Theoretical Investigation of Molecular Properties of the First Excited State of the
Thiophenoxyl Radical
Chi-Wen Cheng, Yuan-Pern Lee, and Henryk A. Witek*
Institute of Molecular Science and Department of Applied Chemistry, National Chiao Tung UniVersity, 30010 Hsinchu, Taiwan
ReceiVed: June 9, 2008; ReVised Manuscript ReceiVed: September 29, 2008
Accurate ab initio study of the lowest excited state (A 2B
2) of the thiophenoxyl radical is presented. The
calculated equilibrium geometries, excitation energies, and harmonic vibrational frequencies show that the A2B
2rX2B1excitation in C6H5S has different characteristics than the analogous transition in the phenoxyl radical. Vertical excitation energies for other low-lying (<4.5 eV) excited states of the thiophenoxyl radical are also presented and compared with available experimental data.
Introduction
Sulfur-containing radicals attracted considerably less attention than their oxygen analogues. Accordingly, the structure and spectroscopy of the thiophenoxyl radical (C6H5S, aka the phenylthiyl, phenylsulfyl, or phenylsulfur radical) is far less studied than the corresponding properties of the phenoxyl radical.
A number of absorption bands were reported in solution,1-11 solid matrix,12-14and gas-phase15-17experiments. No definitive assignment of the observed peaks to particular electronic states of C6H5S was given. Most of the experimental effort was devoted to kinetics and possible reaction pathways of the C6H5S radical, which was produced via flash photolysis,2-6laser flash photolysis,1,7 or pulse radiolysis8,9,11 of liquid or solvated aromatic sulfur compounds. The main spectral tool to monitor this radical was the broad transient UV-vis absorption near 297 and 490 nm. It is noteworthy that the vibrational structure of these bands is not well-defined. Upon photolysis of solid samples of thiophenol or diphenyl disulfide, two bands appeared in the emission spectra. These experiments were performed in 3MP (3-methylpentane) or EPA (a mixture of ethyl ether, isopentane, and ethanol) matrices at 77K. The first band12-14 was observed in the region 626s770 nm (red emission, 1.61s1.98 eV) and had a long lifetime. The second band13,14 had a short lifetime with maximal intensity at 435 nm (broad intense blue emission, 2.85 eV).
Two transient absorption bands were attributed to gaseous C6H5S, one around 310 nm15,18and the other around 470-520 nm.16The laser-induced fluorescence excitation spectrum17of gaseous C6H5S in the region 490-520 nm exhibited a transition origin at 517.38 nm (2.396 eV). The transition origin was tentatively assigned to the2A
2r2B2excitation. (Note that this assignment is erroneous; the lower state should be 2B
1.) Three vibrational frequencies (275, 410, and 483 cm-1) were determined for the2A
2 electronic state. A recent dynamic investigation19-21of photodis-sociation of thiophenol revealed that the lowest excited state (A 2B
2) of C6H5S might be located at 2580 cm-1(0.32 eV). Six fully symmetric vibrational frequencies (436, 724, 991, 1073, 1180, and 1551 cm-1) for the ground electronic state of C6H5S in solution were reported by Tripathi et al. based on their resonance Raman experiment. This study indicated that the CS
bond in C6H5S has essentially a single character with the unpaired electron localized on the sulfur atom.10Three vibra-tional frequencies (430, 610, and 1165 cm-1) were determined for the ground electronic state from observed progressions in the dispersed fluorescence spectrum of gaseous C6H5S.17 No experimental information on the equilibrium structure of C6H5S is available.
Theoretical calculations of equilibrium geometry and har-monic vibrational frequencies were reported for the ground (X 2B
1) state10,22-25 and the first excited (A 2B2) state.24 No reliable quantum chemical predictions are available for higher electronic states of C6H5S.
In a recent study26we presented an analysis of molecular properties of the phenoxyl radical (C6H5O) in its first excited state, A2B
2. A comparison of calculated equilibrium geometries and vibrational frequencies between the ground and excited states showed relatively large structural changes upon the A 2B
2 r X 2B1 transition. The A 2B2 r X 2B1 excitation energy27,28for C
6H5O (1.1 eV) is significantly larger than the corresponding experimental value reported recently for C6H5S (0.32 eV).19-21It is of fundamental interest29to understand how the replacement of oxygen by sulfur alters the energetics, structure, and vibrational spectra upon analogous excitation. A detailed theoretical analysis of various molecular properties of the lowest excited A 2B
2 state of C6H5S, together with a comparison with C6H5O, is presented in this study. The main motivation for our study is a perspective of accurate experi-mental characterization of the optically forbidden A 2B
2 r X2B
1transition of C6H5S using the cavity ringdown absorption technique, which requires detailed information on energetics and vibrational frequencies of the A2B
2state. Computational Details
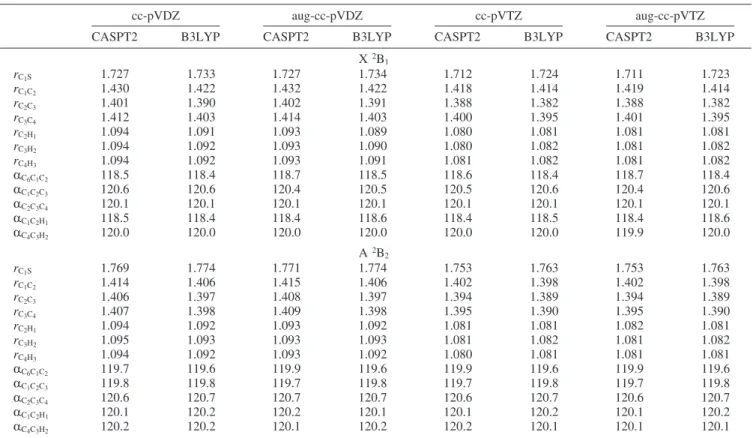
Most of computational details are identical to those in the previous study of the phenoxyl radical.25The molecular model of the thiophenoxyl radical is shown in Figure 1. The symmetry of the ground state is described as a doublet B1 and the symmetry of the first excited state as a doublet B2. The presented energetics and equilibrium geometries have been calculated using complete active space second-order perturbation theory30,31 (CASPT2) and density functional theory (DFT). In addition, the cluster-corrected (Davidson correction) multireference con-figuration interaction (MRCI+Q) vertical and adiabatic
excita-* Corresponding author. E-mail: hwitek@mail.nctu.edu.tw.
10.1021/jp805045s CCC: $40.75 2008 American Chemical Society Published on Web 10/24/2008
tion energetics for the A 2B
2 state has been computed at the optimized CASPT2 geometries in the space of single and double excited configurations using the CASSCF wave function as a reference. Analogous MRCI+Q results for the phenoxyl radical are obtained in a similar way. Vertical excitation energies of low-lying excited states of the thiophenoxyl radical have been computed using the TDDFT32 (time-dependent DFT) and multistate (MS) CASPT233 formalisms. For computing the harmonic frequencies, DFT has been employed. The presented results are obtained with a series of four basis sets: cc-pVDZ, aug-cc-pVDZ, cc-pVTZ, and aug-cc-pVTZ.34,35For the CAS-SCF, CASPT2, and MRCI calculations, we have used the MOLPRO program.36The DFT and TDDFT calculations have been carried out with the Gaussian package37using the B3LYP functional38,39together with the unrestricted Kohn-Sham for-malism.40The complete active space (CAS) in the CASSCF, CASPT2, and MRCI calculations is constructed using all valence π orbitals (two of symmetry a2and five of symmetry b1) and oneσ orbital of symmetry b2corresponding to the lone pair on the sulfur atom. The resulting active space, (0a1, 2a2, 5b1, 1b2), correlates nine active electrons. In the calculation of CASPT2 vertical excitation energies for higher excited states, this CAS space has been augmented with an additional occupiedσ orbital of symmetry a1(the other lone pair on sulfur) to allow for proper description of the 2A
1 state. In the MRCI and CASPT2 calculations, the [He] inner shell of carbon and the [Ne] inner shell of sulfur have not been correlated.
Results
a. Geometrical and Electronic Structure of the A 2B 2 State. Optimized geometrical parameters (see Figure 1) for the ground and first excited state of the thiophenoxyl radical are given in Table 1. The equilibrium bond distances and angles have been calculated using the CASPT2 and DFT approaches. The systematic sequence of four basis sets used in our calculations shows that the computed bond distances depend on the quality of the basis set, giving bond shortening of approximately 0.01 Å upon substituting cc-pVDZ with cc-pVTZ. It is possible that bond distances computed in the complete basis set limit would be even shorter. The quality of the basis set does not affect the equilibrium angles. Augmenting the basis sets with diffuse functions has no effect on equilibrium structures. The difference between the geometrical parameters from DFT and CASPT2 is negligible for the CH bonds and angles, while for rCCand rCS, the difference can be as large as 0.012 Å. This discussion allows for estimating the accuracy of
the calculated structural parameters as (0.01 Å for bond distances and (0.2°for equilibrium angles. The most plausible estimates (in Å) of bond distances in C6H5S are given thus as averaged values of the DFT and CASPT2 parameters at the aug-cc-pVTZ level: rC1S) 1.717, rC1C2) 1.416, rC2C3) 1.385, rC3C4
) 1.398, and rCH) 1.081 for the X2B1state and rC1S) 1.758,
rC1C2) 1.400, rC2C3) 1.392, rC3C4) 1.392, and rCH) 1.081
for the A 2B
2 state. These values are used in the following discussion. The structural parameters reported here should be superior to the previously published theoretical data in terms of the quality of the employed basis set.10,22-25Note that this is especially important for proper description of the structure of the benzene ring in the thiophenoxyl radical, since calculations in smaller basis sets21,24 seem to overestimate the quinoid character of the ground state structure.
In benzene, the experimental length of the CC bond is 1.397 Å.41 In p-benzoquinone, the regular benzene pattern is alternated with rC1C2) rC3C4) 1.477 Å and rC2C3) 1.322 Å.42A
comparison with the calculated rCC values shows that the structure of the benzene ring in C6H5S in both electronic states is aromatic with only little quinoid character for the X2B
1state. This situation is in contrast to that reported for the ground state structure of the phenoxyl radical (rC1C2) 1.448 Å, rC2C3)
1.375 Å, and rC3C4) 1.406 Å), which could be described as
intermediate between aromatic and quinoid.26,43Similar contrast can be observed for the separation between the heteroatom and the benzene ring. The calculated length of the CS bond in both electronic states is intermediate between single and double bond. Compare the calculated values of 1.717 (X2B
1) and 1.758 Å (A 2B
2) with typical lengths of single (1.835 Å in thietane, 1.835 Å in tetrahydrothiophene, 1.818 Å in methanethiol, 1.791 Å in the thiomethoxyl radical, 1.77 Å in thiophenol, and 1.714 Å in thiophene)41,44and double CS bonds (1.681 Å in thioureas, 1.611 Å in thioformaldehyde, 1.610 Å in thioacetal-dehyde, and 1.554 Å in carbon disulfide).41,45 The difference between the CS bond lengths in both studied states of the thiophenoxyl radical (∆rCS) 0.04 Å) is noticeably smaller than the analogous value for the phenoxyl radical (∆rCO) 0.07 Å). It is interesting to note that rCSfor the X2B1state of C6H5S is almost identical to this in thiophene, while for X2B
1of C6H5O, the rCOdistance (1.254 Å)26is noticeably shorter than this in furan (1.362 Å).41 It is clear from this discussion that the molecular structures of the ground states of the phenoxyl and thiophenoxyl radicals are rather distinct, while for the first excited state of both radicals, close structural similarity can be observed. We use this observation in the next two sections to interpret the different spectroscopy of both radicals.
Close analogy can be observed for the electronic structure of both radicals. Similar to the situation for C6H5O, the wave function of both studied states of C6H5S can be adequately described using a single Slater determinant: |σb2 2π b1 2σ a1 2π b1 2π a2 2π b1 o1π a2 /0π b1 /0π b1 /0〉 for the X 2B 1 state and |πb21σa21π2b1πa22πb21σb12πa/02π/0b1πb/01〉 for the A2B2 state. The weight
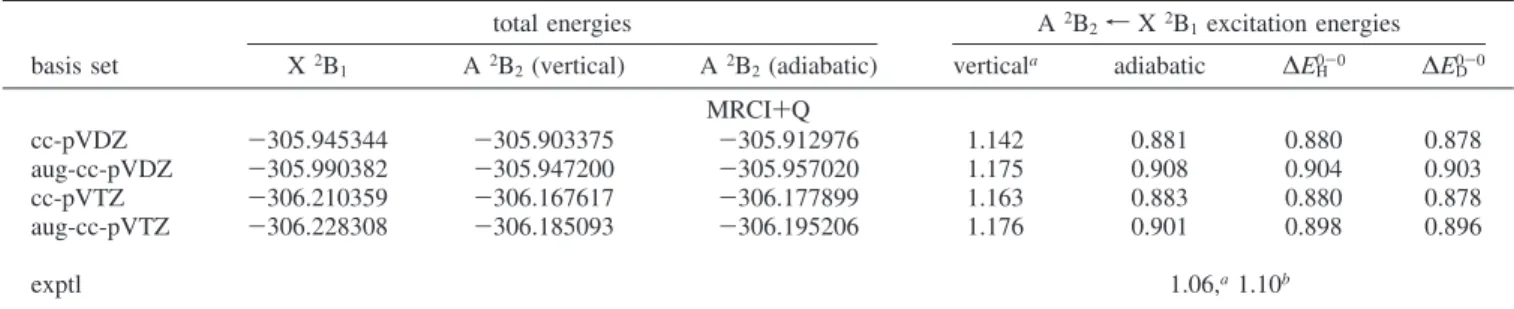
of these Slater determinants in the multiconfigurational CASSCF wave functions is 86% and 89%, respectively. After including dynamic correlation, these values diminish to 63% and 65%, respectively (aug-cc-pVTZ). The weight of other determinants, mostly corresponding to various distributions ofπ electrons in theπ orbitals of the benzene ring, is small (<2% at the CASSCF level). The main chemical difference between the ground states of C6H5O and C6H5S comes thus from the variation of the orbital set used for constructing the wave functions. A schematic energy pattern for the active orbitals for both states of C6H5S is shown in Figure 2. This scheme is almost identical to that presented Figure 1. Molecular structure (C2V) of the thiophenoxyl radical in the
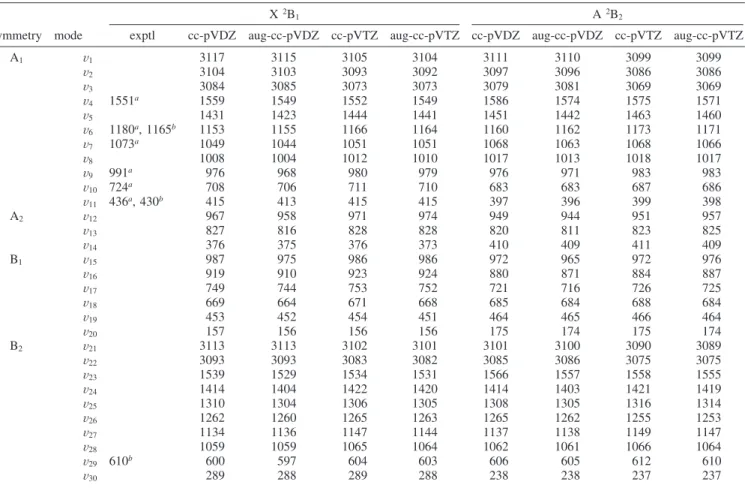
earlier for the phenoxyl radical.26The only important difference concerns the chemical character of the singly occupied b1orbital (SOMO), which is almost completely localized on sulfur in C6H5S and strongly delocalized over oxygen and the aromatic ring in C6H5O. (See also the discussion in refs 9 and 10.) For C6H5S, the delocalization is hinderedsas can be seen from the atomic spin densities shown in Figure 3sowing to a much larger distance between sulfur and the aromatic ring. This larger distance, originating from the larger size of the sulfur atom, does not permit effective overlap of the atomic p orbitals constituting theπ manifold and consequently separates the π
electron densities of the ring and sulfur. In C6H5O, where the distance is approximately 0.5 Å shorter, the coupling of the p orbitals is much larger. (Note that conventional chemical explanation in terms of the large difference between the atomic orbital energies does not apply here. The one-electron energy of the carbon 2p orbital is -0.467 hartree. For the sulfur 3p orbital, one has -0.428 hartree and for oxygen 2p orbital -0.616 hartree.46) Strong localization of the unpaired electron on sulfur for the X 2B
1 state of the thiophenoxyl radical can also be inferred from the experimental ESR and resonance Raman
TABLE 1: Geometrical Parameters for the X2B
1and A2B2States of the Thiophenoxyl Radical Optimized Using CASPT2 and
B3LYPa
cc-pVDZ aug-cc-pVDZ cc-pVTZ aug-cc-pVTZ
CASPT2 B3LYP CASPT2 B3LYP CASPT2 B3LYP CASPT2 B3LYP
X2B1 rC1S 1.727 1.733 1.727 1.734 1.712 1.724 1.711 1.723 rC1C2 1.430 1.422 1.432 1.422 1.418 1.414 1.419 1.414 rC2C3 1.401 1.390 1.402 1.391 1.388 1.382 1.388 1.382 rC3C4 1.412 1.403 1.414 1.403 1.400 1.395 1.401 1.395 rC2H1 1.094 1.091 1.093 1.089 1.080 1.081 1.081 1.081 rC3H2 1.094 1.092 1.093 1.090 1.080 1.082 1.081 1.082 rC4H3 1.094 1.092 1.093 1.091 1.081 1.082 1.081 1.082 RC 6C1C2 118.5 118.4 118.7 118.5 118.6 118.4 118.7 118.4 RC 1C2C3 120.6 120.6 120.4 120.5 120.5 120.6 120.4 120.6 RC 2C3C4 120.1 120.1 120.1 120.1 120.1 120.1 120.1 120.1 RC 1C2H1 118.5 118.4 118.4 118.6 118.4 118.5 118.4 118.6 RC 4C3H2 120.0 120.0 120.0 120.0 120.0 120.0 119.9 120.0 A2B2 rC1S 1.769 1.774 1.771 1.774 1.753 1.763 1.753 1.763 rC1C2 1.414 1.406 1.415 1.406 1.402 1.398 1.402 1.398 rC2C3 1.406 1.397 1.408 1.397 1.394 1.389 1.394 1.389 rC3C4 1.407 1.398 1.409 1.398 1.395 1.390 1.395 1.390 rC2H1 1.094 1.092 1.093 1.092 1.081 1.081 1.082 1.081 rC3H2 1.095 1.093 1.093 1.093 1.081 1.082 1.081 1.082 rC4H3 1.094 1.092 1.093 1.092 1.080 1.081 1.081 1.081 RC 6C1C2 119.7 119.6 119.9 119.6 119.9 119.6 119.9 119.6 RC 1C2C3 119.8 119.8 119.7 119.8 119.7 119.8 119.7 119.8 RC 2C3C4 120.6 120.7 120.7 120.7 120.6 120.7 120.6 120.7 RC 1C2H1 120.1 120.2 120.2 120.1 120.1 120.2 120.1 120.2 RC 4C3H2 120.2 120.2 120.1 120.2 120.2 120.1 120.1 120.1
aAll CASPT2 calculations use the same (0a1, 2a2, 5b1, 1b2) active space described in detail in the text. Distances are given in Å and bonds
in deg. For an explanation of geometrical parameters, see Figure 1.
Figure 2. Orbital energy diagram for the seven activeπ orbitals and one active σ orbital of C6H5S in the X2B1and A2B2states obtained from the CASSCF(0a1, 2a2, 5b1, 1b2)/aug-cc-pVDZ calculations. Occupation numbers are given for every orbital.
studies.10 Another experimental evidence of spin localization is the formation of C6H5SSC6H5as the dominant combination product.10
b. Vertical and Adiabatic A 2B
2 r X 2B1 Excitation Energies and Assignment of Higher Electronic States. The computed vertical, adiabatic, and 0-0 excitation energies for the X 2B
1 f A 2B2transition are given in Table 2. All the employed methods give rather similar theoretical estimates of the excitation energy. The results do not depend strongly on the quality of the basis set used in the calculations. The computed MRCI+Q, CASPT2, DFT, and TDDFT adiabatic and vertical excitation energies do not differ by more than 0.12 eV than the reported experimental value of 0.323 eV.19-21Bearing in mind that the accuracy of the employed computational methods is probably not better than 0.1 eV, the agreement between theory and experiment can be considered as good. The best agreement between theory and experiment is found for the MRCI+Q/aug-cc-pVTZ method with the assumption that the experimentally determined value corresponds to adiabatic excitation energy. Excellent agreement with experiment for the previously reported19,20CASPT2 excitation energy (0.332 eV), which was computed in smaller active space, is probably somewhat fortuitous. The experimental excitation energy was reported by Kim and co-workers, who investigated the pho-tolysis of thiophenol (C6H5SH or C6H5SD) at 243 nm using the H+(D+) velocity map imaging technique.19-21Two distinct anisotropic rings observed in the H+(D+) images were identified to correspond to production of two lowest electronic states of the C6H5S radical upon H (D) detachment. A value of 0.323 ( 0.024 eV (2600 ( 200 cm-1) determined from the difference in threshold translational energy between these two channels was attributed to the energy difference between the A2B
2and
X2B
1states of C6H5S. Is it possible that the experimental value of 0.323 eV corresponds to the energy separation between the X2B
1and A2B2states of the thiophenoxyl radical at the frozen equilibrium geometry of C6H5SD? If this interpretation is correct, the somewhat larger value obtained in our calculations and excellent agreement between MRCI+Q, CASPT2, DFT, and TDDFT can be easily understood. We hope that our future study of the A 2B
2 state of the thiophenoxyl radical using cavity ringdown absorption spectroscopy will help to shed more light on this issue.
In our recent studies26,47 of the first excited state of the phenoxyl radical, we reported theoretical and experimental energy separation between the two lowest electronic states of C6H5O. Unfortunately, the previous experimental values27,28of the separation could not be uniquely assigned as the vertical or adiabatic excitations due to discrepancies in our computed data. We try to compensate for this omission in the present work by comparing the experimental values for the phenoxyl radical with accurate MRCI+Q theoretical data. As can be seen from data shown in Table 3, the MRCI+Q vertical excitation energies lie between the previously reported CASPT2 and B3LYP results and the MRCI+Q adiabatic excitation energies are almost identical to those computed with B3LYP.26 The presented MRCI+Q results suggest that the previously published experi-mental excitation energies should be interpreted as vertical excitations. This is also consistent with the experimental adiabatic separation between these two states that was found by us to be 0.952 eV.47
No definitive assignment of higher lying electronic states of the thiophenoxyl radical is available. Since the only published theoretical study of excited states was performed using a low-level quantum chemical method (configuration interaction with Figure 3. Mulliken charges (a) and spin densities (b) for the X2B1and A2B2states of C6H5S obtained from DFT calculation in four basis set (cc-pVDZ, aug-cc-pvDZ, cc-pVTZ, aug-cc-pVTZ). Values in parentheses are atomic charges with hydrogens summed into heavy atoms.
single excitations only), we use this opportunity to report here the low-lying doublet states of the thiophenoxyl radical. The calculated TDDFT and MS CASPT2 excitation energies are tabulated in Table 4 for all electronic states under 4.5 eV (275 nm). For the TDDFT method, the corresponding transition oscillator strengths are also given. The quality of the basis set does not have a substantial impact on the calculated excitation energies (except for the 2A
1 state at the CASPT2 level). A relatively large number of electronic states are located in the studied spectral region. It is not a difficult task to explain the reported experimental absorption and fluorescence data in terms of the calculated electronic spectrum. The fluorescence excitation signals observed17by Shibuya et al. in the gas phase between 2.38 and 2.58 eV correspond to the2A
2state located by us at 2.44 (TDDFT) and 2.34 eV (CASPT2). This finding confirms the suggested experimental assignment for this state. The 0-0 band was assigned by Shibuya et al.17at 2.40 eV and earlier by Okuyama et al.16at 2.51 eV. The absorption attributed to the 2A
2state is weak; the calculated oscillator strength is only 0.002.
This state is probably also responsible for the weak shoulder band observed11in absorption spectrum in solution at 2.44 eV. The main broad signal in the absorption spectrum in solution has a maximum at 2.70 eV together with intensive sideband at 2.57 eV.10 A similar broad signal is also observed in the emission spectrum.13,14 The position of the maximum in the absorption spectrum depends quite strongly on the solvent: 2.70 eV in water,10,112.79 eV in decalin,12.91 eV in 3-methylpen-tane,13 and 2.92 eV in ethanol.2 It is plausible to assign this band to the calculated 2B
1 state, which is located in our calculations at 3.00 (TDDFT) and 2.66 eV (CASPT2), and the observed sideband to progression associated with some∼1050 cm-1vibrational mode. It is possible that the 0-0 band is weak and both the observed bands correspond to progressions. The calculated oscillator strength for the 2B
1state is large (0.059) and explains well the observed absorption spectrum. The most intensive peak in the absorption spectrum is located at 4.20 eV in aqueous solution10,11 and at 4.17 eV in cyclohexane and ethanol.2It is clear that the state responsible for this peak is
TABLE 2: Total Energies, Vertical Excitation Energies, and Adiabatic Excitation Energies for the X2B
1and A2B2Electronic
States of the Thiophenoxyl Radical Computed Using the CASSCF, CASPT2, MRCI+Q, and B3LYP Methodsd
basis set
total energies A2B2rX2B1excitation energies
X2B1 A2B2(vertical) A2B2(adiabatic) verticala adiabatic ∆EH0-0b
CASSCF cc-pVDZ -627.726063 -627.713627 -627.714759 0.338 0.308 0.303 aug-cc-pVDZ -627.733670 -627.721080 -627.722244 0.343 0.311 0.310 cc-pVTZ -627.795487 -627.783484 -627.784651 0.327 0.295 0.292 aug-cc-pVTZ -627.797276 -627.785374 -627.786538 0.324 0.292 0.290 CASPT2 cc-pVDZ -628.552552 -628.537445 -628.538731 0.411 0.376 0.371 aug-cc-pVDZ -628.598634 -628.582527 -628.583904 0.438 0.401 0.400 cc-pVTZ -628.824789 -628.809950 -628.811244 0.404 0.369 0.366 aug-cc-pVTZ -628.844566 -628.829445 -628.830767 0.412 0.376 0.374 MRCI+Q cc-pVDZ -628.580901 -628.567184 -628.568948 0.373 0.325 0.320 aug-cc-pVDZ -628.619835 -628.605467 -628.607377 0.391 0.339 0.338 cc-pVTZ -628.830279 -628.816726 -628.818590 0.369 0.318 0.315 aug-cc-pVTZ -628.846297 -628.832556 -628.834465 0.374 0.322 0.320 B3LYP cc-pVDZ -629.842725 -629.826715 -629.827936 0.436 (0.403) 0.403 0.398 aug-cc-pVDZ -629.856466 -629.840607 -629.841813 0.432 (0.412) 0.399 0.398 cc-pVTZ -629.924651 -629.909312 -629.910475 0.417 (0.428) 0.386 0.383 aug-cc-pVTZ -629.927282 -629.911999 -629.913156 0.416 (0.433) 0.384 0.382 exptl 0.323 (0.010c
aValues in parentheses have been calculated using the TDDFT method.bIsotope substitution of hydrogen by deuterium does not alter these
values. cReferences 19, 20, and 21. dAll CASSCF, CASPT2, and MRCI+Q calculations use the same (0a1, 2a2, 5b1, 1b2) active space
described in detail in the text.∆EH0-0denotes the 0-0 excitation energy for C6H5S. Total energies are given in hartree and excitation energies in eV.
TABLE 3: Total Energies, Vertical Excitation Energies, and Adiabatic Excitation Energies for the X2B
1and A2B2Electronic
States of the Phenoxyl Radical Computed Using the MRCI+Q Method at the Optimized CASPT2 Geometriesc
total energies A2B2rX2B1excitation energies
basis set X2B1 A2B2(vertical) A2B2(adiabatic) verticala adiabatic ∆EH0-0 ∆ED0-0
MRCI+Q cc-pVDZ -305.945344 -305.903375 -305.912976 1.142 0.881 0.880 0.878 aug-cc-pVDZ -305.990382 -305.947200 -305.957020 1.175 0.908 0.904 0.903 cc-pVTZ -306.210359 -306.167617 -306.177899 1.163 0.883 0.880 0.878 aug-cc-pVTZ -306.228308 -306.185093 -306.195206 1.176 0.901 0.898 0.896 exptl 1.06,a1.10b
aReference 27, in the gas phase.bReference 28, in argon matrix.cAll calculations use the same (0a1, 2a2, 5b1, 1b2) active space described
in detail in ref 26. ∆EH0-0 denotes the 0-0 excitation energy for C6H5O, and ∆ED0-0 for C6D5O. Total energies are given in hartree and excitation energies in eV.
2B
1with the calculated excitation energy of 4.20 (TDDFT) and 4.66 eV (CASPT2), and the largest oscillator strength (0.066) among all theoretically determined electronic states. There exists a large discrepancy between the TDDFT and CASPT2 methods for some higher lying states (the first2A
1and second2A2states). Usually such large discrepancy is attributed to inaccuracies within the TDDFT scheme, but here it also can be caused by deficiencies in the choice of the active space or poor conver-gence of the perturbation series. (Note the large deviation of the 2A
1 r X 2B1 CASPT2 transition energy computed with various basis sets.) The lowest quartet state of the thiophenoxyl radical (4B
1) is found at 4.35 eV in our B3LYP/cc-pVDZ
calculations. Clearly, higher spin states are not going to influence the observed absorption spectrum of C6H5S in the studied spectral region. Our calculations cannot account directly for the red emission observed13,14 in the photolysis of C
6H5SH and C6H5SSC6H5in 3-methylpentane and EPA glasses. (The emis-sion maximum is located at 1.83 eV in 3MP and at 1.92 eV in EPA; well-structured vibrational progression of approximately 1350 cm-1is also observed.) The only electronic state located in this region is2B
1, but the observed emission maxima differ by 0.5-0.6 eV to match this state. It is rather unlikely that there exists another electronic state with such energy, especially given that the positions of the next two observed states,2A
2with2B1,
TABLE 4: Vertical Excitation Energies (in eV) for the Lowest Doublet Excited States of the Thiophenoxyl Radical Computed Using the B3LYP and Multistate (MS) CASPT2 Methodh
TDDFT MS CASPT2
cc-pVDZ aug-cc-pVDZ cc-pVTZ aug-cc-pVTZ cc-pVDZ aug-cc-pVDZ cc-pVTZ aug-cc-pVTZ Exp.
X2B1 -629.842725 -629.856466 -629.924651 -629.927282 -628.549286 -628.594568 -628.820821 -628.840438 2B2 0.403 (0.000) 0.412 (0.000) 0.428 (0.000) 0.433 (0.000) 0.454 0.487 0.451 0.461 0.323a 2A2 2.430 (0.003) 2.383 (0.002) 2.457 (0.003) 2.443 (0.002) 2.547 2.357 2.412 2.344 2.40b, 2.51c 2B1 3.001 (0.054) 2.952 (0.061) 3.013 (0.057) 2.999 (0.059) 2.718 2.633 2.693 2.664 2.70d, 2.79e, 2.91f, 2.92g 2A1 3.947 (0.003) 3.908 (0.002) 3.949 (0.003) 3.944 (0.002) 4.820 4.689 4.751 4.265 2B1 4.186 (0.049) 4.133 (0.061) 4.223 (0.058) 4.197 (0.066) 4.729 4.604 4.746 4.662 4.20d, 4.17g 2B2 4.458 (0.000) 4.379 (0.000) 4.438 (0.000) 4.393 (0.000) 4.265 4.208 4.257 4.225 2A2 4.536 (0.011) 4.424 (0.008) 4.514 (0.010) 4.459 (0.008) 5.262 5.063 5.196 5.084
aReferences 19, 20, and 21. Photoelecton spectroscopy in the gas phase.bReference 13. Fluorescence in the gas phase. cReference 12.
Fluorescence in the gas phase. dReferences 11 and 14. Time-resolved absorption spectroscopy in aqueous solution. eReference 1.
Time-resolved absorption spectroscopy in liquid decalin. fReference 17. Absorption spectrum in 3-methylpentane glass. gReference 2.
Absorption spectrum in cyclohexane and EtOH. hThe lowest excited state in each symmetry is given. Values in parentheses are oscillator
strengths.
TABLE 5: Harmonic Vibrational Frequencies for the X2B
1and A2B2Electronic State of C6H5S Computed Using the B3LYP
Methodc
X2B1 A2B2
symmetry mode exptl cc-pVDZ aug-cc-pVDZ cc-pVTZ aug-cc-pVTZ cc-pVDZ aug-cc-pVDZ cc-pVTZ aug-cc-pVTZ
A1 V1 3117 3115 3105 3104 3111 3110 3099 3099 V2 3104 3103 3093 3092 3097 3096 3086 3086 V3 3084 3085 3073 3073 3079 3081 3069 3069 V4 1551a 1559 1549 1552 1549 1586 1574 1575 1571 V5 1431 1423 1444 1441 1451 1442 1463 1460 V6 1180a, 1165b 1153 1155 1166 1164 1160 1162 1173 1171 V7 1073a 1049 1044 1051 1051 1068 1063 1068 1066 V8 1008 1004 1012 1010 1017 1013 1018 1017 V9 991a 976 968 980 979 976 971 983 983 V10 724a 708 706 711 710 683 683 687 686 V11 436a, 430b 415 413 415 415 397 396 399 398 A2 V12 967 958 971 974 949 944 951 957 V13 827 816 828 828 820 811 823 825 V14 376 375 376 373 410 409 411 409 B1 V15 987 975 986 986 972 965 972 976 V16 919 910 923 924 880 871 884 887 V17 749 744 753 752 721 716 726 725 V18 669 664 671 668 685 684 688 684 V19 453 452 454 451 464 465 466 464 V20 157 156 156 156 175 174 175 174 B2 V21 3113 3113 3102 3101 3101 3100 3090 3089 V22 3093 3093 3083 3082 3085 3086 3075 3075 V23 1539 1529 1534 1531 1566 1557 1558 1555 V24 1414 1404 1422 1420 1414 1403 1421 1419 V25 1310 1304 1306 1305 1308 1305 1316 1314 V26 1262 1260 1265 1263 1265 1262 1255 1253 V27 1134 1136 1147 1144 1137 1138 1149 1147 V28 1059 1059 1065 1064 1062 1061 1066 1064 V29 610b 600 597 604 603 606 605 612 610 V30 289 288 289 288 238 238 237 237
agree well with our calculations. We can see two potential explanations for this phenomenon.
(1) The emission corresponds rather to the B2A
2fA2B2 transition than the B2A
2fX 2B1transition. The calculated energy separation between the B2A
2and A2B2states is 2.01 eV (TDDFT/aug-cc-pVTZ), which matches the experimental emission spectrum more closely than the energy separation between B2A
2and X2B1(2.44 eV). Even better agreement is observed for the associated excitation spectrum (see Figure 2 of ref 14), which has a maximum located at 2.11 eV. This value almost perfectly agrees with the experimentally determined separation between the B2A
2and A2B2states (2.08 eV). (2)The equilibrium geometry of the B 2A
2state computed with DFT is not planar. There exists a close structural similarity with the equilibrium geometry of the phenoxyl radical in the 2A′′state. (For details, see Figure 2 of ref 48.) It is clear that geometry relaxation leads to smaller energy separation between the B2A
2state and the two lower states. However, the energy of the B 2A
2state is lowered only by 0.002 eV (B3LYP/cc-pVDZ) upon the relaxation. At the same time, the energy of the two lower states computed at the optimized geometry of the B 2A
2 state is elevated by 0.36 (X 2B1) and 0.40 eV (A2B
2). The zero-point energy corrections (cc-pVDZ, unscaled) to all three studied states are comparable (2.46, 2.46, and 2.37 eV for X2B
1, A2B2, and B2A2, respectively). The experimental separation between the maxima of emission and absorption signals is 0.28 eV in 3MP and 0.19 eV in EPA. It is rather unlikely that this mechanism could account for the discrepancy of 0.5-0.6 eV for the B 2A
2 f X 2B1transition mentioned above.
Note that none of these hypotheses fully explains the unusually long lifetime of the observed red emission. Jinguji et al.14argue that the long emission lifetime is a consequence of the strongly forbidden character of the corresponding excitation. The computed TDDFT oscillator strengths for the B 2A
2 f A2B
2and B2A2fX2B1transitions are indeed small: 0.002 and 0.000, respectively.
There is no experimental evidence for other electronic states of the thiophenoxyl radical with excitation energies smaller than 4.5 eV.
c. Harmonic Vibrational Frequencies. Harmonic vibra-tional frequencies for the X2B
1and A2B2states of C6H5S are given in Table 5. Analogous data for the isotope-substituted species (C6D5S) are given in Table 6. The presented (scaled) values are computed with B3LYP using a set of four basis sets. The scaling factors (0.971 for cc-pVDZ and aug-cc-pVDZ and 0.970 for cc-pVTZ and aug-cc-pVTZ) have been determined from separate DFT calculations for thiophenol (C6H5SH), for which experimental fundamental frequencies are available.49The frequencies computed with various basis sets are rather similar; the largest deviation observed upon basis set change is 21 cm-1 forν26of C6D5S (X2B1) andν5of C6H5S (A2B2). As discussed in the previous study of the phenoxyl radical,26 the smallest mean absolute deviation from experiment (approximately 25 cm-1) can be expected for the cc-pVTZ basis set. Therefore, for the following discussion, the cc-pVTZ harmonic vibrational frequencies are used. A graphical description of some of the full-symmetric modes can be found in ref 10. Information for the remaining vibrational modes will be made available from the authors upon request.
TABLE 6: Harmonic Vibrational Frequencies for the X2B
1and A2B2Electronic State of C6D5S Computed Using the B3LYP
Methoda
X2B1 A2B2
symmetry mode cc-pVDZ aug-cc-pVDZ cc-pVTZ aug-cc-pVTZ cc-pVDZ aug-cc-pVDZ cc-pVTZ aug-cc-pVTZ
A1 V1 2311 2310 2301 2301 2308 2307 2298 2298 V2 2296 2295 2287 2286 2289 2289 2281 2280 V3 2273 2273 2266 2266 2268 2270 2262 2262 V4 1521 1510 1508 1505 1550 1538 1536 1532 V5 1296 1286 1297 1295 1319 1309 1319 1316 V6 1023 1016 1026 1026 1026 1020 1027 1026 V7 935 928 940 939 936 931 942 941 V8 843 844 853 851 849 850 860 858 V9 807 808 816 815 813 813 821 820 V10 670 668 673 673 650 650 655 654 V11 407 405 408 407 390 389 391 391 A2 V12 786 781 789 793 770 769 772 779 V13 643 634 644 644 639 631 640 642 V14 327 325 327 325 357 355 358 356 B1 V15 826 821 829 827 808 809 810 813 V16 756 751 761 761 725 722 731 732 V17 630 624 632 631 609 604 611 610 V18 520 514 521 520 535 530 536 535 V19 400 398 400 398 408 407 409 407 V20 148 146 147 146 164 163 164 163 B2 V21 2305 2304 2295 2295 2297 2296 2287 2287 V22 2283 2283 2275 2275 2276 2277 2268 2269 V23 1495 1485 1484 1481 1530 1520 1517 1514 V24 1322 1314 1310 1308 1314 1304 1302 1300 V25 1272 1263 1258 1256 1266 1257 1249 1247 V26 998 996 1017 1016 1000 998 1018 1016 V27 824 824 834 832 822 822 832 830 V28 798 801 810 809 806 809 817 815 V29 575 574 580 579 581 580 586 585 V30 275 273 274 274 226 227 226 226
The change in harmonic vibrational frequencies upon the X2B
1fA2B2excitation is small. The difference between the zero-point energies (ZPE) of both states is only 26 cm-1 for C6H5S and 37 cm-1for C6D5S. On average, each vibrational level is displaced by 15 cm-1for C6H5S and 13 cm-1for C6D5S. The largest displacement is observed for the in-plane CS bending (ν30): -51 cm-1 for C6H5S and -48 cm-1 for C6D5S. The change in other vibrational modes involving the sulfur atomsthe out-of-plane CS bending (ν20) and the CS stretch (ν7)sis noticeably smaller. This situation is in contrast with that reported26for the phenoxyl radical, where the corresponding vibrational modes (ν6,ν30, andν20) were displaced by 167, 73, and 38 cm-1 (C6H5O) and 217, 65, and 37 cm-1 (C6D5O), respectively.
The computed harmonic vibrational frequencies of the ground state correspond well to the fundamental modes detected in the resonance Raman spectrum in solution (436, 724, 991, 1073, 1180, and 1551 cm-1)10 and in the gas-phase fluorescence spectrum (430, 610, and 1165 cm-1).17These modes can be easily assigned toν11,ν10,ν9,ν7,ν6, andν4(solution spectrum) and toν11,ν29, andν6(gas-phase spectrum). (For more detailed discussion, see ref 10.) The computed ground-state frequencies are in satisfactory agreement with other theoretical studies.10,22,25 Similar agreement is found for the X2B
1and A2B2frequencies reported by Song et al.,23if one scales these frequencies by an appropriate factor. While no large discrepancies are found with the previous theoretical data, the present study probably can be considered as giving the most accurate set of harmonic vibrational frequencies for both electronic states of the thiophe-noxyl radical owing to the most complete character of employed basis sets.
The presented analysis confirms the observation made previ-ously on the basis of optimized geometrical parameters about structural similarity between both electronic states of the thiophenoxyl radical. The computed force fields are also similar giving an almost identical set of harmonic vibrational frequen-cies for both electronic states: X2B
1and A2B2.
Conclusion
High-level ab initio techniques are employed to study the equilibrium geometry, excitation energies, and harmonic vibra-tional frequencies for the first excited electronic state (A2B
2) of the thiophenoxyl radical. The calculated properties are compared to analogous data for the ground state. The wave functions of both studied electronic states are well represented with single Slater determinants. The A2B
2rX2B1excitation can be concisely described as a transfer of a single electron between two atomic-like p orbitals localized on sulfur. The calculated excitation energies for the A2B
2rX2B1transition correspond well to experimental data. The change in molecular equilibrium geometry and harmonic vibrational frequencies is rather small upon the excitation. The presented results show that the A2B
2rX2B1excitation in C6H5S has a quite different characteristic than the analogous transition in the phenoxyl radical (C6H5O), where the singly occupied orbital in the X2B1 state is largely delocalized over oxygen and the aromatic ring. The delocalization causes energetic stabilization of the X2B
1 state of C6H5O and results in approximately double character of the CO bond. This effect is missing in C6H5S owing to a much larger distance between sulfur and carbon that does not permit large effective overlap of π densities on both atoms. Consequently, both studied electronic states of the thiophenoxyl radical (A2B
2and X2B1) show close energetic and structural similarity.
In addition, a computational study of other low-lying (<4.5 eV) electronic states of the thiophenoxyl radical is presented. The calculated TDDFT and CASPT2 excitation energies cor-respond well to the available experimental absorption and emission data. A reliable assignment of excited states of the thiophenoxyl radical is given in Table 4. The lowest quartet state of C6H5S is found at 4.35 eV (B3LYP/cc-pVDZ).
Acknowledgment. We thank Prof. Soji Tsuchiya for help and the National Center for High-Performance Computing for computer time. The National Science Council of Taiwan is acknowledged for financial support (grants NSC96-2113-M009-022 and NSC96-2113-M009-025). This research has also been supported by Institute of Nuclear Energy Research, Atomic Energy Council, Taiwan, under Contract No. 970147 L, and by the Ministry of Education (MOE-ATU project).
References and Notes
(1) Scott, T. W.; Liu, S. N. J. Phys. Chem. 1989, 93, 1393. (2) Thyrion, F. C. J. Phys. Chem. 1973, 77, 1478. (3) Gaspari, G.; Granzow, A. J. Phys. Chem. 1970, 74, 836. (4) Ito, O.; Matsuda, M. J. Am. Chem. Soc. 1983, 105, 1937. (5) Ito, O.; Matsuda, M. J. Am. Chem. Soc. 1979, 101, 1815. (6) Ito, O.; Matsuda, M. J. Am. Chem. Soc. 1979, 101, 5732. (7) Burkey, T. J.; Griller, D. J. Am. Chem. Soc. 1985, 107, 246. (8) Hermann, R.; Dey, G. R.; Naumov, S.; Brede, O. Phys. Chem. Chem. Phys. 2000, 2, 1213.
(9) Armstrong, D. A.; Sun, Q.; Schuler, R. H. J. Phys. Chem. 1996, 100, 9892.
(10) Tripathi, G. N. R.; Sun, Q.; Armstrong, D. A.; Chipman, D. M.; Schuler, R. H. J. Phys. Chem. 1992, 96, 5344.
(11) Bonifacˇic´, M.; Weiss, J.; Chaudhri, S. A.; Asmus, K.-D. J. Phys. Chem. 1985, 89, 3910.
(12) Feher, F.; Gladden, T.; Kurz, D. Z. Naturforsch. 1970, 25b, 1215. (13) Russell, P. G. J. Phys. Chem. 1975, 79, 1353.
(14) Jinguji, M.; Imamura, T.; Obi, K.; Tanaka, I. Chem. Phys. Lett.
1984, 109, 31.
(15) Porter, G.; Wright, F. J. Trans. Faraday Soc. 1955, 51, 1469. (16) Okuyama, M.; Takakura, T.; Kamada, H. J. Spectrosc. Soc. Jpn.
1977, 26, 164.
(17) Shibuya, K.; Nemoto, M.; Yanagibori, A.; Fukushima, M.; Obi, K. Chem. Phys. 1988, 121, 237.
(18) Norrish, R. G. W.; Zeelenberg, A. P. Proc. R. Soc. London, Ser. A
1967, 240, 293.
(19) Lim, J. S.; Lim, I. S.; Lee, K. S.; Ahn, D. S.; Lee, Y. S.; Kim, S. K. Angew. Chem., Int. Ed. 2006, 45, 6290.
(20) Lim, I. S.; Lim, J. S.; Lee, Y. S.; Kim, S. K. J. Chem. Phys. 2007, 126, 034306.
(21) Lim, J. S.; Lee, Y. S.; Kim, S. K. Angew. Chem., Int. Ed. 2008, 47, 1853.
(22) Kamisuki, T.; Hirose, C. J. Mol. Struct. (THEOCHEM) 2000, 531, 51.
(23) Remacle, F.; Kryachko, E. S. J. Mol. Struct. 2004, 708, 165. (24) Song, L.; Bu, Y; Li, P. Int. J. Quantum Chem. 2005, 105, 186. (25) Xu, W.; Gao, A. J. Phys Chem. A 2006, 110, 997.
(26) Cheng, C.-W.; Lee, Y.-P.; Witek, H. A. J. Phys. Chem. A 2008, 112, 2648.
(27) Radziszewski, J. G.; Gil, M.; Gorski, A.; Spanget-Larsen, J.; Waluk, J.; Mro´z, B. J. J. Chem. Phys. 2001, 115, 9733.
(28) Gunion, R. F.; Gilles, M. K.; Polak, M. L.; Lineberger, W. C. Int. J. Mass Spectrom. Ion Processes 1992, 117, 601.
(29) Foster, S. C.; Miller, T. A. J. Phys. Chem. 1989, 93, 5986. (30) Werner, H.-J. Mol. Phys. 1996, 89, 645.
(31) Celani, P.; Werner, H.-J. J. Chem. Phys. 2000, 112, 5546. (32) Casida, M. E.; Jamorski, C.; Casida, K. C.; Salahub, D. R. J. Phys. Chem. 1998, 108, 4439.
(33) Finley, J.; Malmqvist, P.-Å; Roos, B. O.; Serrano-Andre´s, L. Chem. Phys. Lett. 1998, 288, 299.
(34) Dunning, T. H., Jr J. Chem. Phys. 1989, 90, 1007.
(35) Kendall, R. A.; Dunning, T. H, Jr.; Harrison, R. J. J. Chem. Phys.
1992, 96, 6769.
(36) Werner, H.-J.; Knowles, P. J.; Amos, R. D. et al., MOLPRO, a package of ab initio programs, version 2006.1.
(37) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B. et al. Gaussian 03, Revision A.1; Gaussian Inc.: Pittsburgh, PA,2003.
(38) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
(39) Lee, C.; Yang, W.; Parr, R. G. Phys. ReV. B 1988, 37, 785. (40) Kohn, W.; Sham, L. J. Phys. ReV. 1965, 140, A1133.
(41) NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 101, Release 12, Aug 2005, Russell D. Johnson III, Ed. http://srdata.nist.gov/cccbdb.
(42) Hagen, K.; Hedberg, K. J. Chem. Phys. 1973, 59, 158. (43) Chipman, D. M.; Liu, R.; Zhou, X.; Pulay, P. J. Chem. Phys. 1994, 100, 5023.
(44) Johansson, K. I.; Oldeberg, H.; Selen, H. Ark. Fys. 1967, 33, 313. (45) Allen, F. H.; Kennard, O.; Watson, D. G.; Brammer, L.; Orpen, A. G.; Taylor, R. J. Chem. Soc., Perkin Trans. 1987, 2, S1.
(46) Visscher, L.; Dyall, K. G. At. Data Nucl. Data Tables 1997, 67, 207.
(47) Cheng, C.-W.; Witek, H. A.; Lee, Y.-P. J. Chem. Phys. In press. (48) Liu, R.; Morokuma, K.; Mebel, A. M.; Lin, M. C. J. Phys. Chem.
1996, 100, 9314.
(49) Scott, D. W.; McCullough, J. P.; Hubbard, W. N.; Messerly, J. F.; Hossenlopp, I. A.; Waddington, G. J. Am. Chem. Soc. 1956, 78, 5463. JP805045S