0095-1137/08/$08.00⫹0 doi:10.1128/JCM.02015-07
Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Rapid Differentiation of Influenza A Virus Subtypes and Genetic
Screening for Virus Variants by High-Resolution Melting Analysis
䌤
Jih-Hui Lin,
1† Ching-Ping Tseng,
2,3† Yen-Ju Chen,
4Chy-Yung Lin,
5Shy-Shin Chang,
6,7Ho-Sheng Wu,
1and Ju-Chien Cheng
4*
Center for Disease Control and Prevention, Taipei, Taiwan, Republic of China1; Graduate Institute of Medical Biotechnology2and
Clinical Medical Science,6Chang Gung University, Taoyuan, Taiwan 333, Republic of China; Laboratory of
Molecular Diagnostics, Department of Clinical Pathology,3and Department of Emergency Medicine,7
Chang Gung Memorial Hospital, Taoyuan, Taiwan 333, Republic of China; Department of Medical Laboratory Science and Biotechnology, China Medical University, Taichung,
Taiwan 404, Republic of China4; and Department of Laboratory Medicine,
Changhua Christian Medical Center, Changhua, Taiwan 500, Republic of China5
Received 14 October 2007/Returned for modification 28 November 2007/Accepted 20 December 2007
We assessed the use of high-resolution melting (HRM) analysis for the rapid identification of influenza A virus subtypes and the detection of newly emerging virus variants. The viral matrix gene was amplified by LightCycler real-time reverse transcription-PCR (RT-PCR) in the presence of the LCGreen I fluorescent dye. Upon optimization of the assay conditions, all the major influenza A virus subtypes, including H1N1, H3N2, H5N1, H7N3, and H9N2, were amplifiable by this method and had a PCR product length of 179 bp. Real-time RT-PCR of in vitro-transcribed H3N2 RNA revealed a standard curve for quantification with a linear range (correlation coefficientⴝ 0.9935) across at least 8 log units of RNA concentrations and a detection limit of 103 copies of viral RNA. We performed HRM analysis of the PCR products with the HR-1 instrument and used the melting profiles as molecular fingerprints for virus subtyping. The virus subtypes were identified from the high-resolution derivative plot obtained by heteroduplex formation between the PCR products of the viral isolates tested and those of the reference viral isolates. The melting profiles were consistent with minimal interassay variability. Hence, an HRM database and a working protocol were established for the identification of these five influenza A virus subtypes. When this protocol was used to test 21 clinical influenza A virus isolates, the results were comparable to those obtained by RT-PCR with hemagglutinin-specific primer sets. Sequence variants of the clinical isolates (nⴝ 4) were also revealed by our HRM analytical scheme. This assay requires no multiplexing or hybridization probes and provides a new approach for influenza A virus subtyping and genetic screening of virus variants in a clinical virology laboratory.
Influenza A virus is a medically important viral pathogen that causes significant mortality, morbidity, and financial bur-den throughout the world. Influenza A viruses are classified into subtypes according to the antigenic composition of their hemagglutinin (HA) and neuraminidase (NA) glycoproteins on the viral envelope (35). Of the 15 HA subtypes and 9 NA subtypes, only 3 HA subtypes (subtypes H1, H2, and H3) and 2 NA subtypes (subtypes N1 and N2) have established stable lineages in the human population since the last century (31). Migratory birds and waterfowl usually serve as the reservoir in nature for the other influenza A virus subtypes (31). Although the risk of infection with avian influenza virus (AIV) is gener-ally low for most people, the pathogenic virus can cross the species barrier and acquires the ability to infect and be trans-mitted among the human population. Confirmed cases of hu-man infection caused by different subtypes of AIV, including H5N1, H7N3, and H9N2, have been reported since 1997 (5, 18, 29). Of these AIV subtypes, H5N1 results in the largest
num-ber of detected cases with severe disease and death (19). This scenario makes it important to integrate AIV detection into a human influenza surveillance program.
In the clinical virology laboratory, the diagnosis of influenza A virus infection is typically performed by culturing of clinical specimens, followed by HA analysis and the hemagglutination inhibition assay. Molecular approaches such as multiplex re-verse transcription (RT)-PCR (1, 3), real-time RT-PCR (8), microarray analysis (7, 28), and electrospray ionization mass spectrometry (23) have been developed for the rapid detection and differentiation of influenza A virus subtypes. Due to the high costs of the machines and the complicated procedures associated with microarray analysis and mass spectrometric analysis, RT-PCR-based methods are apparently the most popular methods suitable for use in the clinical laboratory.
In addition to viral subtyping, analysis of the PCR product by restriction fragment length polymorphism analysis (16), sin-gle-strand conformational polymorphism analysis (2), and het-eroduplex mobility assay (HMA) (9, 38) has been used for the genetic screening of influenza A virus variants. Recently, high-resolution melting (HRM) analysis of the PCR amplicon has been used for genotyping and variation scanning, as well as for microbial detection and species determination (4, 10, 12, 13, 15). This technique does not require fluorescently labeled probes or separation steps (33). It differs from LightCycler * Corresponding author. Mailing address: Department of Medical
Laboratory Science and Biotechnology, China Medical University, Taichung, Taiwan 404, Republic of China. Phone: 886-04-22053366, ext. 7220. Fax: 886-04-22022073. E-mail: [email protected].
† J.-H. Lin and C.-P. Tseng contributed equally to this work and are considered co-first authors.
䌤Published ahead of print on 3 January 2008.
SYBR green I melting curve analysis by the incorporation of a novel saturation fluorescent dye, such as LCGreen I; high-resolution instrumentation with a high rate of data acquisition; ideal optics and tight temperature control; and adequate anal-ysis software. Consequently, HRM analanal-ysis with the HR-1 in-strument reliably detects single-base differences in homozy-gous and heterozyhomozy-gous sequences (11). In certain nucleotide compositions, homozygous sequences may not be readily de-tectable by HRM analysis. In this scenario, mixing of the test sample amplicon and a standard PCR amplicon, which results in heteroduplex formation, can be performed to reveal the sequence variant (4). When the amplicon sequence of a test sample differs from the standard PCR amplicon sequence, the melting plot should be changed following heteroduplex forma-tion. No change to the melting plot occurs when the amplicon sequence of a test sample matches the standard PCR amplicon sequence. Hence, HRM analysis is cost-effective, has a high sensitivity and a high specificity, and can be completed with minimal post-PCR handling.
In this study, we report on a novel scheme that combines LightCycler real-time RT-PCR and HRM analysis for the rapid differentiation of influenza A virus subtypes and for the genetic screening of virus variants in clinical specimens. Due to the highly conserved nature of circulating influenza A virus strains and its association with the determination of species specificity and HA subtypes (24, 32), the viral matrix (M) gene was selected as the target for PCR amplification. Without multiplexing or hybridization probes, influenza A virus subtyp-ing and genetic screensubtyp-ing can be completed with one PCR within 4 h from the time of viral RNA isolation.
MATERIALS AND METHODS
Materials.The LCGreen I reagent set and HR-1 instrument were purchased from Idaho Technology (Salt Lake City, UT). The TEMPase Hot Start Taq DNA polymerase was purchased from Ampliqon (Herlev, Denmark). Moloney murine leukemia virus reverse transcriptase, DNase I, and PCR reagents were purchased from Invitrogen (Carlsbad, CA). The HiScribe transcription kit was purchased from New England Biolabs. The LightCycler capillaries and G50 column were purchased from Roche Applied Science (Indianapolis, IN). The QIAamp viral RNA mini kit and the QIAquick PCR purification kit were purchased from Qiagen (Valencia, CA).
Viral specimen and isolation of viral RNA.Clinical influenza A virus strains were isolated from combined nose and/or throat swabs from selected patients who presented to physicians in sentinel practices with influenza-like illnesses. Briefly, the swabs and transportation medium were thoroughly mixed and the medium was filtered through a 0.45-m-pore-size filter. The specimen (100 l) was inoculated into Madin-Darby canine kidney cells for 7 to 14 days. A positive cytopathic effect was confirmed by indirect immunofluorescence assay (Chemi-con Inc. Temecula, CA) and RT-PCR with HA-specific primer sets. The super-natants from positive cultures were harvested and stored at⫺80°C until future analysis. The QIAamp viral RNA mini kit was used to isolate viral RNA.
LightCycler real-time RT-PCR for amplification of viral M gene.A two-step real-time RT-PCR was performed to amplify the viral M-gene fragment. For the RT reaction, viral RNA (2l) was added to a reaction mixture (20 l) containing 125 nM primer IFU-F (5⬘-GCGAGGACTGCAGCGTAGAC-3⬘), 1 mM of each deoxynucleoside triphosphate, 20 U RNaseOUT, and 200 U (final concentration, 10 U/l) Moloney murine leukemia virus reverse transcriptase. The reaction was then carried out at 42°C for 1 h. After cleanup with the QIAquick PCR purifi-cation kit, real-time PCR was then performed with the LightCycler thermal cycler and the primer pair IFU-F and IFU-R (5⬘-TGAGACCCATGCAACTG GCAAG-3⬘). Briefly, viral cDNA (2 l) was added to a reaction mixture con-taining 250 nM of each primer, 2 mM MgCl2, 50 mM Tris-HCl (pH 8.3), 0.2 mM of each deoxynucleoside triphosphate, 250 mg/liter bovine serum albumin, 1⫻ LCGreen I dye, and 2.5 U of TEMPase Hot Start Taq DNA polymerase in 10l. The cycling conditions were 1 cycle of 95°C for 10 min and 40 cycles of 95°C for 10 s, 55°C for 15 s, and 72°C for 30 s, with a transition rate of 20°C/s.
Construction of standard plasmids for influenza A virus subtypes.The viral RNAs of reference isolates A/Taiwan/421/2006 (H1N1), A/Taiwan/482/2005 (H3N2), and A/HongKong/156/1997 (H5N1) were reversed transcribed in the presence of primer IFU-F. PCR was then performed with primer pair IFU-F and IFU-R. The PCR product was subcloned into the pGEM-T Easy vector. For H7N3 and H9N2, synthetic oligonucleotides (Table 1) corresponding to both strands of nucleotides (nt) 238 to 416 of the M gene of A/Canada/rv504/2004 (GenBank accession no. CY015007) and A/HongKong/2108/2003 (GenBank ac-cession no. DQ226095) were annealed and subcloned into the pGEM-T Easy vector, as described by the manufacturer (Promega).
In vitro transcription of viral RNA.The M-gene fragment of A/Taiwan/482/ 2005 in the pGEM-T Easy vector was amplified by PCR with primers IFU-F and T7-IFU-R (5⬘-TAATACGACTCACTATAGGTGAGTCCCATACAACTGGC A-3⬘). After gel purification of the DNA fragment, 1 g of the PCR product was transcribed with the HiScribe transcription kit, in accordance with the instruc-tions provided with the kit. The in vitro-transcribed RNA was treated with 1 U of DNase I and was passed through a G50 column (Roche Applied Sciences) to remove unincorporated free nucleotides. After ethanol precipitation, the in vitro-transcribed RNA was quantified by spectrophotometry.
HRM curve acquisition and analysis.Glass capillaries containing the ampli-fication products were transferred directly from the LightCycler instrument to the HR-1 HRM instrument. The fragments were melted from 70 to 90°C at a rate of 0.1°C/s. The melting profiles were assessed with HR-1 software with fluores-cence normalization and temperature overlay to superimpose the curves at 5 to 20% fluorescence.
Heteroduplex formation. Heteroduplex formation was achieved by mixing equal amounts of the PCR products from the viral strains to be tested and the reference viral strain. The value of the fluorescence intensity after the final run of the PCR was used as a basis to calculate the volume of PCR products required to be mixed. To induce heteroduplex formation, the DNA mixtures were sub-jected to heating at 95°C for 1 s and cooling to 40°C for 10 s at a rate of 20°C/s.
RESULTS
To assess the combined use of real-time RT-PCR and HRM analysis for the rapid detection and differentiation of influenza A virus subtypes, the viral M gene was selected as the target for the design of the PCR amplicon. The corresponding sequences of 502 isolates, including 104 isolates of H1N1, 322 isolates of H3N2, 70 isolates of H5N1, and 1 isolate of H7N3 reported from 2004 to 2007 and 5 isolates of H9N2 reported from 1999 to 2004 were TABLE 1. Oligonucleotide sequences for generation of H7N3 and H9N2 M-gene fragment
Primer Sequence (5⬘–3⬘) H7-Forward...GCGAGGACTGCAGCGTAGACGCTTTGTCCAAAATGCCCTTAATGGGAATGGGGATCCAAACAACATGGA CAGAGCGGTCAAACTGTATAGGAAGCTAAAAAGGGA H7-Reverse ...TGAGGCCCATGCAACTGGCAAGTGCACCAGTTGAATAACTGAGTGCCACTTCTTTTGCCCCATGGAATGT TATTTCCCTTTTTAGCTTCCTATACAGTTTGACC H9-Forward...GCGAGGACTGCAGCGTAGACGGTTTGTCCAAAATGCCCTAAATGGGAATGGAGATCCAAACAACATGG ACAGGGCAGTTAAACTATACAAGAAGCTGAAGAGGGA H9-Reverse ...TGAGACCCATGCAACTGGCAAGCGCACCAGTTGAGTAACTGAGTGCAACTTCCTTTGCTCCATGGAATGT CATTTCCCTCTTCAGCTTCTTGTATAGTTTAACT
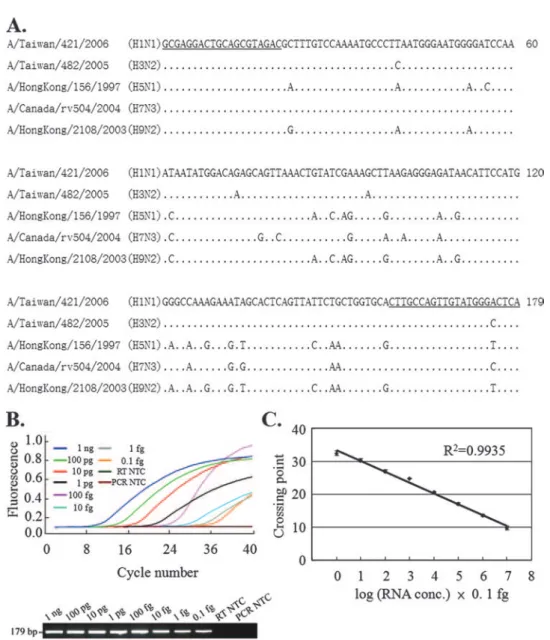
obtained from the Influenza Virus Resource (www.ncbi.nlm.nih .gov/genomes/FLU). After multiple-sequence alignment with the Jemboss software package (22) or the alignment program in the Influenza Virus Resource, nt 238 to 258 and nt 395 to 416 were selected for use in the design of the forward and reverse primers (primers IFU-F and IFU-R, respectively) due to the conserved nature of these nucleotides among various influenza A virus iso-lates. The sequences of the PCR amplicons of our reference isolates A/Taiwan/421/2006 (H1N1), A/Taiwan/482/2005 (H3N2), A/HongKong/156/1997 (H5N1), A/Canada/rv504/2004 (H7N3), and A/HongKong/2108/2003 (H9N2) are shown in Fig. 1A.
A broad-range real-time RT-PCR was then set up with the LightCycler instrument to amplify the 136-bp interprimer
re-gion. The detection limit of this method was determined by using 10-fold serial dilutions of in vitro-transcribed A/Taiwan/ 482/2005 RNA. A typical amplification plot (the change in the fluorescent signal versus the cycle number) and an electro-phoretic analysis of the PCR product are shown in Fig. 1B. The crossing points for the real-time RT-PCR ranged from 9.82⫾ 0.58 to 32.38⫾ 0.69 (n ⫽ 5) for the RNA template when it was present at amounts ranging from 1 ng to 0.1 fg. Hence, we were able to detect RNA at amounts as low as 0.1 fg, equivalent to 103 copies of viral RNA, in a 10-l reaction volume. The
standard curve showed a dynamic linear range for quantifica-tion across at least 8 log units of RNA concentraquantifica-tions and had a correlation coefficient of 0.9935 (Fig. 1C).
FIG. 1. Sequence alignments, real-time RT-PCR, and gel electrophoresis analysis of the M-gene PCR amplicons. (A) Sequence alignments of the PCR amplicons corresponding to nt 238 to 416 of the M genes for the indicated influenza A virus strains. Only parts of the sequences showing differences from the first sequence are shown. Nucleotides identical to the nucleotide in the first sequence are indicated by dots. The underlined sequences were used for the design of primers IFU-F and IFU-R. (B and C) Serial dilution of the in vitro-transcribed H3N2 RNA harboring the M-gene fragment, which was used as the template for real-time RT-PCR with primer pair IFU-F and IFU-R. (B) A typical LightCycler amplification plot and an agarose gel of the PCR product. (C) The crossing points plotted against the RNA concentration. The data represent the means⫾ standard deviations (n ⫽ 5). NTC, no-template control.
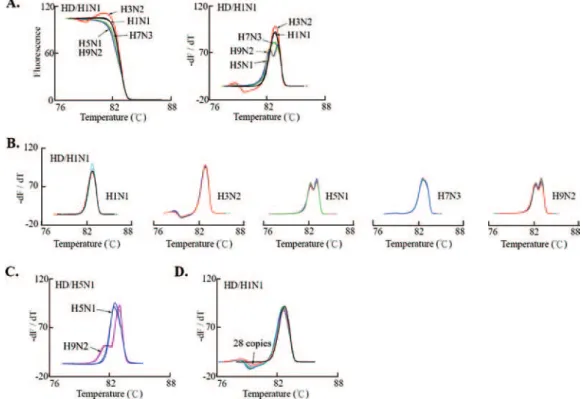
To simplify the scheme for HRM analysis for the differen-tiation of the influenza A virus subtypes, five plasmids contain-ing the M-gene PCR amplicon of the individual reference isolates were generated. All these constructs were amplifiable by our real-time PCR and had a PCR product length of 179 bp (Fig. 2A). We then used the HR-1 instrument to perform HRM analysis. The melting and derivative plots were not dis-tinguishable, with closely overlapping patterns for the five ref-erence isolates and minimal interassay variability (Fig. 2B and C). The melting plot for the no-template control reaction was eliminated automatically by the HR-1 analytical software when the comment of “delete some bad samples” was executed dur-ing fluorescence normalization.
We determined whether heteroduplex formation between the PCR amplicons of the test and the reference isolates pro-duced unique HRM profiles suitable for use for influenza A virus subtyping. By heteroduplexing with the reference H1N1 PCR amplicon, we identified H1N1, H3N2, H5N1/H9N2, and H7N3 by their unique high-resolution melting and derivative plots with minimal interassay variability (Fig. 3A and B). The H5N1 and H9N2 subtypes were further identified by hetero-duplexing of their amplicons with the reference H5N1 PCR amplicon (Fig. 3C). Overall, these results demonstrated that our HR-1 melting-based protocol can be performed without the use of multiplexing or hybridization probes to differentiate influenza A virus subtypes. Using a reference H3N2 virus as an example, we revealed that the patterns of the derivative plots were consistent and ranged from 2.8⫻ 108to 28 copies of the
initial template used for amplification (Fig. 3D). On the basis of these observations, we generated an HRM database and analytical scheme for the rapid differentiation of influenza A virus subtypes (Fig. 4). After the isolates were subtyped ac-cording to their HRM profiles, potential mistyping and new variants could be resolved further by the use of a confirmatory test consisting of HRM analysis of the mixed PCR amplicon between the test sample and a standard strain of the putative subtype (4). The presence of nucleotide variants in the test sample can be identified by the detection of an alteration of the HRM profile upon heteroduplex formation.
We further assessed the ability of our novel approach to differentiate influenza A virus subtypes in clinical samples. The
influenza A viruses infecting 21 nose and/or throat swab spec-imens collected from 2004 to 2007 were well characterized by antigenic and genetic analyses (10 were H1N1 and 11 were H3N2). The viral RNA was isolated from the culture for real-time RT-PCR and HRM analysis, according to our analytical scheme. After heteroduplex formation with the reference H1N1 PCR amplicon, the derivative plots of 18 samples (8 for H1N1 and 10 for H3N2) matched those of the HRM database and had analytical results comparable to those obtained by HA-specific RT-PCR analysis (Fig. 5A). The other three sam-ples revealed two different patterns on the derivative plot, with one for A/Taiwan/286/2004 and A/Taiwan/284/2004 and the other for A/Taiwan/21/2004. We then performed a confirma-tory test by heteroduplex formation of the amplicons of the 10 putative H3N2 samples with the reference H3N2 PCR ampli-con. The derivative plots for nine of the isolates did not change and the isolates were confirmed to be H3N2. In contrast, A/Taiwan/57/2007 had an altered HRM profile upon hetero-duplex formation, indicating that it was not completely identi-cal to the reference H3N2 strain (Fig. 5B). The PCR amplicons of the four isolates with atypical derivative plots were further characterized by sequencing. We found that A/Taiwan/286/ 2004 and A/Taiwan/284/2004 had identical PCR amplicons, with one nucleotide variation in relation to the sequence of the reference H1N1 strain (Fig. 5C). Similarly, A/Taiwan/21/2004 and A/Taiwan/57/2007 had one and two nucleotide variations in relation to the sequence of the reference H3N2 strain (Fig. 5D). These data revealed that our HRM analytical scheme can be used not only for the differentiation of virus subtypes but also for the detection of nucleotide changes that may occur during virus evolution or the formation of a newly emerging viral strain.
DISCUSSION
The risk of pandemic influenza and the recent cross-species spread of AIV H5N1 entail the urgent need for an effective influenza surveillance program for the early identification and classification of the organism responsible for the outbreak. In this study, we describe a “broad-range” real-time RT-PCR technique for the simultaneous detection, quantification, and FIG. 2. Real-time PCR and HRM analysis of five clinically important influenza A virus subtypes. (A) Standard plasmids (1 pg) with the M-gene fragments of the indicated subtypes were subjected to real-time PCR, as described in the Materials and Methods. A typical LightCycler amplification plot and the typical results of agarose gel electrophoresis analysis of the PCR product are presented. Melting (B) and derivative (C) plots for the indicated influenza A virus subtypes were obtained by HRM analysis of the real-time PCR amplicon. Fluorescence normalization and temperature overlay were performed with HR-1 software. For each influenza A virus subtype, the data represent three measurements obtained with the plasmid DNA from the indicated reference isolates.
presumptive differentiation of influenza A virus subtypes. The detection limit of this method is equivalent to 103 copies of
viral RNA and has a dynamic linear range for quantification across at least 8 log units of RNA concentrations. All major HA subtypes, including H1N1, H3N2, H5N1, H7N3, and H9N2, were amplifiable by our real-time RT-PCR with similar efficiencies. Because only one H2N2 isolate was reported in the Influenza Virus Resource from 1999 to 2007, the H2N2
sub-type was not included in this study. However, analysis of the H2N2 M-gene sequences available from the Influenza Virus Resource (n ⫽ 73) revealed that there are only one to two nucleotide variations within the primer region, suggesting that H2N2 viral strains should also be amplifiable by our real-time RT-PCR method.
Most of the methods currently available for influenza A virus subtyping use subtype-specific primers to amplify the HA or the NA gene, which allows the detection of only one specific pathogen at a time (36). For sample screening and subtype identification in an outbreak, multiplexing or multiple reac-tions followed by post-PCR electrophoresis to separate ampli-cons of different sizes are usually required. Although real-time RT-PCR has successfully been applied to the detection of influenza A virus, a TaqMan probe or a hybridization probe is usually needed, and the use of these probes can increase the expense on a cost-per-sample basis (8, 25). Alternatively, anal-ysis of M-gene sequence variants forms a basis for HA subtyp-ing. The principle of this type of assay is based on phylogenetic analysis, which reveals the preferential association between the influenza A virus HA and M genes and the coevolution of these genes (24, 32). This notion allows the design of an assay for analysis of the M-gene nucleotide components and the differentiation of influenza A virus HA subtypes. For instance, restriction fragment length polymorphism analysis of the M-gene PCR product has been applied to the subtyping of human influenza A viruses (6, 17). A real-time LightCycler hybridiza-FIG. 3. HRM plots and derivative plots for influenza A virus subtyping. (A to C) High-resolution melting and derivative plots were obtained by heteroduplex formation (HD) between the PCR products of the indicated influenza A virus subtype and the reference strain of H1N1 (A/Taiwan/421/2006) or H5N1 (A/HongKong/156/97). The melting (A) and derivative (A and C) plots reveal the HRM profile for each virus subtype. The derivative plots for five to six measurements of each influenza A virus subtype reveal the minimal interassay variability (B). Note the consistent derivative plot pattern for each virus subtype. (D) Tenfold serial dilutions of H3N2 plasmid DNAs (from 2.8⫻ 108to 28 copies) were subjected to real-time PCR amplification. After heteroduplex formation with the reference H1N1 PCR amplicon, HRM analysis was performed, and the derivative plots are shown.
FIG. 4. Work flow for rapid detection and differentiation of five clinically important influenza A virus subtypes. An unknown influenza virus isolate was subjected to M-gene real-time RT-PCR and HRM analysis. According to the melting and derivative plots, all five influ-enza A virus subtypes can be identified by heteroduplex formation (HD) between the PCR products of the virus isolate tested and the indicated reference influenza A virus isolate. Potential mistyping and new variants can be resolved further by confirmatory testing by HRM analysis of the mixed PCR amplicon between the test sample and a standard strain of the putative subtype.
tion probe-based assay targeting subtype-specific sequences in the M gene combined with melting temperature analysis was developed for the differentiation of H1N1 and H3N2 (25). Combined PCR-HMA of the M-gene PCR amplicon has also been described for the detection and partial characterization of influenza A viruses from different animal species (9). Although heteroduplex analysis is ideally suited for the differentiation of rapidly evolving RNA viruses (14, 27, 38, 39), post-PCR anal-ysis by electrophoresis on polyacrylamide gels is required. Therefore, it is usually time-consuming, carries an increased risk of carryover contamination, and may not be suitable for use in a routine clinical virology laboratory.
Recent trends in the application of HRM analysis for mi-croorganism identification (4, 12, 21) led us to explore a new avenue for influenza A virus subtyping. This novel approach is based on the same concept used for HMA and has been applied to mutation scanning and genotyping (30, 37). We demonstrate in this study that influenza A virus can be differ-entiated simply through the use of one PCR primer pair for real-time RT-PCR, followed by HRM analysis of the M-gene PCR amplicon. Through bioinformatic analysis, we revealed that the 136-bp interprimer region is relatively conserved within isolates of the same subtype, whereas nucleotide diver-gence is present among different influenza A virus subtypes. Therefore, the PCR amplicon likely contains information for
at least partial phylogenetic characterization and subtyping of clinically important influenza A viruses. Compared to the am-plicons obtained by other subtyping methods, the PCR ampli-con obtained in this study is relatively small (179 bp), which further gives HRM analysis-based subtyping a superior reso-lution power. When it is combined with rapid-cycle PCR, HRM analysis requires minimal time, and the material cost is usually less than $2. The time required for the differentiation of influenza A virus subtypes is considerably shorter when PCR is performed directly with clinical specimens.
Notably, this method is ideally suited not only for the subtyping of current circulating strains but also for the identification of newly emerging strains. Consistent with the findings of previous studies on the application of HRM analysis for genotyping and the identification of genetic variants (13, 20), HRM analysis iden-tified four clinical isolates (A/Taiwan/286/2004, A/Taiwan/284/ 2004, A/Taiwan/21/2004, and A/Taiwan/57/2007) with changes in the nucleotide components of the M gene during our validation assay. Although only one to two nucleotides are different com-pared to the sequence of the reference strain, they are readily detectable by HRM analysis. Such clinical variants may not be unveiled by conventional real-time RT-PCR methods with the TaqMan probe or a hybridization probe. Hence, HRM analysis of viral isolates may also be useful for the early detection of genetic changes that can occur during virus evolution or interspecies FIG. 5. Validation assay with 21 clinical influenza A virus isolates obtained from a clinical virology laboratory. (A and B) The derivative plots were obtained by heteroduplex formation (HD) between the PCR products of the indicated influenza A virus subtypes and the reference strains of H1N1 (A) or H3N2 (B). The clinical strains that did not have derivative plots typical of those in our HRM database are marked. (C and D) Sequence alignment of the PCR amplicons corresponding to nt 238 to 416 of the M genes of the indicated influenza A virus strains. Only parts of the sequences showing differences from the first sequence are shown. Nucleotides that were identical to the nucleotide in the first sequence are indicated by dots.
transmission between animal and human hosts. The novel or unusual influenza A virus isolates identified by HRM analysis can be more extensively characterized by direct sequencing and typing by the hemagglutination inhibition assay.
Because influenza A virus continues to experience nucleo-tide mutations through the mechanism of antigenic drift and antigenic shift, the HRM profile of a specific viral subtype may not be the same when the virus strains are isolated from dif-ferent outbreak areas. Because of this, we compared the HRM profiles of two H5N1 strains, A/Hong Kong/156/1997, which was isolated from the tracheal aspirate from a 3-year-old child in Hong Kong with a fatal illness consistent with influenza (26), and A/Vietnam/1203/2004, which showed evidence of antigenic drift and which was distinct from the AIV circulating before the end of 2003 (34). We found that both isolates had unique HRM profiles that reflected the divergence of nucleotide com-ponents between the two strains (data not shown). Hence, the use of a recently circulating strain, if one is available, as the reference isolate for HRM analysis-based subtyping is recom-mended. With an appropriate HRM profile database, this method should allow the cost-effective differentiation of influ-enza A virus subtypes and the early detection of newly emerg-ing clinical variants in an influenza surveillance program.
ACKNOWLEDGMENTS
This work was supported in part by grants NSC95-2320-B-039-018-MY2 from the National Science Council (to J.-C.C.) and Chang Gung Molecular Medicine Research Center grant EMRPD160131 from the Ministry of Education, Taiwan, Republic of China (to C.-P.T.).
REFERENCES
1. Auewarakul, P., K. Sangsiriwut, K. Chaichoune, A. Thitithanyanont, W.
Wiriyarat, T. Songserm, R. Ponak-nguen, J. Prasertsopon, P. Pooruk, P. Sawanpanyalert, P. Ratanakorn, and P. Puthavathana.2007. Surveillance for reassortant virus by multiplex reverse transcription-PCR specific for eight genomic segments of avian influenza A H5N1 viruses. J. Clin. Microbiol.
45:1889–1892.
2. Cha, T. A., K. Kao, J. Zhao, P. E. Fast, P. M. Mendelman, and A. Arvin. 2000. Genotypic stability of cold-adapted influenza virus vaccine in an effi-cacy clinical trial. J. Clin. Microbiol. 38:839–845.
3. Chaharaein, B., A. R. Omar, I. Aini, K. Yusoff, and S. S. Hassan. 2007. Detection of H5, H7 and H9 subtypes of avian influenza viruses by multiplex reverse transcription-polymerase chain reaction. Microbiol. Res. [Epub ahead of print.] doi:10.1016/j.micres.2007.01.001.
4. Cheng, J. C., C. L. Huang, C. C. Lin, C. C. Chen, Y. C. Chang, S. S. Chang,
and C. P. Tseng.2006. Rapid detection and identification of clinically im-portant bacteria by high-resolution melting analysis after broad-range ribo-somal RNA real-time PCR. Clin. Chem. 52:1997–2004.
5. Class, E. C., A. D. Osterhaus, R. Van Beek, J. C. De Jong, G. F.
Rimmelzwaan, D. A. Senne, S. Krauss, E. K. Shortridge, and R. G. Webster.1998. Human influenza A H5N1 virus related to a highly patho-genic avian influenza virus. Lancet 351:472–477.
6. Cooper, L. A., and K. Subbarao. 2000. A simple restriction fragment length polymorphism-based strategy that can distinguish the internal genes of hu-man H1N1, H3N2, and H5N1 influenza A viruses. J. Clin. Microbiol. 38: 2579–2583.
7. Dawson, E. D., C. L. Moore, D. M. Dankbar, M. Mehlmann, M. B.
Townsend, J. A. Smagala, C. B. Smith, N. J. Cox, R. D. Kuchta, and K. L. Rowlen.2007. Identification of A/H5N1 influenza viruses using a single gene diagnostic microarray. Anal. Chem. 79:378–384.
8. Ellis, J. S., J. W. Smith, S. Braham, M. Lock, K. Barlow, and M. C. Zambon. 2007. Design and validation of an H5 TaqMan real-time one-step reverse transcription-PCR and confirmatory assays for diagnosis and verification of influenza A virus H5 infections in human. J. Clin. Microbiol. 45:1535–1543. 9. Ellis, J. S., and M. C. Zambon. 2001. Combined PCR-heteroduplex mobility assay for detection and differentiation of influenza A viruses from different animal species. J. Clin. Microbiol. 39:4097–4102.
10. Fortini, D., A. Ciammaruconi, R. De Santis, A. Fasanella, A. Battisti, R.
D’Amelio, F. Lista, A. Cassone, and A. Carattoli. 2007. Optimization of high-resolution melting analysis for low-cost and rapid screening of allelic variants of Bacilus anthracis by multiple-locus variable-number tandem re-peat analysis. Clin. Chem. 53:1377–1380.
11. Graham, R., M. Liew, C. Meadows, E. Lyon, and C. T. Wittwer. 2005. Distinguishing different DNA heterozygotes by high-resolution melting. Clin. Chem. 51:1295–1298.
12. Jeffery, N., R. B. Gasser, P. A. Steer, and A. H. Noormohammadi. 2007. Classification of Mycoplasma synoviae strains using single-strand conforma-tion polymorphism and high-resoluconforma-tion melting-curve analysis of the vlhA gene single-copy region. Microbiology 153:2679–2688.
13. Liew, M., R. Pryor, R. Palais, C. Meadows, M. Erali, E. Lyon, and C. T.
Wittwer.2004. Genotyping of single-nucleotide polymorphism by high-res-olution melting of small amplicons. Clin. Chem. 50:1156–1164.
14. Mattick, K. L., J. Green, P. Punia, F. J. Belda, C. I. Gallimore, and D. W.
Brown.2000. The heteroduplex mobility assay (HMA) as a pre-sequencing screen for Norwalk-like viruses. J. Virol. Methods 87:161–169.
15. Odell, I. D., J. L. Cloud, M. Seipp, and C. T. Wittwer. 2005. Rapid species identification within the Mycobacterium chelonae-abscessus group by high-resolution melting analysis of hsp65 PCR products. Am. J. Clin. Pathol.
123:96–101.
16. Offringa, D. P., V. Tyson-Medlock, Z. Ye, and R. A. Levandowski. 2000. A comprehensive systematic approach to identification of influenza A virus genotype using RT-PCR and RFLP. J. Virol. Methods 88:15–24. 17. Park, K. Y., M. G. Lee, and J. C. Ryu. 1997. Evolutionary stasis of M1 gene
of human influenza A viruses and the possibility of their subtyping by re-striction analysis of M1 gene polymerase chain reaction product. Acta Virol.
41:231–239.
18. Peiris, M., K. Y. Yuen, C. W. Leung, K. H. Chan, P. L. Ip, R. W. Lai, W. K.
Orr, and K. F. Shortridge.1999. Human infection with influenza H9N2. Lancet 354:916–917.
19. Peiris, J. S., M. D. de Jong, and Y. Guan. 2007. Avian influenza virus (H5N1): a threat to human health. Clin. Mirobiol. Rev. 20:243–267. 20. Reed, G. H., and C. T. Wittwer. 2004. Sensitivity and specificity of
single-nucleotide polymorphism scanning by high-resolution melting analysis. Clin. Chem. 50:1748–1754.
21. Reischl, U. 2006. Melting of the ribosomal RNA gene reveals bacterial species identity: a step toward a new rapid test in clinical microbiology. Clin. Chem. 52:1985–1987.
22. Rice, P., I. Longden, and A. Bleasby. 2000. EMBOSS: the European molec-ular biology open software suite. Trends Genet. 16:276–277.
23. Sampath, R., T. A. Hall, C. Massire, F. Li, L. B. Blyn, M. W. Eshoo, S. A.
Hofstadler, and D. J. Ecker.2007. Rapid identification of emerging infec-tious agents using PCR and electrospray ionization mass spectrometry. Ann. N. Y. Acad. Sci. 1102:109–120.
24. Scholtissek, C., J. Stech, S. Krauss, and R. G. Webster. 2002. Cooperation between the hemagglutinin of avian viruses and the matrix protein of human influenza A viruses. J. Virol. 76:1781–1786.
25. Stone, B., J. Burrows, S. Schepetiuk, G. Higgins, A. Hampson, R. Shaw, and
T. W. Kok.2004. Rapid detection and simultaneous subtype differentiation of influenza A viruses by real time PCR. J. Virol. Methods 117:103–112. 26. Subbarao, K., A. Klimov, J. Katz, H. Regnery, W. Lim, H. Hall, M. Perdue,
D. Swayne, C. Bender, J. Huang, M. Hemphill, T. Rowe, M. Shaw, X. Xu, K. Fukuda, and N. Cox.1998. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science 279:393– 396.
27. Sun, Z. F., F. F. Huang, P. G. Halbur, S. K. Schommer, F. W. Pierson, T. E.
Toth, and X. J. Meng.2003. Use of heteroduplex mobility assays (HMA) for pre-sequencing screening and identification of variant strains of swine and avian hepatitis E viruses. Vet. Microbiol. 96:165–176.
28. Townsend, M. D., E. D. Dawson, M. Mehlmann, J. A. Smagala, D. M.
Dankbar, C. L. Moore, C. B. Smith, N. J. Cox, R. D. Kuchta, and K. L. Rowlen.2006. Experimental evaluation of the FluChip diagnostic microarray for influenza virus surveillance. J. Clin. Microbiol. 44:2863–2871. 29. Tweed, S. A., D. M. Skowronski, S. T. David, A. Larder, M., Petric, W. Lees,
Y. Li, J. Katz, M. Krajden, R. Tellier, C. Halpert, M. Hirst, C. Astell, D. Lawrence, and A. Mak.2004. Human illness from avian influenza H7N3, British Columbia. Emerg. Infect. Dis. 10:2196–2199.
30. Vandersteen, J. G., P. Bayrak-Toydemir, R. A. Palais, and C. T. Wittwer. 2007. Identifying common genetic variants by high-resolution melting. Clin. Chem. 53:1191–1198.
31. Webster, R. G., W. J. Bean, O. T. Gorman, T. M. Chambers, and Y.
Kawaoka.1992. Evolution and ecology of influenza A viruses. Microbiol. Rev. 56:152–179.
32. Webster, R. G., Y. Kawaoka, and W. J. Bean. 1989. What is the potential of avirulent influenza viruses to complement a cleavable hemagglutinin and generate virulent strains? Virology 171:484–492.
33. Wittwer, C. T., G. H. Reed, C. N. Hundry, J. G. Vandersteen, and R. J. Pryor. 2003. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin. Chem. 49:853–860.
34. World Health Organization Global Influenza Program Surveillance Network. 2005. Evolution of H5N1 avian influenza viruses in Asia. Emerg. Infect. Dis.
11:1515–1521.
35. Wright, P. 2000. Influenza in the family. N. Engl. J. Med. 343:1331–1332. 36. Xie, Z., Y. S. Pang, J. Liu, X. Deng, X. Tang, J. Sun, and M. I. Khan. 2006.
differenti-ation of avian H5, H7, and H9 hemagglutinin subtypes. Mol. Cell. Probes
20:245–249.
37. Zhou, L., L. Wang, R. Palais, R. Pryor, and C. T. Wittwer. 2005. High-resolution DNA melting analysis for simultaneous mutation scanning and genotyping in solution. Clin. Chem. 51:1770–1777.
38. Zou, S. 1997. A practical approach to genetic screening for influenza virus variants. J. Clin. Microbiol. 35:2623–2627.
39. Zou, S., C. Stansfield, and J. Bridge. 1998. Identification of new influenza B virus variants by multiplex reverse transcription-PCR and the heteroduplex mobility assay. J. Clin. Microbiol. 26:1544–1548.