Synthesis and luminescent properties of nano-sized
Y
3
Al
5
O
12
:Eu
3
+
phosphors
Wei-Tse Hsu, Wei-Hong Wu, Chung-Hsin Lu
∗Electronic and Electro-Optical Ceramics Laboratory, Department of Chemical Engineering, National Taiwan University, Taipei, Taiwan, ROC Received 3 February 2003; accepted 9 July 2003
Abstract
Nano-sized Eu3+-activated yttrium aluminum garnet (Y3Al5O12, YAG) phosphors were prepared via a simple sol–gel process at as low a
temperature as 900◦C. In comparison with the conventional solid-state reaction process, this sol–gel process not only reduced the temperature required for synthesizing YAG:Eu3+ powders, but also decreased their particle size to the nanometer range. In the sol–gel process, citric
acid and ethylene glycol formed numerous tiny enclosures that trapped the constituent cations, leading to a reduction of the interdiffusion length and an enhancement of the reactivity of the precursors. The photoluminescence intensity of the sol–gel-prepared YAG:Eu3+phosphors
was markedly greater than that of the samples derived from the solid-state reaction. This is attributed to the increased crystallinity and improved compositional homogeneity in the sol–gel-derived powders. The photoluminescence characteristics of YAG:Eu3+phosphors were
demonstrated to substantially depend on their morphology and particle size. © 2003 Elsevier B.V. All rights reserved.
Keywords: Sol–gel; Synthesis; Nano-sized; Y3Al5O12; Phosphor; Luminescence
1. Introduction
In recent years, inorganic phosphors have been exten-sively investigated for the applications to various types of display panels, such as plasma display panel (PDP), vac-uum fluorescent display (VFD), and field emission display (FED) [1–3]. For enhancing the brightness and resolution of these displays, it is important to develop phosphors with high quantum efficiency, controlled morphology, and small particle sizes. Yttrium aluminum garnet,Y3Al5O12 (abbre-viated as YAG), is a well-known inorganic compound and is also widely utilized in the optical field. YAG single crystal doped with lanthanides, such as Nd3+, is used in solid-state lasers, and the poly-crystals of YAG doped with different color centers are employed as phosphors in the cathode ray tube (CRT) displays.
Traditionally, YAG phosphors are prepared at high tem-peratures (>1600◦C) for a long time via a solid-state tion process. Due to insufficient mixing and the low reac-tivity of raw materials, several intermediate phases such as
∗Corresponding author. Tel.:+886-2-363-5230;
fax:+886-2-362-3040.
E-mail address: [email protected] (C.-H. Lu).
Y4Al2O9(YAM) and YAlO3(YAP) easily exist in the prod-ucts[4]. Repeated grinding and calcination are required to eliminate these intermediate compounds, thus increasing the consumption in time as well as energy. For overcoming the drawbacks of the solid-state reaction process, several wet chemical methods such as co-precipitation [5,6], combus-tion[1,2], and spray pyrolysis[7,8]have been developed to fabricate the YAG-based phosphors. Although YAG can be obtained at low temperatures via these methods, further an-nealing at high temperatures (∼1400◦C) is required to im-prove the luminescent properties of the powders. Recently, a sol–gel process using metal alkoxides has also been used for synthesizing YAG phosphor[9,10]. However, this process is complicated due to the difficulty in handling of alkoxides.
A simple sol–gel process using citric acid and ethy-lene glycol as the gelation agents was adopted to fabricate nano-sized YAG:Eu3+ phosphors in this study. This pro-cess has been used for the preparation of ceramic powders
[11,12]and silica hybrid materials[13,14]. In this process, metal nitrates or carbonates are used as the starting materi-als; therefore, the problems of handling moisture-sensitive alkoxides can be avoided. Furthermore, different cations can be mixed to the quasi-atomic level that allows the pre-cise control of the chemical composition. In this study, the 0921-5107/$ – see front matter © 2003 Elsevier B.V. All rights reserved.
preparation of the nano-sized YAG:Eu3+phosphors via the sol–gel process was investigated. The relation between the photoluminescent properties of the sol–gel-derived powders and the preparation conditions as well as the particle size was discussed. The sol–gel-derived YAG:Eu3+ phosphors and those prepared via the solid-state reaction process were also compared in terms of their emission properties.
2. Experimental
In the sol–gel process for preparing YAG:Eu3+ phos-phors, yttrium nitrate and aluminum nitrate were dissolved in deionized water, and europium oxide was dissolved in dilute nitric acid. The prepared solutions were mixed ac-cording to the chemical formula of Y2.85Eu0.15Al5O12 with 5 at.% europium ions doped with respect to yttrium ions. The molar concentration of all metal ions in the solution was adjusted to 0.2 M. Citric acid and ethylene glycol in a 1:1.5 ratio were employed as gelation reagents, and the molar ra-tio of all metal ions to citric acid was maintained at 1:3. The solutions of metal nitrates and citric acid were mixed and stirred for 1.5 h, followed by adding ethylene glycol, and then the solution was heated to 130◦C on a hot plate for 1.5 h. The solution was evaporated at 300◦C to initi-ate the gelation reaction. A large amount of brownish gas was emitted, and the clear solution became yellowish white dry gels. The precursor powders, obtained by grinding the dry gels, were later calcined in the range of 800–1400◦C for 2 h. On the other hand, Y2.85Eu0.15Al5O12 phosphor was also prepared via the conventional solid-state reaction method. The constituent oxides were mixed in ethanol us-ing a ball-mill for 24 h. The slurry-like mixtures were dried in a vacuum-rotation dryer. The dried powders were cal-cined at 1500◦C for 4 h and then milled. The calcination and milling processes were repeated for three times. The formed phases in the synthesized powders were analyzed using an X-ray diffractometer (MAC science, MXP3). The morphol-ogy and the particle sizes of the powders were examined using a scanning electron microscope (Hitachi, S-800). The photoluminescence properties of the powders were investi-gated using a fluorescence spectrometer (Hitachi, F-4500). A 150 W Xe lamp was used as a multi-wavelength light source.
3. Results and discussion
The precursors prepared from the sol–gel process were yellowish powders. After 800◦C calcination, these precur-sors became white powders, implying that organic com-pounds were burned away during calcination.Fig. 1 illus-trates the X-ray diffraction patterns of YAG:Eu3+calcined at various temperatures from 800 to 1400◦C for 2 h. From the XRD patterns, it was found that the powders were still
Fig. 1. XRD patterns of the sol–gel-derived precursors of Y2.85Eu0.15Al5O12 calcined at (a) 800◦C, (b) 900◦C, (c) 1000◦C, (d)
1200◦C, and (e) 1400◦C for 2 h. The inset shows the corresponding in-tensity of the (420) peak of Y2.85Eu0.15Al5O12 prepared via the sol–gel
process and the solid-state reaction (S.S.).
amorphous at 800◦C. However, when the calcination tem-perature reached 900◦C, the cubic garnet phase of YAG[15]
was formed. The obtained diffraction pattern was well con-sistent with the data reported in ICDD[15], indicating that the single phase of YAG:Eu3+was successfully prepared at a temperature as low as 900◦C. It is noted that no interme-diate phases were formed between 800 and 900◦C, imply-ing that the sol–gel-derived precursors were directly trans-formed into the crystalline YAG phase during calcinations. When the calcination temperature was increased, the diffrac-tion intensities of the calcined powders increased (as shown in the inset ofFig. 1), and the width of the diffraction peaks decreased. The above results reveal that the crystallinity of the powders was enhanced with a rise in the calcination tem-perature. The white color of the prepared powders suggested that they had no absorption in the visible light range.
YAG:Eu3+ phosphors were also prepared via the solid-state reaction process. As shown inFig. 2, after the first calcination at 1500◦C for 4 h, one impure phase, Y4Al2O9 (YAM), coexisted with the YAG phase. After the second milling/calcination process, the impure YAM phase still remained in the sample. When the milling and calcination (1500◦C/4 h) process was repeated three times, pure YAG:Eu3+ phase was obtained. On the contrary, the results in Fig. 1 demonstrate that the required tempera-ture for synthesizing pure YAG:Eu3+ phosphors in the sol–gel process was much lower than that in the solid-state reaction process. This is attributed to the improved com-positional homogeneity and the enhanced reactivity of the sol–gel-derived precursors, thereby reducing the required heating temperature for preparing YAG:Eu3+ phosphors.
The microstructures of YAG:Eu3+powders prepared via the solid-state reaction process and the sol–gel process are shown in Fig. 3. After repeated milling and calcination (1500◦C/4 h), the solid-state-derived YAG:Eu3+ powders
Fig. 2. XRD patterns of Y2.85Eu0.15Al5O12 phosphors obtained via the
repeated solid-state reaction process. Calcination at 1500◦C for 4 h was repeated for (a) one time, (b) two times, and (c) three times.
exhibited a rounded morphology with a size of around 2–2.5m (see Fig. 3(a)). On the other hand, the particle size of 900◦C-calcined powders prepared via the sol–gel process was around 40 nm (as shown inFig. 3(b)). With a rise in the calcination temperatures, the particles of the pre-pared powders enlarged. When the calcination temperature reached 1400◦C, the particle size of YAG:Eu3+ powders increased to 200 nm, and their shape became nearly
spheri-Fig. 3. SEM micrograph of (a) the solid-state reaction derived Y2.85Eu0.15Al5O12 phosphors, and the sol–gel-derived Y2.85Eu0.15Al5O12 phosphors
calcined at (a) 900◦C, (b) 1200◦C, and (c) 1400◦C for 2 h.
Fig. 4. Emission spectra of (a) the solid-state reaction derived Y2.85Eu0.15Al5O12phosphors, and the sol–gel-derived Y2.85Eu0.15Al5O12
phosphors calcined at (b) 900◦C, (c) 1000◦C, (d) 1200◦C, and (e) 1400◦C for 2 h. The excitation wavelength is 250 nm.
cal (seeFig. 3(d)). It was also observed that the particle size of YAG:Eu3+ powders synthesized via the sol–gel process was much smaller than that of the powders prepared from the solid-state reaction process.
Fig. 4depicts the emission spectra of the sol–gel-derived YAG:Eu3+phosphors calcined at temperatures ranging from 900 to 1400◦C under the excitation of 250 nm UV light. The
emission spectrum of the powder prepared at 1500◦C via the solid-state reaction process is also compared in this figure. The powders prepared via these two processes had similar emission spectra except their emission intensity. Three ma-jor emission peaks were observed at 590, 594, and 608 nm, revealing the characteristic emission properties of the Eu3+ activators. The greatest emission at 590 nm occurred from 5D
0 → 7F1 transition of Eu3+. The 5D1 → 7F3 and 5D
0 →7F2 transition processes of Eu3+ were responsible for the emissions at 594 and 608 nm, respectively[16–18]. As for the powders prepared via the sol–gel process, the emission intensity of the obtained powders increased with a rise in the calcination temperature because of the en-hanced crystallinity. On the other hand, the 800◦C-calcined powders obtained from the sol–gel process did not exhibit any characteristic emission spectrum since the crystalline YAG phase was not formed in this sample, as shown in
Fig. 1.
It was found that the emission intensity of the phosphors prepared via the sol–gel process was much higher than that of the powders prepared via the solid-state reaction process. In the sol–gel process, citric acid polymerizes with the ethy-lene glycol, and these cross-linked organics form a large amount of tiny enclosures that effectively trap the constituent metal ions. Since the sol–gel process provides a more homo-geneous environment than the solid-state mixing, the crys-tallinity of YAG:Eu3+powders is improved (as shown in the inset inFig. 1) and the distribution of Eu3+ions in the YAG matrix is enhanced. The high crystallinity and good distribu-tion of Eu3+ activators reduce the non-radiative relaxation and results in an increase in the emission intensity. The ex-citation spectra of the emission at 608 nm of YAG:Eu3+ phosphors prepared via the sol–gel process are illustrated in
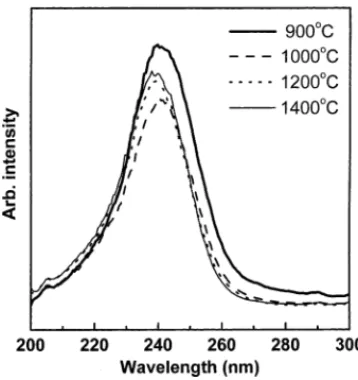
Fig. 5. The spectrum of 900◦C-calcined YAG:Eu3+ phos-phors exhibited a broad band on the long-wavelength side,
Fig. 5. Excitation spectrum of Y2.85Eu0.15Al5O12 phosphors monitored
the emission at 608 nm. These phosphors were prepared via the sol–gel process and calcined at various temperatures.
which is possibly related to the charge transfer process in the phosphors. Since the 900◦C-calcined sample had a low crystallinity, its crystal periodicity tended to be disorderly. This led to the production of more possible energy states for the Eu3+–O2− pairs, thereby resulting in the broaden-ing of the excitation band. The above results indicate that the precise control of the morphology, particle size, and crystallinity of YAG:Eu3+ is required for improving their photoluminescence properties.
4. Conclusions
Nano-sized YAG:Eu3+ phosphors were successfully ob-tained via a sol–gel process at a temperature as low as 900◦C. The synthesis temperature was much lower than that in the conventional solid-state reaction process. During the gelation process, citric acid and ethylene glycol formed nu-merous tiny enclosures that trapped the constituent cations, leading to a shortening of the interdiffusion length and an increase in the reactivity of the precursors. The particle size of the sol–gel-derived YAG:Eu3+phosphors was 40 nm (900◦C), which was greatly smaller than that of the pow-ders prepared via the solid-state reaction process. The pho-toluminescence intensity of the sol–gel-derived phosphors was markedly stronger than that of the powders prepared via the solid-state reaction process. This is considered to be attributed to the increased crystallinity and improved dis-tribution of activators in the sol–gel-derived powders. The photoluminescence properties of YAG:Eu3+phosphors were found to be significantly dependent on their morphology and particle size.
References
[1] L.E. Shea, J. McKittrick, O.A. Lopez, J. Am. Ceram. Soc. 79 (1996) 3257.
[2] J. McKittrick, L.E. Shea, C.F. Bacalski, E.J. Bosze, Displays 19 (1999) 169.
[3] Y.C. Kang, I.W. Lenggoro, S.B. Park, K. Okuyama, J. Phys. Chem. Solids 60 (1999) 1855.
[4] I. Matsubara, M. Parathaman, S.W. Allison, M.R. Cates, D.L. Beshears, D.E. Holcomb, Mater. Res. Bull. 35 (2000) 217. [5] J.G. Li, T. Ikegami, J.H. Lee, T. Mori, Y. Yajima, J. Mater. Res. 15
(2000) 1864.
[6] N. Matsushita, N. Tsuchiya, K. Nakatsuka, J. Am. Ceram. Soc. 82 (1999) 1977.
[7] Y.C. Kang, Y.S. Chung, S.B. Park, J. Am. Ceram. Soc. 82 (1999) 2056.
[8] Y.C. Kang, I.W. Lenggoro, S.B. Park, K. Okuyama, Mater. Res. Bull. 35 (2000) 789.
[9] J.Y. Choe, D. Ravichandran, S.M. Blomquist, K.W. Kirchner, E.W. Forsythe, D.C. Morton, J. Lumin. 93 (2001) 119.
[10] J.Y. Choe, D. Ravichandran, S.M. Blomquist, D.C. Morton, K.W. Kirchner, M.H. Ervin, U. Lee, Appl. Phys. Lett. 78 (2001) 3800. [11] C.H. Lu, S.K. Saha, Mater. Res. Bull. 35 (2000) 2135.
[12] A. Kahoul, P. Nkeng, A. Hammouche, F. Naamoune, G. Poillerat, J. Solid State Chem. 161 (2001) 379.
[13] A. Campero, J. Cardoso, S. Pacheco, J. Sol–Gel Sci. Tech. 8 (1997) 535.
[14] M. Yabuki, R. Takahashi, S. Sato, T. Sodesawa, K. Ogura, Phys. Chem. Chem. Phys. 4 (2002) 4830.
[15] X-ray Powder Data File, Card No. 33-0040, International Centre of Diffraction Data, 1997.
[16] S.K. Ruan, J.G. Zhou, A.M. Zhong, J.F. Duan, X.B. Yang, M.Z. Su, J. Alloys Compd. 275–277 (1998) 72.
[17] S. Shikao, W. Jiye, J. Alloys Compd. 327 (2001) 82.
[18] D. Ravichandran, R. Roy, A.G. Chakhovskoi, C.E. Hunt, W.B. White, S. Erdei, J. Lumin. 71 (1997) 291.