigens in DNA vaccines also suggests that HSP60 may be broadly applicable as a strategy to enhance DNA vaccines encoding a variety of antigens. This possibility will be explored further in future DNA vaccination studies, in which HSP60 will be linked to other viral or tumor-specific antigens.
七、計畫成果自評
本 論 文 已 經 刊 登 於 國 際 學 術 期 刊
Gynecol Oncol. 2007 Sep 28; [Epub ahead of
print]
References
[1] D.M. Pardoll, Exposing the immunology of naked DNA vaccines, Immunity 3 (1995), pp. 165–169.
[2] R. Wang, J. Epstein, Y. Charoenvit, F.M. Baraceros, N. Rahardjo and T. Gay et al., Induction in humans of CD8+ and CD4+ T cell and antibody responses by sequential immunization with malaria DNA and recombinant protein, J Immunol 172 (2004), pp. 5561–5569.
[3] S.J. Ha, D.J. Kim, K.H. Baek, Y.D. Yun and Y.C. Sung, IL-23 induces stronger
sustained CTL and Th1 immune responses than IL-12 in hepatitis C virus envelope protein 2 DNA immunization, J Immunol 172 (2004), pp. 525–531.
[4] F. Rodriguez, J. Zhang and J.L. Whitton, DNA immunization: ubiquitination of a viral protein enhances cytotoxic
T-lymphocyte induction and antiviral protection but abrogates antibody induction, J Virol 71 (1997), pp. 8497–8503.
[5] J.S. Boyle, J.L. Brady and A.M. Lew, Enhanced responses to a DNA vaccine encoding a fusion antigen that is directed to sites of immune induction, Nature 392 (1998), pp. 408–411.
[6] C.A. King, M.B. Spellerberg, D. Zhu, J. Rice, S.S. Sahota and A.R. Thompsett et al., DNA vaccines with single-chain Fv fused to fragment C of tetanus toxin induce protective immunity against lymphoma and myeloma, Nat Med 4 (1998), pp. 1281–1286.
[7] K. Suzue, X. Zhou, H.N. Eisen and R.A. Young, Heat shock fusion proteins as vehicles for antigen delivery into the major histocompatibility complex class I presentation pathway, Proc Natl Acad Sci U S A 94 (1997), pp. 13146–13151.
[8] K. Suzue and R.A. Young, Adjuvant-free hsp70 fusion protein system elicits
humoral and cellular immune responses to HIV-1 p24, J Immunol 156 (1996), pp. 873–879.
[9] Y. Enomoto, A. Bharti, A.A. Khaleque, B. Song, C. Liu and V. Apostolopoulos et al., Enhanced immunogenicity of heat shock protein 70 peptide complexes from dendritic cell-tumor fusion cells, J Immunol 177 (2006), pp. 5946–5955. [10] P.K. Srivastava, A. Menoret, S. Basu, R.J.
Binder and K.L. McQuade, Heat shock proteins come of age: primitive functions acquire new roles in an adaptive world, Immunity 8 (1998), pp. 657–665. [11] D. Przepiorka and P.K. Srivastava, Heat
shock protein–peptide complexes as immunotherapy for human cancer, Mol Med Today 4 (1998), pp. 478–484. [12] T. Chan, Z. Chen, S. Hao, S. Xu, J. Yuan
and A. Saxena et al., Enhanced T-cell immunity induced by dendritic cells with phagocytosis of heat shock protein 70 gene-transfected tumor cells in early phase of apoptosis, Cancer Gene Ther 14 (2007), pp. 409–420.
[13] S. Peng, H. Ji, C. Trimble, L. He, Y.C. Tsai and J. Yeatermeyer et al.,
Development of a DNA vaccine targeting human papillomavirus type 16 oncoprotein E6, J Virol 78 (2004), pp. 8468–8476. [14] N.E. Blachere, H. Udono, S. Janetzki, Z.
Li, M. Heike and P.K. Srivastava, Heat shock protein vaccines against cancer, J Immunother 14 (1993), pp. 352–356. [15] C.W. Liao, C.A. Chen, C.N. Lee, Y.N. Su,
M.C. Chang and M.H. Syu et al., Fusion
protein vaccine by domains of bacterial exotoxin linked with a tumor antigen generates potent immunologic responses and antitumor effects, Cancer Res 65 (2005), pp. 9089–9098.
[16] W.F. Cheng, C.F. Hung, C.Y. Chai, K.F. Hsu, L. He and M. Ling et al.,
Tumor-specific immunity and
antiangiogenesis generated by a DNA vaccine encoding calreticulin linked to a tumor antigen, J Clin Invest 108 (2001), pp. 669–678.
[17] W.F. Cheng, C.F. Hung, C.A. Chen, C.N. Lee, Y.N. Su and C.Y. Chai et al.,
Characterization of DNA vaccines encoding the domains of calreticulin for their ability to elicit tumor-specific immunity and antiangiogenesis, Vaccine 23 (2005), pp. 3864–3874.
[18] C.F. Hung, W.F. Cheng, L. He, M. Ling, J. Juang and C.T. Lin et al., Enhancing major histocompatibility complex class I antigen presentation by targeting antigen to centrosomes, Cancer Res 63 (2003), pp. 2393–2398.
[19] C.F. Hung, W.F. Cheng, C.Y. Chai, K.F. Hsu, L. He and M. Ling et al., Improving vaccine potency through intercellular spreading and enhanced MHC class I presentation of antigen, J Immunol 166 (2001), pp. 5733–5740.
[20] K.Y. Lin, F.G. Guarnieri, K.F. Staveley-O'Carroll, H.I. Levitsky, T. August and D.M. Pardoll et al., Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen, Cancer Res 56 (1996), pp. 21–26.
[21] M.B. Bloom, D. Perry-Lalley, P.F. Robbins, Y. Li, M. el-Gamil and S.A. Rosenberg et al., Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma, J Exp Med 185 (1997), pp. 453–459.
[22] C.H. Chen, H. Ji, K.W. Suh, M.A. Choti, D.M. Pardoll and T.C. Wu, Gene
gun-mediated DNA vaccination induces antitumor immunity against human papillomavirus type 16 E7-expressing murine tumor metastases in the liver and lungs, Gene Ther 6 (1999), pp.
1972–1981.
[23] W.F. Cheng, L.K. Chen, C.A. Chen, M.C. Chang, P.N. Hsiao and Y.N. Su et al., Chimeric DNA vaccine reverses morphine-induced immunosuppression and tumorigenesis, Molec Ther 13 (2006), pp. 203–210.
[24] M.C. Feltkamp, H.L. Smits, M.P. Vierboom, R.P. Minnaar, B.M. de Jongh and J.W. Drijfhout et al., Vaccination with cytotoxic T lymphocyte
epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells, Eur J Immunol 23 (1993), pp. 2242–2249.
[25] W.F. Cheng, C.F. Hung, K.F. Hsu, C.Y. Chai, L. He and J.M. Polo et al., Cancer immunotherapy using Sindbis virus replicon particles encoding a
VP22-antigen fusion, Hum Gene Ther 13 (2002), pp. 553–568.
[26] C.F. Hung, W.F. Cheng, K.F. Hsu, C.Y. Chai, L. He and M. Ling et al., Cancer immunotherapy using a DNA vaccine encoding the translocation domain of a
bacterial toxin linked to a tumor antigen, Cancer Res 61 (2001), pp. 3698–36703. [27] W.F. Cheng, C.F. Hung, C.Y. Chai, K.F.
Hsu, L. He and C.M. Rice et al., Enhancement of Sindbis virus
self-replicating RNA vaccine potency by linkage of Mycobacterium tuberculosis heat shock protein 70 gene to an antigen gene, J Immunol 166 (2001), pp.
6218–6226.
[28] W.F. Cheng, C.F. Hung, C.N. Lee, Y.N. Su, M.C. Chang and L. He et al., Naked RNA vaccine controls tumors with
down-regulated MHC class I expression through NK cells and perforin-dependent pathways, Eur J Immunol 34 (2004), pp. 1892–1900.
[29] D.P. Dialynas, Z.S. Quan, K.A. Wall, A. Pierres, J. Quintans and M.R. Loken et al., Characterization of the murine T cell surface molecule, designated L3T4, identified by monoclonal antibody GK1.5: similarity of L3T4 to the human Leu-3/T4 molecule, J Immunol 131 (1983), pp. 2445–2451.
[30] M. Sarmiento, A.L. Glasebrook and F.W. Fitch, IgG or IgM monoclonal antibodies reactive with different determinants on the molecular complex bearing Lyt 2 antigen block T cell-mediated cytolysis in the absence of complement, J Immunol 125 (1980), pp. 2665–2772.
[31] G.C. Koo, F.J. Dumont, M. Tutt, J. Hackett Jr. and V. Kumar, The NK-1.1(−) mouse: a model to study differentiation of murine NK cells, J Immunol 137 (1986), pp. 3742–3747.
targets with epitope-linked beta
2-microglobulin constructs, J Immunol 160 (1998), pp. 1598–1605.
[33] S.B. Flohe, J. Bruggemann, S. Lendemans, M. Nikulina, G. Meierhoff and S. Flohe et al., Human heat shock protein 60 induces maturation of dendritic cells versus a Th1-promoting phenotype, J Immunol 170 (2003), pp. 2340–2348.
[34] A. Porgador, K.R. Irvine, A. Iwasaki, B.H. Barber, N.P. Restifo and R.N. Germain, Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization, J Exp Med 188 (1998), pp. 1075–1082. [35] P.K. Srivastava, H. Udono, N.E. Blachere
and Z. Li, Heat shock proteins transfer peptides during antigen processing and CTL priming, Immunogenetics 39 (1994), pp. 93–98.
[36] F. Castellino, P.E. Boucher, K. Eichelberg, M. Mayhew, J.E. Rothman and A.N. Houghton et al.,
Receptor-mediated uptake of antigen/heat shock protein complexes results in major histocompatibility complex class I antigen presentation via two distinct processing pathways, J Exp Med 191 (2000), pp. 1957–1964.
[37] R. Suto and P.K. Srivastava, A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides, Science 269 (1995), pp. 1585–1588. [38] A. Kol, A.H. Lichtman, R.W. Finberg, P.
Libby and E.A. Kurt-Jones, Cutting edge: heat shock protein (HSP) 60 activates the innate immune response: CD14 is an essential receptor for HSP60 activation of
mononuclear cells, J Immunol 164 (2000), pp. 13–17.
[39] K. Ohashi, V. Burkart, S. Flohe and H. Kolb, Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex, J Immunol 164 (2000), pp. 558–561.
[40] R.M. Vabulas, P. Ahmad-Nejad, C. da Costa, T. Miethke, C.J. Kirschning and H. Hacker et al., Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells, J Biol Chem 276 (2001), pp.
31332–31339.
[41] M.F. Tsan and B. Gao, Cytokine function of heat shock proteins, Am J Physiol Cell Physiol 286 (2004), pp. C739–C744. [42] R.J. Binder, D.K. Han and P.K. Srivastava,
CD91: a receptor for heat shock protein gp96, Nat Immunol 1 (2000), pp. 151–155.
[43] R. Milan, R. Alois, C. Josef, B. Jana and W. Evzen, Recombinant protein and DNA vaccines derived from hsp60 Trichophyton mentagrophytes control the clinical course of trichophytosis in bovine species and guinea-pigs, Mycoses 47 (2004), pp. 407–417.
[44] M. Radwanska, S. Magez, A. Michel, B. Stijlemans, M. Geuskens and E. Pays, Comparative analysis of antibody responses against HSP60, invariant surface glycoprotein 70, and variant surface glycoprotein reveals a complex antigen-specific pattern of
immunoglobulin isotype switching during infection by Trypanosoma brucei, Infect
Immun 68 (2000), pp. 848–860. [45] H. Amir-Kroll, G. Nussbaum and I.R.
Cohen, Proteins and their derived peptides as carriers in a conjugate vaccine for Streptococcus pneumoniae: self-heat shock protein 60 and tetanus toxoid, J Immunol 170 (2003), pp. 6165–6171. [46] S. Konen-Waisman, A. Cohen, M. Fridkin
and I.R. Cohen, Self heat-shock protein (hsp60) peptide serves in a conjugate vaccine against a lethal pneumococcal infection, J Infect Dis 179 (1999), pp. 403–413.
[47] F.J. Quintana, P. Carmi, F. Mor and I.R. Cohen, DNA fragments of the human 60-kDa heat shock protein (HSP60) vaccinate against adjuvant arthritis: identification of a regulatory HSP60 peptide, J Immunol 171 (2003), pp. 3533–3541.
[48] F.J. Quintana, A. Rotem, P. Carmi and I.R. Cohen, Vaccination with empty plasmid DNA or CpG oligonucleotide inhibits diabetes in nonobese diabetic mice: modulation of spontaneous 60-kDa heat shock protein autoimmunity, J Immunol 165 (2000), pp. 6148–6155.
[49] A.R. Lussow, C. Barrios, J. van Embden, R. Van der Zee, A.S. Verdini and A. Pessi et al., Mycobacterial heat-shock proteins as carrier molecules, Eur J Immunol 21 (1999), pp. 2297–2302.
[50] M.C. Feltkamp, M.P. Vierboom, R.E. Toes, F. Ossendorp, J. ter Schegget and C.J. Melief et al., Competition inhibition of cytotoxic T-lymphocyte (CTL) lysis, a more sensitive method to identify
candidate CTL epitopes than induction of
antibody-detected MHC class I
stabilization, Immunol Lett 47 (1995), pp. 1–8.
Figure Legends
Fig. 1. The protein expression levels of respective constructs in transfected 293 DbKb cells.
293 DbKb cells were transfected with
respective DNA. The E6 or E7 protein expression levels in various constructs are shown as follows: E6 (MW 18 kDa), E7 (MW 18 kDa), HSP60/E6 (MW 78 kDa), HPS60/E7 (MW 78 kDa), and HSP60/E6/E7 (MW 96 kDa).
Fig. 2. Immunologic profiles of vaccinated mice using intracellular cytokine staining and flow cytometry analysis and ELISA. (A) Representative figures of flow cytometric
analysis. (B) Bar graph depicting the number of E6-specific IFN-γ-secreting CD8+ T cell
precursors/3.5 × 105 splenocytes
(mean ± SEM). Mice vaccinated with HSP60/E6 or HSP60/E6/E7 DNA generated higher numbers of E6-specific IFN-γ-secreting CD8+ T cell precursors than other vaccination
groups (p < 0.01, one-way ANOVA). (C) Bar graph depicting the number of E7-specific
IFN-γ-secreting CD8+ T cell
precursors/3.5 × 105 splenocytes
(mean ± SEM). Mice vaccinated with HSP60/E7 or HSP60/E6/E7 DNA generated higher numbers of E7-specific IFN-γ-secreting CD8+ T cell precursors than other vaccination groups (p < 0.01, one-way ANOVA). (D) Bar graph depicting the number of E6-specific
IFN-γ-secreting CD4+ T cell
precursors/3.5 × 105 splenocytes
(mean ± SEM). None of the vaccinated groups generated significantly higher number of E6-specific CD4+ T cell precursors when compared with naive mice (p>0.05, one-way ANOVA). (E) Bar graph depicting the number of E7-specific IFN-γ-secreting CD4+ T cell
precursors/3.5 × 105 splenocytes
(mean ± SEM). None of the vaccinated groups generated significantly higher number of E7-specific CD4+ T cell precursors when compared with naive mice (p > 0.05, one-way ANOVA)..
Fig. 3. CTL assays demonstrate enhanced presentation of E6 or E7 through the MHC class I pathway directly by 293 DbKb cells transfected with HSP60/E6, HSP60/E7, or HSP60/E6/E7 DNA constructs. (A) CTL
assays with various E/T ratios (E/T = 1:1, 5:1, 15:1, 45:1) by using 293 DbKb cells transfected with various DNA constructs served as target cells and Db-restricted E6-specific CD8+ T cells as effector cells. (B) CTL assays with
various E/T ratios (E/T = 1:1, 5:1, 15:1, 45:1) by using 293 DbKb cells transfected with various DNA constructs served as target cells and Db-restricted E7-specific CD8+ T cells as effector cells. CTL assays demonstrate enhanced presentation of E6, or E7 through the MHC class I pathway indirectly by bone marrow-derived DCs pulsed with cell lysates containing chimeric HSP60/E6, HSP60/E7, or HSP60/E6/E7 protein. (C) CTL assays at fixed E/T ratio (9:1) using bone marrow-derived DCs pulsed with different concentrations of cell lysates from various DNA-transfected 293 Db cells, and Db-restricted E6-specific CD8+ T cells as effector cells. (D) CTL assays at fixed E/T ratio (9:1) using bone marrow-derived DCs pulsed with different concentrations of cell lysates from various DNA-transfected 293 DbKb cells and Db-restricted E6-specific CD8+ T cells as effector cells.
Fig. 4. In vivo tumor protection experiments in mice vaccinated with various protein vaccines and in vivo Ab depletion experiments in mice vaccinated with HSP60/E6 or HSP60/E7 DNA vaccines. (A)
In vivo tumor protection experiments. 100% of
mice receiving HSP60/E6, HSP60/E7, or HSP60/E6/E7 remained tumor-free 60 days after TC-1 challenge. (B) In vivo Ab depletion experiments of mice vaccinated with HSP60/E6 DNA vaccine. (C) In vivo Ab depletion experiments of mice vaccinated with HSP60/E7 DNA vaccine. All of the HSP60/E6 or HSP60/E7 vaccinated mice depleted with CD8+ T lymphocytes, as well as naive mice, developed tumors within 14 days after TC-1
tumor challenge. All of the HSP60/E6 or HSP60/E7 vaccinated mice depleted of CD4+ T lymphocytes or NK1.1+ cells were tumor-free after 60 days of TC-1 tumor challenge.
Fig. 5. In vivo tumor treatment experiments in mice at a high therapeutic dose. Mice
treated with DNA encoding HSP60/E6, HSP60/E7, or HSP70/E6/E7 showed similar
numbers of tumor nodules, all significantly lower than those in mice treated with DNA encoding E6, E7, or E6/E7. Data are expressed as mean number of pulmonary tumor nodules + SEM
Fig. 6. In vivo tumor treatment experiments in mice with various therapeutic conditions.
(A) Representative pulmonary tumor nodules in various DNA vaccinated groups. (1) HSP60/E6 group, (2) HSP60/E7 group, (3) HSP60/E6 mixed with HSp60/E7 group, (4) HSP60/E6/E7 group. (B) Mean lung weights in various DNA vaccinated groups. Mice treated with DNA encoding HSP60/E6/E7 showed significantly lower lung weights than those treated with either HSP60/E6 or HSP60/E7 DNA only. Mice vaccinated with HSP60/E7 DNA had lower lung weights than those vaccinated with HSP60/E6 DNA. Data are expressed as mean number of pulmonary tumor nodules ± SEM.