Baicalein inhibition of oxidative-stress-induced apoptosis
via modulation of ERKs activation and induction of
HO-1 gene expression in rat glioma cells C6
Yen-Chou Chen
a,⁎
, Jyh-Ming Chow
b, Cheng-Wei Lin
a, Chin-Yen Wu
a, Shing-Chuan Shen
c,da
Graduate Institute of Pharmacognosy, School of Pharmacy, Taipei Medical University, Taipei, Taiwan
b
Section of Hematology-Oncology, Department of Internal, Medicine, Taipei Municipal Wan-Fang Hospital, Taipei Medical University, Taipei, Taiwan
c
Department of Dermatology, School of Medicine, Taipei Medical University, Taipei, Taiwan
d
Department of Dermatology, Taipei Municipal Wan-Fang Hospital, Taipei, Taiwan Received 13 January 2006; revised 9 May 2006; accepted 14 May 2006
Available online 19 May 2006
Abstract
In the present study, we examined the protective mechanism of baicalein (BE) and its glycoside, baicalin (BI), on hydrogen-peroxide (H
2O
2)-induced cell death in rat glioma C6 cells. Results of the MTT assay, LDH release assay, and morphological observation showed that H
2O
2addition
reduced the viability of C6 cells, and this was prevented by the addition of BE but not BI. Incubation of C6 cells with BE significantly decreased
the intracellular peroxide level induced by H
2O
2according to flow cytometric analysis using DCHF-DA as a fluorescent substrate. Suppression of
H
2O
2-induced apoptotic events including DNA ladders, hypodiploid cells, and activation of caspases 3, 8, and, 9 by BE but not BI was identified
in C6 cells. The cytotoxicity and phosphorylation of ERK proteins induced by H
2O
2were blocked by the ERK inhibitor PD98059. Catalase
addition prevented H
2O
2-induced ROS production, ERKs protein phosphorylation, and cell death, and BE dose-dependently inhibited H
2O
2-induced ERK protein phosphorylation in C6 cells. These data suggest that ROS-scavenging activity is involved in BE prevention of H
2O
2-induced
cell death via blocking ERKs activation. Additionally, BE but not BI induced heat shock protein 32 (HSP32; HO-1) protein expression in both
time- and dose-dependent manners, but not heme oxygenase 2 (HO-2), heat shock protein 70 (HSP70), or heat shock protein 90 (HSP90) protein
expression. In the absence of H
2O
2, BE induces ERKs protein phosphorylation, and HO-1 protein expression induced by BE was blocked by the
addition of cycloheximide, actinomycin D, and the ERK inhibitor PD98059. The addition of the HO inhibitor ZnPP inhibited the protective effect
of BE against H
2O
2-induced cytotoxicity in C6 cells according to the MTT assay and apoptotic morphology under microscopic observation,
accompanied by blocking the ROS-scavenging activity of BE in C6 cells. However, BE treatment was unable to protect C6 cells from
C2-ceramide-induced cell death. These data indicate that BE possesses abilities to inhibit ROS-mediated cytotoxic effects through modulation of
ERKs activation and induction of HO-1 protein expression. The role of HO-1 in ROS-scavenging activity of BE is proposed.
© 2006 Elsevier Inc. All rights reserved.
Keywords: Baicalein; Hydrogen peroxide; Apoptosis; ERKs; HO-1
Introduction
Baicalein (BE) is one of the major flavonoids in Scutellaria
baicalensis, which has long been extensively used in Chinese
herbal medicine. Several biological effects of BE such as
anti-viral, anti-inflammation, anti-hepatotoxicity, and anti-tumor
properties have been reported (
Ahn et al., 2001; Huang et al.,
2005; Hwang et al., 2005
). Most activities of BE are attributed to
its antioxidant and prooxidant capacities.
Wang et al. (2004)
indicated that BE induced apoptosis in tumor cells via the
Abbreviations: BE, baicalein; BI, baicalin; LPS, lipopolysaccharide; TPA,12-O-tetradecanoylphorbol 13-acetate; H2O2, hydrogen peroxide; HO-1, heme
oxygenase 1; HSP70, heat shock protein 70; HSP90, heat shock protein 90; HO-2, heme oxygenase 2; LDH, lactate dehydrogenase; ZnPP, zinc protoporphyrin; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; NBT, nitroblue tetrazolium; BCIP, 5-bromo-4-chloro-3-indolyl phosphate; ERKs, extracellular regulated kinases; JNKs, c-Jun N-terminal kinases; ROS, reactive oxygen species.
⁎ Corresponding author. Fax: +1 886 2 23787139. E-mail address:[email protected](Y.-C. Chen).
0041-008X/$ - see front matter © 2006 Elsevier Inc. All rights reserved. doi:10.1016/j.taap.2006.05.008
production and accumulation of intracellular reactive oxygen
species (ROS). There is additional evidence showing that BE
exhibits protective effects against ischemia-induced apoptosis in
cardiomyocytes via reducing hydrogen peroxide production
(
Shao et al., 1999, 2002
). Increasing evidence indicates that BE
possesses the ability to protect against cellular damage induced
Fig. 1. BE but not BI protects glioma C6 cells from H2O2-induced cytotoxicity. (A) Chemical structures of baicalein (BE) and baicalin (BI). BI is a BE glycoside and
contains a glucoside moiety at C7 of BE. (B) C6 cells were treated with BE or BI (100μM) for 30 min followed by H2O2(400μM) incubation for a further 12 h. The
morphological changes were detected under microscopic observation via Giemsa staining. (C) As described in (B), C6 cells were treated with BE or BI (50 and 100μM) for 30 min followed by H2O2(400μM) incubation for a further 12 h. The viability of C6 cells under different treatments was examined by an MTT assay as
described in Materials and methods. (D) Under conditions described in (C), the amount of LDH released from cells after different treatments was detected, and data are expressed as the percentage of cytotoxicity as described in Materials and methods. C or CON, control group. Data are expressed as the mean ± SE. **pb 0.01 indicates a significant difference from the H2O2-treated group.
by ROS and chemotoxic agents.
Shieh et al. (2000)
indicated
that BE and BI possess potent eliminative activities on free
radical production via suppressing xanthine oxidase activity. In
the presence of lipopolysaccharide (LPS) treatment, BE prevents
the degeneration of dopaminergic neurons induced by LPS, and
the suppression of nitric oxide (NO)-induced cytotoxicity by BE
in microglia cells via blocking NF-
κB activation was identified
(
Chen et al., 2004a,b; Li et al., 2005
). Although several
biological activities of BE have been reported, the mechanism
by which BE prevents oxidative-stress-induced cell death in
glioma cells is still unclear.
Structural modification of flavonoids occurs extensively in
plants, and glycosylation, one type of structural modification,
commonly appears in the metabolism of flavonoids (
Hollman et
al., 1999; Rivera et al., 2004
). Several previous studies indicated
that glycosylation of flavonoids increases their hydrophilicity and
induces resistance to enzyme oxidation in plants (
Birt et al., 2001;
Chen et al., 2001; Regev-Shoshani et al., 2003
). Evidence related
to the effects of glycoside addition on the biological activities of
flavonoids is still lacking. Our previous study demonstrated that
aglycon flavonoids such as quercetin showed more significant
NO inhibitory activity and apoptosis-inducing activities than its
glycosides, rutin and quercitrin, in RAW264.7 macrophages and
human leukemia HL-60 cells, respectively (
Chen et al., 2001;
Shen et al., 2003
). Additionally, the aglycons, hesperitin and
naringenin, but not their respective glycosides, hesperidin and
naringin, inhibit NO production in macrophages via inducing
heme oxygenase 1 (HO-1) protein expression, as well as inducing
apoptosis in leukemia cells (
Chen et al., 2003; Lin et al., 2005
).
These data indicate that glycosides may play as a negative moiety
in the biological actions of flavonoids. However, the effect of
glycoside in BE prevention of oxidative-stress-induced cell death
in glioma cells has yet to be investigated.
It has been reported that ROS participate in causing such
human diseases as diabetes, tumors, atherosclerosis, stroke, and
neurodegenerative diseases such as Parkinson's disease, stroke,
amyotrophic lateral sclerosis, and Alzheimer's disease (
Crack
and Taylor, 2005; Henze et al., 2005; Laufs et al., 2005; Park
et al., 2005
). Glial cells play a major role in maintaining the
functions of neurons in the brain, and several studies have
indicated that ROS accumulation in glial cells leads to cell death
and dysfunction of the surrounding neurons (
de Bernardo et al.,
2004; Juravleva et al., 2005; Qian et al., 2005
). Therefore, agents
with the ability to protect glial cells from ROS-dependent cell
death may possess the potential to treat neurodegenerative
diseases. Heme oxygenases (HOs) are enzymes which catalyze
heme to bilirubin, biliverdin, carbon monoxide (CO), and
ferrous iron (Fe
2+). At least three HOs including HO-1, HO-2,
and HO-3 have been identified. HO-1 is inducible in response to
several stimuli such as heme, heavy metals, LPS, and
inflammatory cytokines. Previous studies showed that induction
of HO-1 gene expression protects cells from cell death (
Chen
et al., 2000, 2002; Choi et al., 2004; Petrache et al., 2000
), and
upregulation of the HO-1 protein by the HO-1 inducers, hemin
and cadmium, induces resistance to apoptotic stimuli in human
gastric cancer cells (
Liu et al., 2004
). Overexpression of the
HO-1 protein protects vascular smooth muscle cells from
angio-tensin-II-induced damages (
Morita et al., 2005
). These data
support the protective function of the HO-1 protein; however,
the role of the HO-1 protein in the biological action of BE is still
undefined.
In the present study, we investigated the mechanism of BE
and its glycoside, baicalin, on oxidative-stress (H
2O
2)-induced
apoptosis in rat glioma C6 cells, and the roles of the HO-1
protein, ERK protein, and ROS-scavenging activity in the
pre-ventive mechanism of BE were investigated.
Materials and methods
Cell culture. Rat glioma C6 cells from ATCC (American Type Culture Collection; Rockville, MD) were incubated in RPMI-1640 medium supplemented with 2 mM glutamine, antibiotics (100 U/ml of penicillin A and 100 U/ml of streptomycin), and 10% heat-inactivated fetal bovine serum and maintained at 37 °C in a humidified incubator containing 5% CO2.
Chemicals. The structurally related flavonoids including baicalein and baicalin were obtained from Sigma Chemical (St. Louis, MO). (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) (MTT), hydrogen peroxide (H2O2), zinc
protophyrin (ZnPP), actinomycin D, cycloheximide, 2 ′,7′-dichlorodihydrofluor-escein-diacetate (DCHF-DA), and propidium iodine (PI) were also obtained from Sigma. The antibodies of anti-HO-1, anti-α-tubulin, pERKs, HO-2, anti-iNOS, and anti-HSP90 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). PD98059, Ac-DEVD-pNA, Ac-IETD-pNA, and Ac-LEHD-pNA were obtained from Calbiochem (La Jolla, CA).
Cell viability. MTT was used as an indicator of cell viability as determined by its mitochondrial-dependent reduction to formazone. Cells were plated at a density of 4 × 105cells/well in 24-well plates for 12 h followed by treatment with different concentrations of BE for a further 12 h. Cells were washed with PBS three times, and MTT (50 mg/ml) was added to the medium for 4 h. Then, the supernatant was removed, and the formazone crystals were dissolved using 0.04 N HCl in isopropanol. The absorbance was read at 600 nm with an ELISA analyzer (Dyna-tech MR-7000; Dyna(Dyna-tech Laboratories).
LDH release assay. The percentage of LDH release was expressed as the proportion of LDH released into the medium compared to the total amount of LDH present in cells treated with 2% Triton X-100. The activity was monitored as the oxidation of NADH at 530 nm by an LDH assay kit (Roche Applied Science). The cytotoxicity (%) was determined by the equation [(OD530 of the treated group − OD530 of the control group)/(OD530 of the Triton X-100-treated group− OD530 of the control group)] × 100%.
Caspase activity assay. After different treatments, glioma C6 cells were collected and washed three times with PBS and resuspended in 50 mM Tris– HCl (pH 7.4), 1 mM EDTA, and 10 mM ethyleneglycoltetraacetic acid (EGTA). Cell lysates were clarified by centrifugation at 20,000 × g for 3 min, and clear lysates containing 100μg of protein were incubated with 100 μM enzyme-specific colorimetric substrates including Ac-DEVD-pNA for caspase 3/CPP32, Ac-IETD-pNA for caspase 8, and Ac-LEHD-pNA for caspase 9 at 37 °C for 1 h. Alternative activity of the indicated caspases was described as the cleavage of colorimetric substrate by measuring the absorbance at 405 nm.
Western blotting. Total cellular extracts were prepared, and an equal amount of proteins from each group was separated on 8%–12% SDS-polyacrylamide minigels followed by transfer to immobilon polyvinylidenedifluoride membranes (Millipore, Bedford, MA). Membranes were incubated with 1% bovine serum albumin and then incubated with specific antibodies overnight at 4 °C. Protein expression was detected by staining with nitroblue tetrazolium (NBT) and 5-bromo-4-chloro-3-indolyl phosphate (BCIP) (Sigma).
DNA gel electrophoresis. Cells under different treatments were collected, washed twice with PBS, lysed in 80μl of lysis buffer (50 mM Tris (pH 8.0), 10 mM ethylenediaminetetraacetic acid (EDTA), 0.5% sodium sarkosinate, and 1 mg/ml proteinase K) for 3 h at 56 °C, and then treated with 0.5 mg/ml RNase A for an additional 1 h at 56 °C. DNA was extracted with phenol/chloroform/
isoamyl alcohol (25/24/1, v/v) before loading. Samples were mixed with loading buffer (50 mM Tris, 10 mM EDTA, 1% (w/w) low-melting point agarose, and 0.025% (w/w) bromophenol blue) and loaded onto a pre-solidified 2% agarose gel containing 0.1 mg/ml ethidium bromide. The agarose gels were run at 50 V for 90 min in TBE buffer, after which they were observed and photographed under UV light.
Determination of ROS production by flow cytometry analysis and fluorescent microscopic observation. The production of ROS was monitored by flow cytometry using DCHF-DA. This dye is a stable compound that readily diffuses into cells and is hydrolyzed by intracellular esterase to yield DCHF, which is trapped within cells. Hydrogen peroxide or low-molecular-weight hydroperoxides produced by cells oxidize DCHF to the highly fluorescent compound, 2 ′,7′-dichlorofluorescein (DCF). Thus, the fluorescence intensity is proportional to the amount of peroxide produced by cells. In the present study, cells were treated with BE for 30 min followed by H2O2addition for 1 h. Then, compound-treated cells
were washed twice with PBS to remove the extracellular compounds, and DCHF-DA (100μM) green fluorescence was added, excited using an argon laser, and detected using a 525-nm (FL1-H) band-pass filter by flow cytometric analysis. As the same part of experiments, the green fluorescence in cells was observed by fluorescent microscope.
Statistical analysis. All experiments were performed in triplicate. ANOVA was used to determine differences among groups. If a significant difference was found, t test was used to determine the location of the difference between indicated two groups; values of pb 0.05 and p b 0.01 were considered statistically significant.
Results
BE but not BI protects glioma C6 cells from H
2O
2-induced cell
death
The structures of BE and BI are depicted in
Fig. 1
A; BI
contains a glycoside at C7 of BE (
Fig. 1
A). In order to
investigate if BE or BI exhibits the ability to protect glioma
cells from ROS-induced cell death, C6 cells were treated with
H
2O
2(400
μM) in the presence or absence of BE or BI
incubation, and the viability of cells was examined by
morphological observation and MTT and LDH release assays.
Results of the morphological observation via Giemsa staining
indicated that H
2O
2induced the appearance of condensed cells
(dark ones) in C6 cells, which was prevented by the addition of
BE but not BI (100
μM) (
Fig. 1
B). Data from the MTT and
LDH release assays showed that a decrease in the viability of
C6 cells was detected in H
2O
2-treated C6 cells and that this
was blocked by adding BE but not BI (
Figs. 1
C, D). These
data revealed that BE but not BI protected C6 cells from H
2O
2-induced cytotoxicity.
Fig. 2. BE inhibits H2O2-induced apoptotic events in C6 cells. (A) BE prevention of DNA fragmentation induced by H2O2in C6 cells. C6 cells were treated with BE or BI
(50 or 100μM) for 30 min followed by H2O2treatment for a further 12 h. The integrity of the DNA in cells under different treatments was determined via agarose
electrophoresis. (B) BE inhibition of H2O2-induced hypodiploid cells (sub-G1) in C6 cells. As described in (A), the ratio of hypodiploid cells was measured by flow
cytometric analysis. (C–E) BE inhibition of H2O2-induced caspase 3, 8, and 9 enzyme activity in glioma C6 cells. Activities of caspase 3 (C), caspase 8 (D), and caspase 9 (E)
enzymes stimulated by H2O2in the presence or absence of BE or BI (100μM) were detected using specific peptidyl substrates as described in Materials and methods. (F) BE
reversed H2O2-induced decreases in the expressions of pro-caspases 3, 8, and 9 and pro-PARP protein. As described previously, the expression of the pro-form of caspases 3,
8, and 9 and the PARP protein was detected by Western blotting. The expression ofα-tubulin protein was used as an internal control. Each value is presented as the mean ± SE. **pb 0.01 indicates a significant difference from the control group; and##pb 0.01 indicates a significant difference between indicated groups.
BE inhibits H2O2-induced apoptotic events in rat glioma C6
cells
We further investigated the effect of BE and BI on H
2O
2-induced apoptotic characteristics including DNA ladders,
hypo-diploid cells (sub-G1), and caspase activation in rat glioma cells
C6. Results in
Fig. 2
A show that H
2O
2induced a loss in the
integrity of DNA in accordance with the appearance of
frag-mented DNA in C6 cells, and this was prevented by the addition
of BE but not BI to C6 cells (
Fig. 2
A). Data of the flow cytometric
analysis showed that the ratio of hypodiploid cells induced by
H
2O
2was attenuated by the addition of BE but not BI (
Fig. 2
B).
An increase in the activation of the indicated caspases, including
caspases 3, 8, and 9, was detected in H
2O
2-treated C6 cells using
specific colorimetric peptidyl substrates (Ac-DEVD-pNA for
caspase 3, Ac-IETD-pNA for caspase 8, and Ac-LEHD-pNA for
caspase 9), and those effects were significantly attenuated by BE
but not BI (
Figs. 2
B–D). Data from Western blotting showed that
H
2O
2treatment induced decreases in the levels of pro-caspases 3,
8, and 9 and caspase 3 substrate PARP protein in C6 cells, and
these were blocked by BE but not BI (
Fig. 2
E). A similar level of
α-tubulin protein expression in each lane was used as an internal
control. This suggests that BE prevents H
2O
2-induced cell death
via blocking cells from undergoing apoptosis.
Activation of ERKs protein via phosphorylation induction is
involved in H2O2-induced C6 cell death which was blocked by
BE
Since activation of ERKs by H
2O
2has extensively been
shown, we investigated the role of ERK activation in the
pre-ventive mechanism of BE. Results in
Fig. 3
A show that the
Fig. 3. Involvement of ERK activation in H2O2-induced cell death. (A) H2O2-induced ERK protein phosphorylation in a time-dependent manner. C6 cells were treated
with H2O2(400μM) for different times (10, 20, 40, 60, and 80 min), and the expressions of phosphorylated ERK (p-ERK) and total ERK (t-ERK) proteins were
detected by Western blotting using specific antibodies. (B) (Lower panel) PD98059, a specific ERK activity inhibitor, suppressed H2O2-induced cytotoxicity in C6
cells according to MTT assay. Cells were treated with different doses of PD98059 (2.5, 5, 10, and 20μM) for 30 min followed by H2O2treatment for a further 12 h. The
viability of cells was measured by the MTT assay. (Upper panel) PD98059 dose-dependently attenuated H2O2-induced phosphorylation of ERK protein. Cells were
treated with different doses of PD98059 (2.5, 5, 10, and 20μM) for 30 min followed by H2O2treatment for a further 20 min. The expressions of phosphorylated ERK
(p-ERK) and total ERK (t-ERK) proteins were detected by Western blotting using specific antibodies. (C) Catalase addition inhibited H2O2-induced ERK protein
phosphorylation in accordance with suppression of the H2O2-induced cytotoxic effects in C6 cells. (Upper panel) C6 cells were treated with catalase (25, 50, and
100 U/ml) for 30 min followed by H2O2(400μM) treatment for 20 min (ERK protein detection) and 12 h (MTT assay). The expressions of p-ERK and t-ERK proteins
(upper panel) and the viability of C6 cells (lower panel) under different treatments were examined by Western blotting and the MTT assay, respectively. (D) BE dose-dependently inhibited ERK activation induced by H2O2. Cells were treated with different doses of BE (25, 50, and 100μM) for 30 min followed by H2O2treatment for
a further 20 min. The expressions of p-ERK and t-ERK proteins were detected by Western blotting using specific antibodies. Each value is presented as the mean ± SE. **pb 0.01 indicates a significant difference from the control group; and##pb 0.01 indicates a significant difference from H2O2-treated groups.
expression of phosphorylated ERKs proteins was induced in
H
2O
2-treated C6 cells, and the time course of phosphorylated
ERKs proteins was appeared in 10 to 60 min after H
2O
2treat-ment. Incubation of C6 cells with the chemical ERK inhibitor,
PD98059, dose-dependently protected C6 cells from H
2O
2-induced cytotoxicity according to the MTT assay (
Fig. 3
B; lower
panel), accompanied by suppression of H
2O
2-induced ERKs
protein phosphorylation (
Fig. 3
B; upper panel). Catalase addition
dose-dependently prevented H
2O
2-induced cytotoxicity according to
the MTT assay by blocking ERKs protein phosphorylation in C6
cells (
Fig. 3
C). Furthermore, results in
Fig. 3
D show that BE
dose-dependently inhibited H
2O
2-induced ERK protein phosphorylation
in C6 cells. These data indicated that blocking ERK protein
phosphorylation induced by H
2O
2is involved in the action of BE in
rat glioma C6 cells.
ROS-scavenging activity participates in BE protection against
H2O2-induced cell death in rat glioma C6 cells
It is important to examine if BE protects C6 cells from H
2O
2-induced apoptosis via reducing intracellular ROS production.
DCHF-DA has been used to examine changes in intracellular
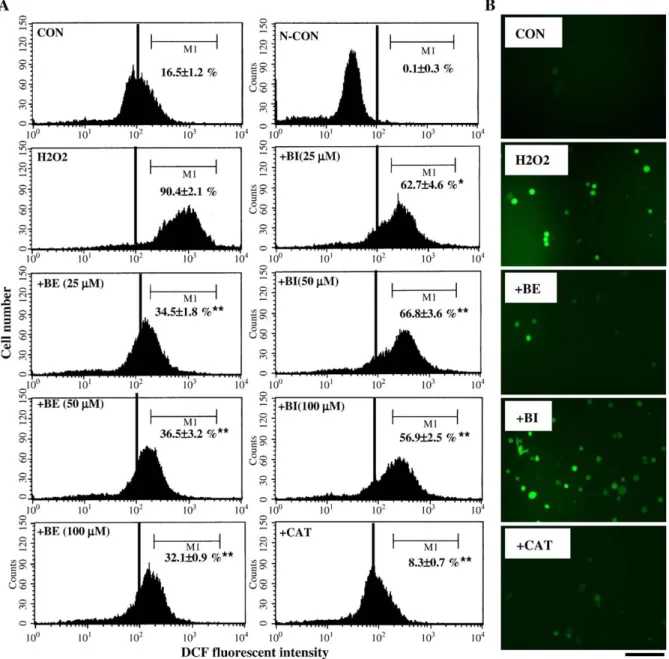
peroxide levels through flow cytometric analysis. Results in
Fig. 4
A indicate that H
2O
2(400
μM) treatment significantly
induced intracellular peroxide production, and the percentage of
ROS-overexpressed C6 cells (M1) is 90.4 ± 2.1%. A decrease
Fig. 4. Free radical-scavenging activities of BE and BI by the DCHF-DA assay. (A) C6 cells were treated with different doses of BE and BI (25, 50, and 100μM) or catalase (50 U/ml) for 30 min followed by adding H2O2(400μM) for a further 1 h. The level of intracellular peroxide was detected by the DCHF-DA assay using flow cytometric
analysis. The percentage of cells under different treatments in M1 was detected and expressed as the mean ± SE from three independent experiments. *pb 0.05, **p b 0.01 indicate a significant difference between indicated groups, as analyzed by Student's t test. (B) As described in (A), cells were treated with or without BE (100μM), BI (100μM), or catalase (CAT; 50 U/ml) for 30 min followed by H2O2(400μM) treatment. The fluorescence in cells was detected by fluorescent microscopic observation.
in intracellular fluorescent intensity was detected in C6 cells
without DCHF-DA addition as a negative control to verify the
specificity of the reaction (N-CON). Incubation of C6 cells with
different doses (25, 50, and 100
μM) of BE significantly
re-duced H
2O
2(400
μM)-induced intracellular peroxide
produc-tion, and the percentages of M1 in 25, 50, and 100
μM of
BE-treated cells are 34.5 ± 1.8%, 36.5 ± 3.2%, and 32.1 ± 0.9%,
respectively. BI performs significant, but less effective than BE,
inhibitory effect on H
2O
2-induced ROS production, and the
percentages of M1 in 25, 50, and 100
μM of BI-treated cells are
62.7 ± 4.6%, 66.8 ± 3.6%, and 56.9 ± 2.5%, respectively.
Catalase (CAT) reduction of H
2O
2-induced intracellular
perox-ide production to the control level was used as a positive
control. As the same part of experiment, data of fluorescent
microscopic observation indicated that an increase in
intracel-lular fluorescent intensity was detected in H
2O
2-treated C6 cells
and was blocked by BE and catalase. These data suggest the
ROS-scavenging activity of BE in C6 cells.
BE but not BI induces HO-1 protein expression in rat glioma
C6 cells
Induction of HSPs has been shown to protect cells from
oxidative-stress-induced cell death, therefore we investigated
the effect of BE on the expressions of HO-1, HO-2, HSP70, and
HSP90 in C6 cells. As illustrated in
Fig. 5
A, BE but not BI
dose-dependently induced HO-1, but not HO-2, HSP70, or
HSP90, protein expression in C6 cells. In the same part of the
experiment, BE induced HO-1 protein expression in a
time-dependent manner (
Fig. 5
B). Both with and without FBS, BE
expressed similar inductive effects on HO-1 protein expression
in C6 cells (
Fig. 5
C). An increase in HO-1 protein expression
by the traditional HO-1 inducer hemin was used as a positive
control here.
Induction of ERKs, but not p38 or JNKs, protein phosphorylation
is involved in BE induction of HO-1 protein expression
Data from
Fig. 6
A show that the induction of ERKs, but not
p38 or JNKs, protein phosphorylation was detected in
BE-treated glioma C6 cells by Western blotting using specific
antibodies. No change in the expression of total ERKs, p38, and
Fig. 5. BE but not BI induced HO-1 protein expression in C6 cells. (A)Dose-dependent induction of HO-1 protein by BE (but not BI) in C6 cells. Cells were treated with BE or BI (50, 100, and 200μM) for 12 h, and the expressions of HO-1, HO-2, HSP70, and HSP90 protein were detected by Western blotting. (B) BE and hemin (HEM) time-dependently induced HO-1 protein expression in C6 cells. Cells were treated with BE (100μM) or hemin (HEM, 10 μM) for different times (2, 4, 8, and 12 h), and the expressions of HO-1, HO-2, and HSP90 protein were detected. (C) A similar inductive pattern of HO-1 protein stimulated by BE (50, 100, and 200μM) was detected in the presence (FBS) or absence (SF) of 10% FBS. Cells were treated with different doses of BE with or without FBS, and the expressions of HO-1, HO-2, and HSP90 proteins were detected by Western blotting.
Fig. 6. Induction of phosphorylated ERK (but not p38 or JNK) proteins is involved in BE-induced HO-1 protein expression. (A) BE induced ERK but not p38 or JNK protein phosphorylation in C6 cells. C6 cells were treated with BE or BI (100μM) for different times (20, 40, and 60 min), and the expressions of phosphorylated and total ERK, p38, and JNK proteins were detected by Western blotting using specific antibodies. (B) PD98059 dose-dependently inhibited BE-induced HO-1 protein expression in C6 cells. Cells were treated with PD98059 (5, 10, and 20μM) for 30 min followed by the addition of BE (100 μM) for a further 12 h. The expressions of HO-1 and HO-2 proteins were detected by Western blotting using specific antibodies. (C) Addition of the translational inhibitor cycloheximide (CHX; 1 μg/ml) or the transcriptional inhibitor actinomycin D (Act D; 1μg/ml) reduced HO-1 protein expression induced by BE (100μM). C6 cells were treated with CHX or Act D for 30 min followed by BE treatment for a further 12 h (for detecting HO-1, HO-2, and HSP90 proteins) or 20 min (for detecting phosphorylated and total ERK proteins).
JNKs protein was detected to verify similar amount of proteins
loaded in each lane. Adding PD98059 blocked HO-1 (but not
HO-2) protein expression induced by BE (
Fig. 6
B).
Incuba-tion of C6 cells with the translaIncuba-tional inhibitor cycloheximide
(CHX) and the transcriptional inhibitor actinomycin D (Act D)
significantly inhibited HO-1, but not HO-2, HSP70, or HSP90,
protein expression in BE-treated C6 cells. However, CHX and
Act D were unable to block ERK protein phosphorylation
induced by BE (
Fig. 6
C). These data suggest that HO-1 protein
expression induced by BE occurs via de novo protein synthesis
and that it is located downstream of ERK activation in C6 cells.
The HO-1 activity inhibitor zinc protoporphyrin suppresses the
inhibitory effect of BE on H2O2-induced cell death
Zinc protoporphyrin (ZnPP) has been used as an HO inhibitor.
In order to delineate if HO-1 induction participates in BE protection
of C6 cells against H
2O
2-induced cytotoxicity, cells were treated
with BE (100
μM) for 8 h to induce HO-1 protein expression
followed by adding different doses (0.5, 1, 2
μM) of ZnPP for
30 min and incubated with H
2O
2for a further 12 h. Results of the
MTT assay showed that the protective effect of BE on H
2O
2-induced cytotoxicity was dose-dependently reversed by the
addi-tion of ZnPP (
Fig. 7
A). Data of the morphological observations
indicated that condensed cells reappeared in ZnPP-treated cells in
the presence of BE and H
2O
2treatments (
Fig. 7
B). Analysis of
intracellular peroxide production shows that ZnPP suppresses the
ROS-scavenging activity of BE, and the percentages of M1 in
control, H
2O
2, ZnPP, H
2O
2plus BE, H
2O
2plus ZnPP, and H
2O
2plus BE plus ZnPP-treated C6 cells are 6.2 ± 0.9%, 92.5 ± 2.7%,
17.2 ± 1.8%, 22.8 ± 3.1%, 75.5 ± 4.3%, and 41.6 ± 1.5%,
respectively (
Fig. 7
C). It suggests that induction of HO-1 gene
expression participates in BE against H
2O
2-induced cell death and
ROS production in C6 cells.
Fig. 7. The chemical HO-1 enzyme inhibitor ZnPP reversed the protective effect and ROS-scavenging activity of BE against H2O2-induced cell death. (A) C6 cells
were treated with BE (100μM) for 8 h followed by incubating with different doses of ZnPP (0.5, 1, and 2 μM) for 30 min and addition of H2O2(400μM) into the cells
for a further 12 h. The viability of cells was detected by the MTT assay. (B) As described in (A), morphological changes in the condition with or without BE (100μM) or ZnPP (2μM) followed by H2O2stimulation for 12 h were observed microscopically using Giemsa staining. (C) Alternations in intracellular peroxide production
were detected by DCHF-DA assay. C6 cells were treated with BE (100μM) for 8 h followed by incubating with or without ZnPP (2 μM) for 30 min. H2O2(400μM)
BE expresses no protective effect against C2-ceramide-induced
cell death in rat glioma C6 cells
We examined if BE possesses an inhibitory effect on
intracellular ROS-induced cell death in C6 cells. C2-ceramide
has been shown to induce apoptosis in several cells via inducing
intracellular ROS production. Therefore, we investigated the
effect of BE on the C2-ceramide-induced cytotoxic effect in C6
cells. As illustrated in
Fig. 8
A, the addition of BE and BI
produced no protective effect on the C2-ceramide-induced
cytotoxic effect in C6 cells according to the MTT assay. Loss of
DNA integrity was detected in C2-ceramide-treated C6 cells;
however, neither BE nor BI inhibited C2-ceramide-induced
DNA fragmentation (
Fig. 8
B). In addition, condensed and
rounded cells appeared in C2-ceramide-treated C6 cells under
microscopic observation, and their formation was not prevented
by BE or BI treatment (
Fig. 8
C).
Discussion
Several studies have indicated that the protective effects of
flavonoids are derived from their free radical-scavenging
acti-vities, whereas the antioxidant properties are insufficient to
explain the protective mechanism of flavonoids (
Horvathova
et al., 2003; Singh and Chopra, 2004
). BE have been
demon-strated to prevent ROS-induced cell damage by acting as a free
radical scavenger due to its high trolox equivalent antioxidant
capacity (TEAC) and DPPH free radical scavenging activity
(
Pietta, 2000; Ishige et al., 2001
). In the present study, BE
inhibited apoptosis induced by H
2O
2with a reduction in
intra-cellular peroxide levels. This suggests that the antioxidative
activity may participate in BE protection of C6 cells against
H
2O
2-induced cytotoxic events. C2-ceramide has been shown
to induce apoptosis via accumulating intracellular ROS in
mi-tochondria (
Kannan et al., 2004
), therefore C2-ceramide was
Fig. 8. Neither BE nor BI suppressed C2-ceramide-induced cell death in glioma C6 cells. (A) Cells were treated with BE or BI (50 or 100μM) for 30 min followed by the addition of C2-ceramide (20μM) for a further 24 h. The viability of C6 cells under different treatments was detected by the MTT assay. (B) As described in (A), the DNA integrity in cells under different treatments was analyzed by agarose electrophoresis. (C) The morphological changes induced by C2-ceramide were not altered by the addition of BE or BI (100μM). As described in (A), the morphology of cells was observed microscopically via Giemsa staining.applied to examine the role of BE in intracellular ROS-mediated
cell death in the present study. Data of the present study
indi-cated no protective effect of BE against C2-ceramide-induced
cell death in C6 cells. Differential protective effects of BE
against H
2O
2and intracellular ROS (C2-ceramide)-mediated
cytotoxicity were observed. The reason for the ineffectiveness
of BE against C2-ceramide-induced cell death is still unclear.
Several studies reported that ceramide-mediated cell death is
Ca
2+-dependent (
Pinton et al., 2001; Townley et al., 2005; Wu
et al., 2005
).
Maher and Hanneken (2005)
delineated that BE
protected against oxidative stress induced by GSH depletion,
tBOOH, and H
2O
2toxicity, but not Ca
2+influx. Induction of
intracellular ROS and cytosolic Ca
2+and long-lasting lose of
Ca
2+in mitochondria were detected in ceramide-treated cells,
and long-lasting lose of calcium in mitochondria, but not others,
led to cell death (
Darios et al., 2003
). These data supported an
important role of Ca
2+on ceramide-induced apoptosis. It
sug-gests that no inhibitory effect of BE on C2-ceramide-induced
cell death may in part attribute to its ineffectiveness on Ca
2+-induced cytotoxicity in cells.
Flavonoids have been shown to regulate the activity of
protein kinases such as mitogen activated protein kinases
(MAPKs), tyrosine kinases, and protein kinase C (
Shen et al.,
2004; Ko et al., 2005
).
Nakahata et al. (2003)
indicated that BE
inhibited histamine- and A23187-induced ERK activation in C6
cells. In the present study, activation of ERKs was detected in
H
2O
2-treated C6 cells, and blocking ERK activation by
PD98059 or catalase significantly attenuated H
2O
2-induced
cell death. Suppression of H
2O
2-induced ERK protein
phos-phorylation by the addition of BE was identified in glioma C6
cells. This suggests that the protective effect of BE against H
2O
2-induced apoptosis is via suppression of ERK activation.
However, BE induced HO-1 protein expression via stimulation
of ERK protein phosphorylation in the absence of H
2O
2treatment. Therefore, a contradictory effect of ERK activation
was observed in BE-treated C6 cells in conditions with and
without H
2O
2. This suggests that BE possesses the ability to
induce intracellular kinase cascades such as ERKs to activate
cellular protective genes such as HO-1. In response to oxidative
stress, BE may directly or indirectly scavenge ROS production
and reduce oxidative-stress-induced kinase cascades such as
ERKs via its antioxidant activity. A double action of flavonoids
on the activation and inhibition of ERK activity, depending on
the extracellular oxidative condition, is proposed.
HO-1 protein has been shown to protect cells from ischemia/
reperfusion-induced damage (
Szabo et al., 2004
). Our previous
study indicated that induction of HO-1 protein attenuated
LPS-induced inducible nitric oxide synthase (iNOS) protein
expres-sion and NO production (
Lin et al., 2003
).
Chow et al. (2005)
indicated that HO-1 induction participates in quercetin protection
against cell death in RAW264.7 macrophages.
Chen et al. (2005)
demonstrated that resveratrol induction of HO-1 via activation of
Nrf2-ARE signaling participated in augmenting cellular
antiox-idant defense capacity to protect PC12 cells from
oxidative-stress-induced cell death. However, the role of the HO-1 protein in the
protective effect of BE against H
2O
2-induced cell death is still
undefined. An increase in HO-1 protein expression was detected
in BE (but not BI)-treated C6 cells. The chemical HO enzyme
inhibitor ZnPP suppressed the protective effect of BE against
H
2O
2-induced cell death. We found that ZnPP addition was able
to reverse the ROS-scavenging activity of BE. This suggests that
induction of HO-1 protein expression by BE may in part
participate in BE protection against H
2O
2-induced cell death.
Although several biological activities of BE have been
re-ported, data of the present study provide additional evidence to
suggest that BE possesses beneficial effects which protect against
H
2O
2-induced cell death, and modulation of ERKs activation and
induction of HO-1 protein expression via reduction of ROS
production were demonstrated. The potential for using BE in
experimental and practical application is highlighted for further
investigation.
Acknowledgments
This study was supported by the National Science Council of
Taiwan (NSC93-2321-B-038-009 and
NSC94-2320-B-038-049), Center of Excellence for Clinical Trial and Research in
Neurology Specialty, and Topnotch Stroke Research Center
Grant, Ministry of Education.
References
Ahn, H.C., Lee, S.Y., Kim, J.W., Son, W.S., Shin, C.G., Lee, B.J., 2001. Binding aspects of baicalein to HIV-1 integrase. Mol. Cells 12, 127–130. Birt, D.F., Hendrich, S., Wang, W., 2001. Dietary agents in cancer prevention:
flavonoids and isoflavonoids. Pharmacol. Ther. 90, 157–177.
Chen, K., Gunter, K., Maines, M.D., 2000. Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J. Neurochem. 75, 304–313.
Chen, Y.C., Shen, S.C., Lee, W.R., Hou, W.C., Yang, L.L., Lee, T.J., 2001. Inhibition of nitric oxide synthase inhibitors and lipopolysaccharide induced inducible NOS and cyclooxygenase-2 gene expressions by rutin, quercetin, and quercetin pentaacetate in RAW 264.7 macrophages. J. Cell. Biochem. 82, 537–548.
Chen, Y.C., Shen, S.C., Lee, W.R., Lin, H.Y., Ko, C.H., Lee, T.J., 2002. Nitric oxide and prostaglandin E2 participate in lipopolysaccharide/interferon-gamma-induced heme oxygenase 1 and prevent RAW264.7 macrophages from UV-irradiation-induced cell death. J. Cell. Biochem. 86, 331–339. Chen, Y.C., Shen, S.C., Lin, H.Y., 2003. Rutinoside at C7 attenuates the
apoptosis-inducing activity of flavonoids. Biochem. Pharmacol. 66, 1139–1150. Chen, C.J., Raung, S.L., Liao, S.L., Chen, S.Y., 2004a. Inhibition of inducible
nitric oxide synthase expression by baicalein in endotoxin/cytokine-stimu-lated microglia. Biochem. Pharmacol. 67, 957–965.
Chen, T.J., Shen, S.C., Lin, H.Y., Chien, L.L., Chen, Y.C., 2004b. Lipopolysac-charide enhancement of 12-o-tetradecanoylphorbol 13-acetate-mediated transformation in rat glioma C6, accompanied by induction of inducible nitric oxide synthase. Toxicol. Lett. 147, 1–13.
Chen, C.Y., Jang, J.H., Li, M.H., Surh, Y.J., 2005. Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem. Biophys. Res. Commun. 331, 993–1000.
Choi, B.M., Pae, H.O., Jeong, Y.R., Oh, G.S., Jun, C.D., Kim, B.R., Kim, Y.M., Chung, H.T., 2004. Overexpression of heme oxygenase (HO)-1 renders Jurkat T cells resistant to fas-mediated apoptosis: involvement of iron released by HO-1. Free Radic. Biol. Med. 36, 858–871.
Chow, J.M., Shen, S.C., Huan, S.K., Lin, H.Y., Chen, Y.C., 2005. Quercetin, but not rutin and quercitrin, prevention of H2O2-induced apoptosis via
anti-oxidant activity and heme oxygenase 1 gene expression in macrophages. Biochem. Pharmacol. 69, 1839–1851.
Crack, P.J., Taylor, J.M., 2005. Reactive oxygen species and the modulation of stroke. Free Radic. Biol. Med. 38, 1433–1444.
Darios, F., Lambeng, N., Troadec, J.D., Michel, P.P., Ruberg, M., 2003. Ceramide increases mitochondrial free calcium levels via caspase 8 and Bid: role in initiation of cell death. J. Neurochem. 84, 643–654.
de Bernardo, S., Canals, S., Casarejos, M.J., Solano, R.M., Menendez, J., Mena, M.A., 2004. Role of extracellular signal-regulated protein kinase in neuronal cell death induced by glutathione depletion in neuron/glia mesencephalic cultures. J. Neurochem. 91, 667–682.
Henze, C., Earl, C., Sautter, J., Schmidt, N., Themann, C., Hartmann, A., Oertel, W.H., 2005. Reactive oxidative and nitrogen species in the nigrostriatal system following striatal 6-hydroxydopamine lesion in rats. Brain Res. 1052, 97–104.
Hollman, P.C., Bijsman, M.N., van Gameren, Y., Cnossen, E.P., de Vries, J.H., Katan, M.B., 1999. The sugar moiety is a major determinant of the absorption of dietary flavonoid glycosides in man. Free Radic. Res. 31, 569–573. Horvathova, K., Novotny, L., Vachalkova, A., 2003. The free radical scavenging
activity of four flavonoids determined by the comet assay. Neoplasma 50, 291–295.
Huang, W.H., Lee, A.R., Chien, P.Y., Chou, T.C., 2005. Synthesis of baicalein derivatives as potential anti-aggregatory and anti-inflammatory agents. J. Pharm. Pharmacol. 57, 219–225.
Hwang, J.M., Tseng, T.H., Tsai, Y.Y., Lee, H.J., Chou, F.P., Wang, C.J., Chu, C.Y., 2005. Protective effects of baicalein on tert-butyl hydroperoxide-induced hepatic toxicity in rat hepatocytes. J. Biomed. Sci. 12, 389–397.
Ishige, K., Schubert, D., Sagara, Y., 2001. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic. Biol. Med. 30, 433–446. Juravleva, E., Barbakadze, T., Mikeladze, D., Kekelidze, T., 2005. Creatine enhances survival of glutamate-treated neuronal/glial cells, modulates Ras/ NF-kappaB signaling, and increases the generation of reactive oxygen species. J. Neurosci. Res. 79, 224–230.
Kannan, R., Jin, M., Gamulescu, M.A., Hinton, D.R., 2004. Ceramide-induced apoptosis: role of catalase and hepatocyte growth factor. Free Radic. Biol. Med. 37, 166–175.
Ko, C.H., Shen, S.C., Lee, T.J., Chen, Y.C., 2005. Myricetin inhibits matrix metalloproteinase 2 protein expression and enzyme activity in colorectal carcinoma cells. Mol. Cancer Ther. 4, 281–290.
Laufs, U., Wassmann, S., Czech, T., Munzel, T., Eisenhauer, M., Bohm, M., Nickenig, G., 2005. Physical inactivity increases oxidative stress, endothelial dysfunction, and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 25, 809–814. Li, F.Q., Wang, T., Pei, Z., Liu, B., Hong, J.S., 2005. Inhibition of microglial activation by the herbal flavonoid baicalein attenuates inflammation-media-ted degeneration of dopaminergic neurons. J. Neural Transm. 112, 331–347. Lin, H.Y., Juan, S.H., Shen, S.C., Hsu, F.L., Chen, Y.C., 2003. Inhibition of lipopolysaccharide-induced nitric oxide production by flavonoids in RAW264.7 macrophages involves heme oxygenase-1. Biochem. Pharmacol. 66, 1821–1832. Lin, H.Y., Shen, S.C., Chen, Y.C., 2005. Anti-inflammatory effect of heme oxygenase 1: glycosylation and nitric oxide inhibition in macrophages. J. Cell. Physiol. 202, 579–590.
Liu, Z.M., Chen, G.G., Ng, E.K., Leung, W.K., Sung, J.J., Chung, S.C., 2004. Upregulation of heme oxygenase-1 and p21 confers resistance to apoptosis in human gastric cancer cells. Oncogene 23, 503–513.
Maher, P., Hanneken, A., 2005. Flavonoids protect retinal ganglion cells from oxidative stress-induced death. Invest. Ophthalmol. Visual Sci. 46, 4796–4803.
Morita, T., Imai, T., Sugiyama, T., Katayama, S., Yoshino, G., 2005. Heme oxygenase-1 in vascular smooth muscle cells counteracts cardiovascular damage induced by angiotensin II. Curr. Neurovasc. Res. 2, 113–120.
Nakahata, N., Tsuchiya, C., Nakatani, K., Ohizumi, Y., Ohkubo, S., 2003. Baicalein inhibits Raf-1-mediated phosphorylation of MEK-1 in C6 rat glioma cells. Eur. J. Pharmacol. 461, 1–7.
Park, L., Anrather, J., Zhou, P., Frys, K., Pitstick, R., Younkin, S., Carlson, G.A., Iadecola, C., 2005. NADPH-oxidase-derived reactive oxygen species med-iate the cerebrovascular dysfunction induced by the amyloid beta peptide. J. Neurosci. 25, 1769–1777.
Petrache, I., Otterbein, L.E., Alam, J., Wiegand, G.W., Choi, A.M., 2000. Heme oxygenase-1 inhibits TNF-alpha-induced apoptosis in cultured fibroblasts. Am. J. Physiol., Lung Cell. Mol. Physiol. 278, L312–L319.
Pietta, P.G., 2000. Flavonoids as antioxidants. J. Nat. Prod. 63, 1035–1042. Pinton, P., Ferrari, D., Rapizzi, E., Di Virgilio, F., Pozzan, T., Rizzuto, R., 2001.
The Ca2+concentration of the endoplasmic reticulum is a key determinant of
ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 20, 2690–2701.
Qian, Y., Zheng, Y., Ramos, K.S., Tiffany-Castiglioni, E., 2005. GRP78 compart-mentalized redistribution in Pb-treated glia: role of GRP78 in lead-induced oxidative stress. Neurotoxicology 26, 267–275.
Regev-Shoshani, G., Shoseyov, O., Bilkis, I., Kerem, Z., 2003. Glycosylation of resveratrol protects it from enzymic oxidation. Biochem. J. 374, 157–163. Rivera, F., Urbanavicius, J., Gervaz, E., Morquio, A., Dajas, F., 2004. Some aspects of the in vivo neuroprotective capacity of flavonoids: bioavailability and structure–activity relationship. Neurotoxicol. Res. 6, 543–553. Shao, Z.H., Li, C.Q., Vanden Hoek, T.L., Becker, L.B., Schumacker, P.T., Wu,
J.A., Attele, A.S., Yuan, C.S., 1999. Extract from Scutellaria baicalensis Georgi attenuates oxidant stress in cardiomyocytes. J. Mol. Cell. Cardiol. 31, 1885–1895.
Shao, Z.H., Vanden Hoek, T.L., Qin, Y., Becker, L.B., Schumacker, P.T., Li, C.Q., Dey, L., Barth, E., Halpern, H., Rosen, G.M., Yuan, C.S., 2002. Baicalein attenuates oxidant stress in cardiomyocytes. Am. J. Physiol. 282, H999–H1006.
Shen, S.C., Chen, Y.C., Hsu, F.L., Lee, W.R., 2003. Differential apoptosis-inducing effect of quercetin and its glycosides in human promyeloleukemic HL-60 cells by alternative activation of the caspase 3 cascade. J. Cell. Biochem. 89, 1044–1055.
Shen, S.C., Ko, C.H., Hsu, K.C., Chen, Y.C., 2004. 3-OH flavone inhibition of epidermal growth factor-induced proliferation through blocking prostaglan-din E2 production. Int. J. Cancer 108, 502–510.
Shieh, D.E., Liu, L.T., Lin, C.C., 2000. Antioxidant and free radical scavenging effects of baicalein, baicalin and wogonin. Anticancer Res. 20, 2861–2865. Singh, D., Chopra, K., 2004. The effect of naringin, a bioflavonoid on ischemia–reperfusion induced renal injury in rats. Pharmacol. Res. 50, 187–193.
Szabo, M.E., Gallyas, E., Bak, I., Rakotovao, A., Boucher, F., de Leiris, J., Nagy, N., Varga, E., Tosaki, A., 2004. Heme oxygenase-1-related carbon monoxide and flavonoids in ischemic/reperfused rat retina. Invest. Ophthalmol. Visual Sci. 45, 3727–3732.
Townley, H.E., McDonald, K., Jenkins, G.I., Knight, M.R., Leaver, C.J., 2005. Ceramides induce programmed cell death in Arabidopsis cells in a calcium-dependent manner. Biol. Chem. 386, 161–166.
Wang, J., Yu, Y., Hashimoto, F., Sakata, Y., Fujii, M., Hou, D.X., 2004. Baicalein induces apoptosis through ROS-mediated mitochondrial dysfunction pathway in HL-60 cells. Int. J. Mol. Med. 14, 627–632.
Wu, Z., Tandon, R., Ziembicki, J., Nagano, J., Hujer, K.M., Miller, R.T., Huang, C., 2005. Role of ceramide in Ca2+-sensing receptor-induced apoptosis. J. Lipid Res. 46, 1396–1404.