Synthesis, Structures, Solution Behavior, and Reactions
of Thiolato-Bridged Diruthenium Carbonyl Phosphine
Complexes
Kom-Bei Shiu*
Department of Chemistry, National Cheng Kung University, Tainan, Taiwan 701
Sue-Lein Wang and Fen-Ling Liao
Department of Chemistry, National Tsing Hua University, Hsinchu, Taiwan 300
Michael Y. Chiang
Department of Chemistry, National Sun Yat-Sen University, Kaohsiung, Taiwan 804
Shie-Ming Peng and Gene-Hsiang Lee
Department of Chemistry, National Taiwan University, Taipei, Taiwan 106
Ju-Chun Wang and Lin-Shu Liou
Department of Chemistry, Soochow University, Taipei, Taiwan 111 Received November 13, 1997
Dinuclear thiolato-bridged complexes [Ru2(CO)4(µ-SR)2(PR′3)2] (R′) Ph, Me) have been readily prepared from the reaction of [Ru2(CO)4(MeCN)4(PR′3)2][BF4]2(R′) Ph (1), Me (2)) with RSH (R )tBu,iPr, Ph) and Et
3N at ambient temperature. Although only the syn form of [Ru2(CO)4(µ-StBu)2(PPh3)2] (3) and only the anti form of [Ru2(CO)4(µ-SPh)2(PMe3)2] (6) were found, an equilibrium mixture of both the syn (isomer A) and anti (isomer B) forms was present in solution for [Ru2(CO)4(µ-SiPr)2(PPh3)2] (4) and [Ru2(CO)4(µ-SPh)2(PPh3)2] (5). The spectral data support that the syn form (4A) is the major isomer of 4, while the anti form (5B) is the major isomer of 5. The two thiolato bridges are located cis to the two phosphine ligands in solid-state structures 3, 4A, and 5B, but they are located cis to only one of the two ligands whereas they are trans to the other in structure 6. Iodination of 3-6 gave one identical product [Ru2(CO)4(µ-SR)2I2(PR′3)2] (R′) Ph, R )tBu (7),iPr (8), Ph (9); R′) Me, R ) Ph (10)). Both spectral and structural evidence shows that 7-10 exist in the syn form with no Ru-Ru bonding interaction. The relative orientation of the two thiolato bridges with respect to the two phosphine ligands present in 6 remains in structure 10, in spite of the change from anti to syn. The reactions of the diiodide complexes [Ru2(CO)4( µ-SR)2I2(PR′3)2] with R′′S anions give either [Ru2(CO)4(µ-SR)2(PR′3)2] and R′′SSR′′ or [Ru2 -(CO)4(µ-SR)2(SR′′)2(PR′3)2].
Introduction
Transition-metal complexes with sulfur ligands are of significant interest not only because they often display unusual structures and novel reactivity1but also because they can serve as synthetic analogues for the active sites of metalloprotein,2and show some relevance to metal sulfide hydrodesulfurization and demercura-tion catalysts.3
Diiron thiolato-bridged complexes, [Fe2(CO)4(µ-SR)2L2] (L ) CO, phosphine, phosphite, one or one-half diphos-phine or -arsine ligand; R ) alkyl, phenyl, or aryl), have been studied extensively in terms of synthesis, reactiv-ity, and spectroscopic measurements.4 The existence of two isomers, syn and anti, with a common4-6 arrange-ment for the two axially coordinated ligands L ) phosphine, trans to the metal-metal bond (Chart 1),
* To whom correspondence should be addressed. Fax: (+886) 6 274 0552. E-mail: kbshiu@mail.ncku.edu.tw.
(1) For recent reviews, see: (a) Holm, R. H.; Ciurli, S.; Weigel, J. A. Prog. Inorg. Chem. 1990, 38, 1. (b) Krebs, B.; Henkel, G. Angew. Chem., Int. Ed. Engl. 1991, 30, 769. (c) Shibahara, T. Coord. Chem. Rev. 1993, 123, 73. (d) Saito, T. In Early Transition Metal Clusters withπ-Donor Ligands; Chisholm, M. H., Ed.; VCH: New York, 1995; Chapter 3. (e) Dance, I.; Fisher, K. Prog. Inorg. Chem. 1994, 41, 637. (f) Stiefel, E. I. Ed. Transition Metal Sulfur Chemistry; American Chemical Society: Washington, DC, 1996.
(2) (a) Coucouvanis, D. Adv. Inorg. Chem. 1992, 38. 1. (b) Coucou-vanis, D. In Molybenum Enzymes, Cofactors, and Model systems; Stiefel, E. I., Coucouvanis, D., Newton, W. E., Eds.; American Chemical Society: Washington, DC, 1993; p 304. (c) Rees, D. C.; Chan, M. K.; Kim, J. Adv. Inorg. Chem. 1994, 40, 89.
(3) (a) Angelici, R. J. Acc. Chem. Res. 1988, 21, 387. (b) Chen, J.; Daniels, L. M.; Angelici, R. J. J. Am. Chem. Soc. 1990, 112, 199. (c) Wiegand, B. C.; Friend, C. M. Chem. Rev. 1992, 92, 491. (d) Riaz, V.; Curnow, O. J.; Curtis, M. D. J. Am. Chem. Soc. 1994, 116, 4357. (e) Bianchini, C.; Meli, A. J. Chem. Soc., Dalton Trans. 1996, 801. (f) Hill, A. F.; Wilton-Ely, J. D. E. T. Organometallics 1997, 16, 4517. S0276-7333(97)01002-9 CCC: $15.00 © 1998 American Chemical Society
Publication on Web 03/31/1998
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
and equilibrated isomerization favoring one isomer or the other were recognized for some types of compounds. Thermal oxidative and reductive decompositions yield-ing sulfides, disulfides, and thiols were also reported. In contrast the chemistry of diruthenium thiolato-bridged complexes are still relatively unexplored.5
In this paper, we present the following new informa-tion: (1) the reactions between [Ru2(CO)4(MeCN)4 -(PR′3)2][BF4]2 (R′ ) Ph (1), Me (2)) and two thiolate anions via RSH/Et3N can occur even at ambient tem-perature to afford the Ru-Ru singly bonded compounds [Ru2(CO)4(µ-SR)2(PR′3)2] (3-6) (R )tBu,iPr, Ph; R′) Ph, Me); (2) the facile interconversion between syn and anti forms of [Ru2(CO)4(µ-SiPr)2(PPh3)2] (4) and [Ru2 -(CO)4(µ-SPh)2(PPh3)2] (5) is observed at ambient tem-perature in solution; (3) the first crystallographically characterized syn and anti forms of the diruthenium thiolato-bridged carbonyl phosphine complexes are pre-sented, adopting either a common geometry in 3-5, with the two thiolato bridges cis to the two phosphine groups observed previously in diiron thiolato-bridged complexes,4i or an unprecedented geometry, with the two thiolato bridges cis to one but trans to the other phosphine ligand in 6; (4) iodination of 3-6 affords the single diiodide adducts (7-10) all in the syn form, where the unique geometry observed in 6 is retained in its diiodide adduct 10; and (5) the reactions of [Ru2(CO)4 -(µ-SR)2I2(PR′3)2] with R′′S- via R′′SH/Et3N produce either the substituted product [Ru2(CO)4(µ-SR)2(SR′′)2 -(PR′3)2] or the reductive-deiodination products [Ru2 -(CO)4(µ-SR)2(PR′3)2] with apparently external R′′S -being oxidized into R′′SSR′′.
Experimental Section
All solvents were dried and purified by standard methods (ethers, paraffins, and arenes from potassium with benzophe-none as indicator; halocarbons and acetonitrile from CaH2and
alcohols from the corresponding alkoxide) and were freshly distilled under nitrogen immediately before use. All reactions and manipulations were carried out in standard Schlenk ware, connected to a switchable double manifold providing vacuum and nitrogen. Reagents were used as supplied by Aldrich.1H
and31P NMR spectra were measured on a Brueker AMC-400
(1H, 400 MHz;31P, 162 MHz;13C, 100 MHz) NMR
spectrom-eter. 1H chemical shifts (δ in ppm, J in hertz) are defined as
positive downfield relative to internal MeSi4 (TMS) or the
deuterated solvent, while31P chemical shifts are referred to
external 85% H3PO4. The IR spectra were recorded on a
Bio-Rad FTS 175 instrument. The following abbreviations were used: s, strong (IR); m, medium; s, singlet (NMR); d, doublet; h, heptet; m, multiplet. Microanalyses were carried out by the staff of the Microanalytical Service of the Department of Chemistry, National Cheng Kung University.
Synthesis of [Ru2(CO)4(µ-SR)2(PR′3)2] (R′) Ph, R )tBu (3),iPr (4), Ph (5); R′) Me, R ) Ph (6)). To a solution of [Ru2(CO)4(MeCN)4(PR′3)2][BF4]25b(0.26 mmol) dissolved in 20
mL of MeCN were added carefully deaerated HSR (1 mL) and Et3N (2 mL). The solution was stirred for 2 h at ambient
temperature (ca. 28 °C), forming an orange-yellow precipitate for starting compounds with R′ ) Ph and solution for the compounds with R′) Me. The solvent and volatiles were then removed under vacuum. Recrystallization from CH2Cl2/MeOH
gave pure product.
3: Anal. Calcd for C48H48O4P2Ru2S2: C, 56.68; H, 4.75.
Found: C, 56.61; H, 4.75. Yield: 87%. IR (CH2Cl2): vCO, 2001 s, 1965 m, 1931 s cm-1. 1H NMR (25 °C, acetone-d 6, 400 MHz): δ 0.64 (s, 18 H), 7.53 (m, 30 H). 31P{1H}NMR (25 °C, 162 MHz): δ 27.21 (s, 2 P) in acetone-d6and 17.78 (s, 2 P) in CDCl3.
4: Anal. Calcd for C46H44O4P2Ru2S2: C, 55.86; H, 4.48.
Found: C, 55.68; H, 4.47. Yield: 88%. IR (CH2Cl2): vCO, 2010
s, 2001 s, 1974 m, 1964 m, 1945 s, 1935 s cm-1. 1H NMR (25
°C, CDCl3, 400 MHz): δ 0.32 (d, 12 H, J ) 6.6), 2.54 (h, 2 H)
for 4A; andδ 0.31 (d, 6 H, J ) 6.2), 1.03 (d, 6 H, J ) 6.2), 2.39
(h, 1 H), 2.71 (h, 1 H) for 4B. 31P{1H}NMR (25 °C, CDCl 3,
162 MHz): δ 19.97 (s, 2 P) for 4A, and δ 26.54 (s, 2 P) for 4B.
5: Anal. Calcd for C52H40O4P2Ru2S2: C, 59.08; H, 3.81.
Found: C, 59.15; H, 3.92. Yield: 84%. IR (CH2Cl2): vCO, 2016
s, 1979 m, 1951 s cm-1. 1H NMR (25 °C, CDCl
3, 400 MHz): δ
6.68 (m, 10 H), 7.38 (m, 30 H). 31P{1H}NMR (25 °C, CDCl 3,
162 MHz): δ 21.98 (s, 2 P) for 5A, and 26.15 (s, 2 P) for 5B.
6: 89% yield. Anal. Calcd for C16H32O4P2Ru2S2: C, 31.16;
H, 5.23. Found: C, 31.07; H, 5.22. IR (CH2Cl2): vCO, 2043 s, 1983 s, 1970 sh, 1937 m cm-1. 1H NMR (25 °C, acetone-d 6, 400 MHz): δ 1.84 (d, 9 H, J ) 10.1), 1.70 (d, 9 H, J ) 10.1), 7.04 (m, 10 H). 31P{1H}NMR (298 or 220 K, acetone-d 6, 162 MHz): δ 6.42 (s, 1 P), 15.17 (s, 1P).
Iodination of 3-6. To a solution of [Ru2(CO)4(µ-SR)2
-(PR′3)2] (0.12 mmol) dissolved in 20 mL of CH2Cl2was added
dropwise 4 mL (ca. 0.13 mmol) of an I2solution prepared by
dissolving 0.129 g (0.51 mmol) of I2in 15 mL of CH2Cl2. The
solution was stirred for 10 min at ambient temperature. The product, 7-10, was separated as a major yellow band by thin-layer chromatography (silica gel, CH2Cl2:hexane ) 1:1), using
TLC plates (Kieselguhr 60 F254, E. Merck), and recrystallized
from CH2Cl2/MeOH.
[Ru2(CO)4(µ-StBu)2I2(PPh3)2] (7): 77% yield. Anal. Calcd
for C48H48I2O4P2Ru2S2: C, 45.36; H, 3.81. Found: C, 45.21; H, 3.83. 1H NMR (25 °C, acetone-d 6, 400 MHz): δ 0.76 (s, 18 H), 7.73 (m, 30 H). 31P{1H}NMR (300 or 220 K, acetone-d 6, 162 MHz): δ 44.58 (s, 2 P). IR (CH2Cl2): vCO, 2057 s, 2049 s, 2003 s cm-1. (4) (a) Dahl, L. F.; Wei, C.-H. Inorg. Chem. 1963, 2, 328. (b) Hieber,
W.; Kaiser, K. Chem. Ber. 1969, 102, 4043. (c) Crow, J. P.; Cullen, W. R. Can. J. Chem. 1971, 49, 2948. (d) De Beer, J. A.; Haines, R. J. J. Chem. Soc. A 1971, 3271. (e) Maresca, L.; Greggio, F.; Sbrignadello, G.; Bor, G. Inorg. Chim. Acta 1971, 5, 667. (f) De Beer, J. A., Haines, R. J. J. Organomet. Chem. 1972, 36, 297. (g) De Beer, J. A.; Haines, R. J. J. Organomet. Chem. 1972, 37, 173. (h) Ellgen, P. C.; Gerlach, J. N. Inorg. Chem. 1973, 12, 2526. (i) Borgne, G. L.; Grandjean, D.; Mathieu, R.; Poilblanc, R. J. Organomet. Chem. 1977, 131, 429. (j) Nametkin, N. S.; Tyurin, V. D.; Kukina, M. A. J. Organomet. Chem. 1978, 149, 355. (k) Shriver, D. F.; Whitmire, K. H. In Comprehensive Organometallic Chemistry; Wilkinson, G., Stone, F. G. A., Abel, E. W., Eds.; Pergamon: Oxford, England, 1982; Vol. 4, Chapter 31, p 280. (l) Hawker, P. H.; Twigg, M. V. In Comprehensive Coordination Chem-istry; Wilkinson, G. W., Gillard, R. D., Eds.; Pergamon: Oxford, England, 1987; Vol. 4. Chapter 44.1, p 1238. (m) Whitmire, K. H, In Comprehensive Organometallic Chemistry II; Abel, E. W., Stone, F. G. A., Wilkinson, G., Eds.; Pergamon: Oxford, England, 1995; Vol. 7, Chapter 1, p 65.
(5) (a) Andreu, P. L.; Cabeza, J. A.; Riera, V.; Robert, F.; Jeannin, Y. J. Organomet. Chem. 1989, 372, C15. (b) Shiu, K.-B.; Li, C.-H.; Chan, T.-J.; Peng, S.-M.; Cheng, M.-C.; Wang, S.-L.; Liao, F.-L. Organome-tallics 1995, 14, 524. (c) Soler, J.; Ros, J.; Carrasco, M. R.; Ruiz, A.; Alvarez-Larena, A.; Piniella, J. F. Inorg. Chem. 1995, 34, 6211.
(6) Andreu, P. L.; Cabeza, J. A.; Miguel, D.; Riera, V.; Villa, M. A.; Garcia-Granda, S. J. Chem. Soc., Dalton Trans. 1991, 533.
Chart 1
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
[Ru2(CO)4(µ-SiPr)2I2(PPh3)2] (8): 83% yield. Anal. Calcd for C46H44I2O4P2Ru2S2: C, 44.45; H, 3.57. Found: C, 44.21; H, 3.60. 1H NMR (27 °C, acetone-d 6, 400 MHz): δ 0.95 (d, 12 H, J ) 6.7), 2.99 (h, 2 H), 7.60 (m, 30 H). 31P{1H}NMR (300 or 220 K, acetone-d6, 162 MHz): δ 46.65 (s, 2 P). IR (CH2 -Cl2): vCO, 2059 s, 2051 s, 2005 s cm-1.
[Ru2(CO)4(µ-SPh)2I2(PPh3)2] (9): 85% yield. Anal. Calcd
for C52H40I2O4P2Ru2S2: C, 47.64; H, 3.08. Found: C, 47.43; H, 3.11. 1H NMR (27 °C, acetone-d 6, 400 MHz): δ 7.47 (m, 40 H). 31P{1H}NMR (300 or 220 K, acetone-d 6, 162 MHz): δ 50.97 (s, 2 P). IR (CH2Cl2): vCO, 2070 sh, 2062 s, 2017 s cm-1.
[Ru2(CO)4(µ-SPh)2I2(PMe3)2] (10): 82% yield. Anal. Calcd
for C22H28I2O4P2Ru2S2: C, 28.16; H, 3.01. Found: C, 28.11; H, 3.01. 1H NMR (27 °C, acetone-d 6, 400 MHz): δ 1.74 (d, 9 H, J ) 10.8), 1.84 (d, 9 H, J ) 10.8), 7.53 (m, 10 H). 31P{1H} NMR (300 or 220 K, acetone-d6, 162 MHz): δ 18.47 (s, 1 P), 27.77 (s, 1 P). IR (CH2Cl2): vCO, 2047 s, 2008 sh, 1991 s, 1960 m cm-1.
Reaction of [Ru2(CO)4(µ-SR)2I2(PR′3)2] with R′′SH (R′′
)tBu,iPr, Ph) and Et
3N. (a) Reactions between 7 and R′′SH/
Et3N and that between 9 andtBuSH/Et3N are quite similar,
and only one typical example is shown below. Compound 7 (445 mg, 0.35 mmol) was dissolved in 20 mL of MeCN, and to this solution were then added 1 mL of PhSH and 2 mL of Et3N.
The solution was stirred for 2 h, forming a orange-yellow precipitate. The solvent and volatiles were then removed under vacuum, and the residue was taken up in a minimum amount of CH2Cl2. The products were separated by thin-layer
chromatography using CH2Cl2/hexane mixed solvents to give
63 mg of PhSSPh (83%) and 274 mg of 3 (77%). (b) Reaction between 9 and PhSH/Et3N: Compound 9 (203 mg, 0.155 mmol)
was dissolved in 10 mL of MeCN to form a clear yellow solution, and to this solution were then added 1 mL of Et3N
and 0.5 mL of PhSH. Upon addition of PhSH, the orange-yellow color developed immediately and an orange-orange-yellow precipitate appeared within 3 min. The suspension was stirred for an additional 30 min. The precipitate was collected, washed with 5 mL of MeOH three times, and dried under
vacuum to afford 148 mg of the product [Ru2(CO)4(µ-SPh)2
-(SPh)2(PPh3)2] (11) in 75% yield. Anal. Calcd for
C64H50O4P2Ru2S4: C, 60.27; H, 3.95. Found: C, 60.17; H, 3.95.
IR (CH2Cl2): vCO, 2045 s, 2032 sh, 1985 s cm-1. 1H NMR (27
°C, CDCl3, 400 MHz): δ 7.39 (m, 50 H). 31P{1H}NMR (300
K, CDCl3, 162 MHz): δ 19.47 (s, 2 P).
Single-Crystal X-ray Diffraction Studies of 3, 4A, 5B, 6, and 10. Suitable single crystals were grown from CH2Cl2/
MeOH or CH2Cl2/hexane at room temperature and chosen for
single crystal structure determinations. The X-ray diffraction data of 4A, 5B, and 6 were measured on a four-circle diffrac-tometer, and those of 3 and 10 were measured in frames with increasingω (0.3 deg/frame) and with the scan speed at 10.00
s/frame on a Siemens SMART-CCD instrument, equipped with a normal focus and 3 kW sealed-tube X-ray source. For data collected on the four-circle diffractometer, three standard reflections were monitored every hour or every 50 reflections throughout the collection. The variation was less than 2%. Empirical absorption corrections were carried out based on an azimuthal scan. For 6, the structure was solved by direct methods and refined by a full-matrix least-squares procedure using TEXSAN.7 For 5B, the structures were solved by the
heavy-atom method and refined by a full-matrix least-squares procedure using NRCVAX.8 For 3, 4A, and 10, the structures
were solved by direct methods and refined by a full-matrix least-squares procedure using SHELXTL-PLUS.9 Neutral
atom scattering factors for non-hydrogen atoms and the values for∆f′and∆f′′described in each software7-9were used. The
other essential details of single-crystal data measurement and refinement are listed in Table 1. One molecule of MeOH and two molecules of H2O were found in the asymmetric unit of
(7) Crystal Structure Analysis Package, Molecular Structure Cor-poration, Texas, 1985 and 1992.
(8) Gabe, E. J.; Le page, Y.; Charland, J.-P.; Lee, F. L.; White, P. S. J. Appl. Crystallogr. 1989, 22, 384.
(9) (a) Sheldrick, G. M. SHELXTL-Plus Crystallographic System, release 4.21; Siemens Analytical X-ray Instruments: Madison, WI, 1991. (b) Siemens Analytical X-ray Instruments Inc.: Karlsruhe, Germany, 1991.
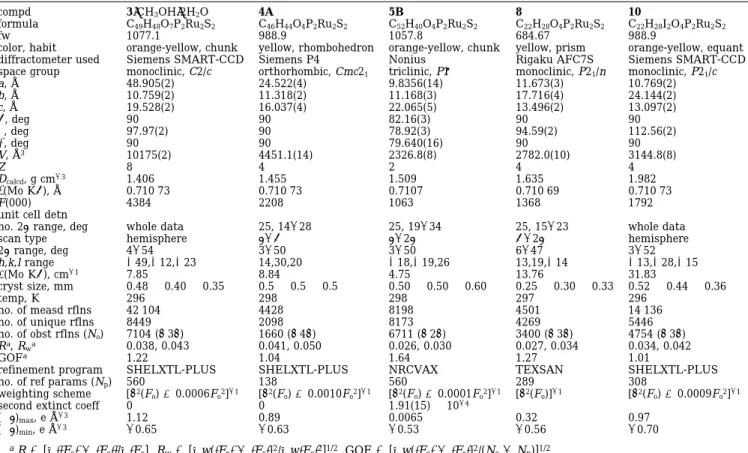
Table 1. Crystal Data
compd 3‚CH3OH‚2H2O 4A 5B 8 10
formula C49H48O7P2Ru2S2 C46H44O4P2Ru2S2 C52H40O4P2Ru2S2 C22H28O4P2Ru2S2 C22H28I2O4P2Ru2S2
fw 1077.1 988.9 1057.8 684.67 988.9
color, habit orange-yellow, chunk yellow, rhombohedron orange-yellow, chunk yellow, prism orange-yellow, equant diffractometer used Siemens SMART-CCD Siemens P4 Nonius Rigaku AFC7S Siemens SMART-CCD space group monoclinic, C2/c orthorhombic, Cmc21 triclinic, P1h monoclinic, P21/n monoclinic, P21/c
a, Å 48.905(2) 24.522(4) 9.8356(14) 11.673(3) 10.769(2) b, Å 10.759(2) 11.318(2) 11.168(3) 17.716(4) 24.144(2) c, Å 19.528(2) 16.037(4) 22.065(5) 13.496(2) 13.097(2) R, deg 90 90 82.16(3) 90 90 β, deg 97.97(2) 90 78.92(3) 94.59(2) 112.56(2) γ, deg 90 90 79.640(16) 90 90 V, Å3 10175(2) 4451.1(14) 2326.8(8) 2782.0(10) 3144.8(8) Z 8 4 2 4 4 Dcalcd, g cm-3 1.406 1.455 1.509 1.635 1.982 λ(Mo KR), Å 0.710 73 0.710 73 0.7107 0.710 69 0.710 73 F(000) 4384 2208 1063 1368 1792
unit cell detn
no. 2θ range, deg whole data 25, 14-28 25, 19-34 25, 15-23 whole data
scan type hemisphere θ-ω θ-2θ ω-2θ hemisphere
2θ range, deg 4-54 3-50 3-50 6-47 3-52 h,k,l range (49,(12,(23 14,30,20 (18,(19,26 13,19,(14 (13,(28,(15 µ(Mo KR), cm-1 7.85 8.84 4.75 13.76 31.83 cryst size, mm 0.48× 0.40 × 0.35 0.5× 0.5 × 0.5 0.50× 0.50 × 0.60 0.25× 0.30 × 0.33 0.52× 0.44 × 0.36 temp, K 296 298 298 297 296 no. of measd rflns 42 104 4428 8198 4501 14 136 no. of unique rflns 8449 2098 8173 4269 5446 no. of obst rflns (No) 7104 (>3σ) 1660 (>4σ) 6711 (>2σ) 3400 (>3σ) 4754 (>3σ) Ra, R wa 0.038, 0.043 0.041, 0.050 0.026, 0.030 0.027, 0.034 0.034, 0.042 GOFa 1.22 1.04 1.64 1.27 1.01
refinement program SHELXTL-PLUS SHELXTL-PLUS NRCVAX TEXSAN SHELXTL-PLUS
no. of ref params (Np) 560 138 560 289 308
weighting scheme [σ2(F
o) + 0.0006Fo2]-1 [σ2(Fo) + 0.0010Fo2]-1 [σ2(Fo) + 0.0001Fo2]-1 [σ2(Fo)]-1 [σ2(Fo) + 0.0009Fo2]-1
second extinct coeff 0 0 1.91(15)× 10-4
(∆F)max, e Å-3 1.12 0.89 0.0065 0.32 0.97
(∆F)min, e Å-3 -0.65 -0.63 -0.53 -0.56 -0.70
aR ) [∑||F
o| - |Fc||/∑|Fo]. Rw) [∑w(|Fo| - |Fc|)2/∑w|Fo|2]1/2. GOF ) [∑w(|Fo| - |Fc|)2/(No- Np)]1/2.
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
the crystals used for 3. The solvent hydrogen positions in this structure were not included in the structure refinement.
Results and Discussion
Synthesis, Structures, and Solution Behavior of [Ru2(CO)4(µ-SR)2(PR′3)2] (3-6). Following one of our
recent reports that the acetonitrile ligands of 1 and 2 can be easily replaced by weak anions such as NO3-,10 we rediscovered that the temperature for reaction between 1 and 1,2-benzenedithiol to produce [Ru2(CO)4 -(µ,η2-S
2C6H4)(PPh3)2]5bcan be reduced to the ambient temperature. Likewise the reaction of 1 and 2 with a thiol in the presence of a base such as Et3N can occur readily at ambient temperature to give 3-6 in satisfac-tory yield (Scheme 1). The diruthenium trimethylphos-phine derivative was found to be more air-sensitive than the triphenylphosphine analogue. Both the thiol and Et3N should be carefully deaerated before addition to compounds 1 and 2, especially for the preparation of 6. This compound is orange-yellow. However, when aer-ated thiol and Et3N were used to react with 2, the resulted orange-red to red products contained obviously multiple components displaying more than 1531P{1H} NMR singlets betweenδ -20 and 33 in CDCl3.
Compound 3 was previously reported to be formed either from the thermal degradation of [Ru3(CO)9(PPh3)3] withtBuSH by Cabeza et al. in 19895aor from a long (20 h) thermal displacement reaction between [Ru2(CO)4(µ-O2CH)2(PPh3)2] and the thiol in refluxing toluene (bp 110 °C) by Ros et al. in 1995.5c Although it is not unreasonable that both syn and anti isomers are produced under these reaction conditions, the31P{1H} chemical shifts of Ros’s two isomers are rather high at δ 45.3 for syn and 39.5 for anti, compared with the values of 23.87 reported for Cabeza’s compound5aand 17.78 measured for 3 in our laboratory. The31P{1H} singlet observed for this compound remains at 220 K
in acetone-d6, or at 420 K in acetophenone-d3, indicating probably the presence of only one isomer at either high or low temperature. On the basis of the three-carbonyl-stretching-band pattern observed for 3 dissolved in CH2 -Cl2, the structure is not in the anti form (Cssymmetry), but in the syn geometry (C2v symmetry), which was confirmed by X-ray diffraction methods (Figure 1). The high-temperature31P NMR evidence reflects that the syn geometry is more stable thermodynamically than the anti form, if this form can be prepared in some way. It is hence quite surprising that Ros’s isolated “syn” isomer was found to be a minor product with “syn”/“anti” ) 1:8.5c
The isolated 4 and 5 exist in solution as equilibrium mixtures of syn ( isomer A) and anti (isomer B) isomers, as evidenced by the following: (1) similar NMR and IR spectra were obtained by dissolving either the products 4 and 5 or their single crystals (4A and 5B); (2) the syn: anti ratio was found to be changeless within experi-mental error, even after a toluene solution of 4 (or 5) was heated under reflux for more than 80 h; and (3) two exchange cross peaks, indicating clearly PsynTPanti, were shown in the31P{1H}NOESY NMR experiments11 for 4 and 5 in CDCl3 at 298 K. The spectral data support that 4A (the syn form) is the major isomer for 4, whereas 5B (the anti form) is the major component for 5. The observed syn/anti (or 4A:4B) integration ratio of 1.16:1 based on the 1H NMR doublets assigned for the methyl groups ofiPr of 4 is close to that of 1.14:1 based on the inverse-gated31P{1H}NMR singlets (i.e., without NOE) in CDCl3. Similarly, the calculated integration ratio between one carbonyl13C NMR singlet for 5A (the syn form) and two carbonyl singlets for 5B (the anti form) is 1.14:1:1, based on the observed 5A: 5B integration ratio of 1:1.766 found as two inverse-gated 31P{1H}NMR singlets, is close to the observed integration ratio of 1.13:1:1 based on the three broad (10) Shiu, K.-B.; Yang, L.-T.; Jean, S.-W.; Li, C.-H.; Wu, R.-L.; Wang,
J.-C.; Liou, L.-S.; Chiang, M. Y. Inorg. Chem. 1996, 35, 7845.
(11) Bodenhausen, G.; Ernst, R. R. J. Am. Chem. Soc. 1982, 104, 1304.
Scheme 1
Figure 1. ORTEP plot of [Ru2(CO)4(µ-StBu)2(PPh3)2] (3).
Selected bond distances (Å): Ru(1)-Ru(2) ) 2.640(1), Ru-(1)-S(1) ) 2.319(1), Ru(1)-S(2) ) 2.422(1), Ru(1)-P(1) ) 2.449(1), Ru(1)-C(1) ) 1.867(4), Ru(1)-C(2) ) 1.867(5), Ru(2)-S(1) ) 2.564(1), Ru(2)-S(2) ) 2.466(1), Ru(2)-P(2) ) 2.265(1), Ru(2)-C(3) ) 1.887(4), Ru(2)-C(4) ) 1.945(5). Selected bond angles (deg): Ru(1)-Ru(2)-S(1) ) 52.9(1), Ru(1)-Ru(2)-S(2) ) 56.5(1), Ru(1)-Ru(2)-P(2) ) 148.6-(1), Ru(2)-Ru(1)-S(1) ) 61.9148.6-(1), Ru(2)-Ru(1)-S(2) ) 58.1-(1), Ru(2)-Ru(1)-P(1) ) 150.358.1-(1), Ru(1)-S(1)-Ru(2) ) 65.2(1), Ru(1)-S(2)-Ru(2) ) 65.4(1), P(1)-Ru(1)-S(1) ) 96.6(1), P(1)-Ru(1)-S(2) ) 97.9(1), P(2)-Ru(2)-S(1) ) 100.8(1), P(2)-Ru(2)-S(2) ) 100.6(1).
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
13C NMR carbonyl singlets measured in CDCl 3. Both the 31P{1H}NMR singlet and the 13C NMR carbonyl singlet for the syn form were observed at an upfield position relative to those for the anti form (31P{1H}NMR δ 19.97 for 4A vs 26.54 for 4B and 21.98 for 5A vs. 26.15 for 5B;13C{1H}NMR 202.0 for 5A vs 203.4 and 204.6 for 5B). The single-crystal structures were determined by X-ray diffraction methods, confirming the stereo-chemistry with the syn form for 4A (Figure 2) and the anti form for 5B (Figure 3). Quite coincidentally, the major isomer is the one giving the structures. The spectral and the X-ray crystal structural evidence apparently explain why the two isomers of 4 and 5 cannot be separated in our hands by either silica or alumina column chromatography by CH2Cl2/hexane mixtures, although such a separation method was
previously reported in obtaining one “syn” and two “anti” isomers of a similar compound, [Ru2(CO)4(µ-SBz)2 -(PPh3)2] (Bz ) benzyl), by Ros’s group.5c The syn-anti equilibria of the organothio-bridged deriviatives of iron carbonyl, [Fe2(CO)6(µ-SR)2] (with R ) Me, Et), and of their monosubstitution products, [Fe2(CO)5(µ-SMe)2(Pn -Bu3)], were reported previously to have the predominant form in anti geometry for the former but in syn geometry for the latter.4e The disubstitution products of a related selenium compound, [Ru2(CO)6(µ-SePh)2], with PPh3were also found to consist of both forms, with the anti mode favored.6 Apparently both steric and electronic factors should be combined to account for the different ratio between the two forms.4g,h
Dinuclear thiolato-bridged phosphine carbonyl com-plexes [M2(CO)4(µ-SR′)2(PR3)2] (M ) Fe, Ru) were rarely characterized by X-ray diffraction methods and limited in the syn form of [Fe2(CO)4(µ-SMe)2(PMe3)2] in 1977,4i and no crystal structures containing diruthenium ana-logues have been characterized so far. Hence, 3, 4A, and 5B are the first crystallographically characterized syn and anti structures of diruthenium thiolato-bridged carbonyl phosphine complexes. The Ru-Ru distances of 2.640(1) Å in 3, 2.694(1) Å in 4A, and 2.679(1) Å in 5B are similar to each other and fall within the range 2.558-2.873 Å of other singly Ru-Ru bonded complexes with ligation of PPh3 ligands.10,12 Structure 4A has a crystallographically imposed mirror plane containing the midpoint of the Ru-Ru bond, two sulfur atoms, and four carbon atoms, C(3)-C(6) (Figure 2). Like the previously reported structure with two thiolato bridges, [Fe2(CO)4(µ-SMe)2(PMe3)2],4iand those with one dithi-olato bridge, [Ru2(CO)4(µ,η2-S2(C6H4)(PPh3)2]5band [Ru2 -(CO)4(µ,η2-S2(CH2)3)(PPh3)2],5cthe three thiolato-bridged structures 3 (Figure 1), 4A (Figure 2), and 5B (Figure 3) follow to have a common structure type with the two thiolato bridges all being located cis to the two phos-phine groups.
Like 4 and 5, product 6 exhibited two31P{1H}NMR singlets at δ 6.42 and 15.17 at ambient temperature (298 K) and at a low temperature (220 K). However, the single-crystal X-ray structure evidence of 6 did not support the presence of both syn and anti isomers in solution, but revealed an unprecedented structure type for only one anti isomer. The two thiolato bridges are located cis to one but trans to the other phosphine ligand, resulting in two inequivalent phosphine atoms and two31P{1H}NMR singlets in the NMR spectra, with d(Ru-Ru) ) 2.6923(8) Å in 6 (Figure 4). The four-band vCOpattern displayed by a CH2Cl2 solution of 6 in an IR spectrum is consistent with the C1symmetry of this structure. However, the vCO pattern should not be overemphasized without consideration of other evidence such as NMR results in the characterization of the related structures, for the bands may be overlapped into a different pattern or may not be resolved by the IR instrument, explaining a six-band and a three-band pattern observed for 4 and 5, respectively, each contain-ing an equilibrium mixture of two differently weighted (12) (a) Sherlock, S. J.; Cowie, M.; Singleton, E.; Steyn, M. M. de V. J. Organomet. Chem. 1989, 361, 353. (b) Garcia-Granda, S.; Obeso-Rosete, R.; Gonzalez, J. M. R.; Anillo, A. Acta Crystallogr. 1990, C46, 2043. (c) Shiu, K.-B.; Peng, S.-M.; Cheng, M.-C. J. Organomet. Chem. 1993, 452, 143. (d) Klemperer, W. G.; Bianxia, Z. Inorg. Chem. 1993, 32, 5821.
Figure 2. ORTEP plot of [Ru2(CO)4(µ-SiPr)2(PPh3)2] (4A).
Selected bond distances (Å): Ru(1)-Ru(1a) ) 2.694(1), Ru-(1)-S(1) ) 2.424(3), Ru(1)-S(2) ) 2.422(3), Ru(1)-P(1) ) 2.375(3), Ru(1)-C(1) ) 1.874(10), Ru(1)-C(2) ) 1.855(10). Selected bond angles (deg): Ru(1a)-Ru(1)-S(1) ) 56.3-(1), Ru(1a)-Ru(1)-S(2) ) 56.256.3-(1), Ru(1a)-Ru(1)-P(1) ) 148.7(1), Ru(1)-S(1)-Ru(1a) ) 67.5(1), P(1)-Ru(1)-S(1) ) 97.5(1), P(1)-Ru(1)-S(2) ) 102.6(1).
Figure 3. ORTEP plot of [Ru2(CO)4(µ-SPh)2(PPh3)2] (5B).
Selected bond distances (Å): Ru(1)-Ru(2) ) 2.6788(10), S(1) ) 2.4285(9), S(2) ) 2.4358(11), Ru(1)-P(1) ) 2.3897(11), Ru(1)-C(1) ) 1.868(3), Ru(1)-C(2) ) 1.861(3), Ru(2)-S(1) ) 2.4260(8), Ru(2)-S(2) ) 2.4323(9), P(2) ) 2.3762(11), C(3) ) 1.874(3), Ru(2)-C(4) ) 1.870(3). Selected bond angles (deg): Ru(2)-(1)-S(1) ) 51.463(24), Ru(2)-Ru(1)-S(2) ) 56.55(3), Ru-(2)-Ru(1)-P(1) ) 159.917(24), Ru(1)-Ru(2)-S(1) ) 56.55(3), Ru(1)-Ru(2)-S(2) ) 56.68(3), Ru(1)-Ru(2)-P(2) ) 156.929-(24), Ru(1)-S(1)-Ru(2) ) 66.98(3), Ru(1)-S(2)-Ru(2) ) 66.77(3), P(1)-Ru(1)-S(1) ) 109.04(4), P(1)-Ru(1)-S(2) ) 109.96(4), S(1) ) 106.57(4), P(2)-Ru(2)-S(2) ) 107.81(4).
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
syn and anti isomers. A mechanism probably involving conversion of a bridging into a terminal thiolato group with concomitant formation of multiple Ru-Ru bonding interactions is proposed to account for the equilibrium process between the syn and anti forms of 4 and 5 in solution (Scheme 2).
Synthesis, Structures, and Reactions of [Ru2
-(CO)4(µ-SR′)2I2(PR3)2] (7-10). The electrophilic
ad-dition of I2to CH2Cl2solutions of 3-6 generates almost immediately at ambient temperature the diiodide ad-ducts [Ru2(CO)4(µ-SR′)2I2(PR3)2] (7-10) (Scheme 3). It appears that there are two different classes of products, as shown in the three-band and the four-band vCO patterns observed for 7-9 and 10, respectively. Quite coincidentally, the difference between the highest vCO bands of 7-10 and their respective precursor com-pounds 3-6 is as high as about 50 cm-1for 7-9 and as low as 4 cm-1for 10 (2057 in 7 vs 2001 in 3; 2059 in 8 vs 2001 in 4; 2070 in 9 vs 2016 in 4; 2047 in 10 vs 2043 cm-1in 6). The single crystals of 10 were easily grown from CH2Cl2/hexane at ambient temperature, and the structure was soon found to keep the unique structure just like the precursor 6 but change the geometry from anti in 6 into syn in 10 (Figure 5). This novel structure with inequivalent phosphine atoms remains not only in the solid state but also in solution, as shown with two observed31P{1H}NMR singlets atδ 18.47 and 27.77 in
acetone-d6 at 300 and 220 K. The long distance with d(Ru‚‚‚Ru) ) 3.743 Å between two Ru atoms indicates no Ru-Ru single bond in 10 and probably also in 7-9. The two iodides are trans to each other, reflecting a trans addition of diiodine to 6 and probably also to 3-5. If the resulting structure 7-9 has two trans iodide atoms with each at one Ru atom, the observed31P{1H} NMR singlet atδ 44.58 for 7, 46.65 for 8, and 50.97 for 9 in acetone-d6 at 300 and 220 K may suggest a very facile process, probably involving conversion of two terminal into bridging iodides concomitantly with open-ing two bridgopen-ing into terminal thiolato groups while not permitting rotation of two carbonyls and one phosphine attached to each Ru atom along a pseudo-C3 axis (Scheme 4), resulting in two equivalent and inequivalent phosphine atoms for 7-9 and 10, respectively, at the NMR time scale. The evidence of the three-band vCO pattern and the one31P{1H}NMR singlet described for Figure 4. ORTEP plot of [Ru2(CO)4(µ-SPh)2(PMe3)2] (6).
Selected bond distances: Ru(2) ) 2.6923(8), Ru(1)-S(1) ) 2.423(1), Ru(1)-S(2) ) 2.433(1), Ru(1)-P(1) ) 2.357-(1), Ru(1)-C(1) ) 1.862(4), Ru(1)-C(2) ) 1.868(5), Ru(2)-S(1) ) 2.417(1), Ru(2)-S(2) ) 2.407(1), Ru(2)-P(2) ) 2.327(1), Ru(2)-C(3) ) 1.891(4), Ru(2)-C(4) ) 1.880(5) Å. Selected bond angles: Ru(2)-Ru(1)-S(1) ) 56.09(3), Ru-(2)-Ru(1)-S(1) ) 55.76(3), Ru(2)-Ru(1)-P(1) ) 151.95-(3), Ru(1)-Ru(2)-S(1) ) 56.32151.95-(3), Ru(1)-Ru(2)-S(2) ) 56.65(3), Ru(1)-Ru(2)-P(2) ) 102.74(3), Ru(1)-S(1)-Ru-(2) ) 67.60(3), Ru(1)-SRu(1)-S(1)-Ru-(2)-RuRu(1)-S(1)-Ru-(2) ) 67.60(3), P(1)-Ru(1)-S(1) ) 99.71(4), P(1)-Ru(1)-S(2) ) 109.13(4), P(2)-Ru(2)-S(1) ) 159.04(4), P(2)-Ru(2)-S(2) ) 90.81(4)o. Scheme 2
Figure 5. ORTEP plot of [Ru2(CO)4(µ-SPh)2I2(PMe3)2] (10).
Selected bond distances (Å): Ru(1)-I(1) ) 2.799(1), Ru-(1)-S(1) ) 2.466(1), Ru(1)-S(2) ) 2.458(1), Ru(1)-P(1) ) 2.337(1), Ru(1)-C(1) ) 1.886(6), Ru(1)-C(2) ) 1.869(6), Ru(2)-S(1) ) 2.471(1), Ru(2)-S(2) ) 2.463(1), Ru(2)-P(2) ) 2.360(1), Ru(2)-C(3) ) 1.865(9), Ru(2)-C(4) ) 1.859(7). Selected bond angles (deg): I(1)-Ru(1)-P(1) ) 171.6(1), I(2)-Ru(2)-C(4) ) 172.7(2), Ru(1)-S(1)-Ru(2) ) 98.6(1), Ru(1)-S(2)-Ru(2) ) 99.1(1), P(1)-Ru(1)-S(1) ) 91.5(1), P(1)-Ru(1)-S(2) ) 90.2(1), P(2)-Ru(2)-S(1) ) 94.5(1), P(2)-Ru(2)-S(2) ) 168.3(1).
Scheme 3
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
7-9 and that of one1H NMR singlet atδ 0.76 for the twotBu groups in 7, one doublet at 0.95 and one heptet at 2.99 for twoiPr groups in 8, and one31C NMR doublet at 192.5 with JP,C ) 11.0 Hz13 for the four carbonyl groups in 9 suggest that 7-9 should adopt probably the syn form rather than the anti form.
The reported reaction chemistry of [Fe2(CO)4(µ-SR)2 -(PPh3)2] (R ) Et, Ph) with X2 (X ) Cl, Br, I) is quite rich, producing either [Fe2(CO)4(µ-SR)2(PPh3)2I]I3 or [Fe3(CO)4(µ-SR)2(PPh3)2X4]X with no Fe-Fe metal bond.4b However, decarbonylation occurs quite readily for iodi-nation of 7-9, resulting in intractable products, appar-ently without carbonyl groups attached to the metal center on the basis of the IR spectra obtained. Reactions of 7-9 with nucleophiles such as thiolate anions were then carried out. The reaction results between com-plexes 7 and 9 and the anions via R′′SH/Et3N (R′′)t -Bu,iPr, Ph) appear somewhat unexpectedly to have two reaction pathways. Among the five reactions studied, only the one between 9 and PhS- gave the expected substitution product, [Ru2(CO)4(µ-SPh)2(SPh)2(PPh3)2] (11). Other reactions regenerated reductively 3 and 5 with the apparently external R′′S anions being oxidized into R′′SSR′′in high yield (Scheme 3). Disulfide forma-tion was previously reported for the internally ligated thiolato groups of [Fe2(CO)6(µ-SR)2] but under oxidation conditions with O2,4j different from our examples. Under similar reaction conditions with excess R′′SH/ Et3N relative to the complex (e.g., 1 mL of R′′SH, 2 mL of Et3N, and ca. 0.30 mmol of the complex), we found that the time required for a complete redox reaction is ca. 2 h, much longer than that of 0.5 h needed for a complete substitution reaction. Further, on the basis of the IR spectra measured sequentially for the longer redox reaction, no intermediate compounds were ob-served with typical IR spectra recorded in an overlaid mode for the reaction between 7 and PhSH/Et3N in CH2 -Cl2. These facts led us to propose a mechanism favoring substitution (route a) rather than a redox process (route b) for the first step of the reaction.15 It probably involves the replacement of one iodide at the less sterically congested site (cf. Scheme 5) with one thiolate anion to form the intermediate [Ru2(CO)4(µ-SR)2(SR′′)I(PR′3)2]. For PhS-, the steric hindrance of this intermediate with R ) R′ ) R′′ ) Ph is not large, enabling a further replacement reaction (route c) to give 11. However, when the hindrance of the intermediate with one of the other combinations of R, R′, and R′′(i.e., R ) R′) Ph, R′′)tBu; or R ) Ph, R′)tBu, R′′) Ph,iPr, ortBu) is too high to allow a second replacement reaction, a redox reaction (route d) occurs with regeneration of complexes 3 and 5 and R′′SSR′′.
Like 9, 11 displays one31P{1H}NMR singlet for the two PPh3ligands in this compound dissolved in CH2
-Cl2at 300 or 220 K, suggesting a similar facile process involving conversion of two terminal into bridging thiolato groups concomitantly with opening two bridging into terminal thiolato groups (cf. Scheme 4) to make two phosphine atoms equivalent at the NMR time scale. However, the two structures are different, with a syn form for 9 and an anti form for 11, based on the two observed13C NMR doublets atδ197.2 (J ) 8.3 Hz)13and 197.5 (J ) 9.7 Hz)13 for the four carbonyls in this compound dissolved in CDCl3. Apparently, it is rather difficult to predict the structure preference for any diruthenium compounds with two thiolato bridges prior to examining the characterization data. The two thi-olato bridges in a dinuclear system may adopt a syn form like 3 and 7-10 or an anti form like 6 and 11, or they may involve an equilibrium mixture of syn and anti forms with the syn form favored for 4 and the anti form for 5 in solution. This nonspecific feature is probably more associated with steric effects than electronic effects, and the one or two forms adopted may represent nonbonded interactions of low, if not the least, repul-siveness.
Conclusion
Our investigation into diruthenium carbonyl phos-phine complexes containing two µ-thiolato linkages resulted in the facile synthesis of 3-6 from 1 and 2 in satisfactory yield (Scheme 1). Electrophilic additions of 3-6 with I2produce diiodide adducts 7-10. Further reactions of 7 and 9 with thiolate anions give either expected nucleophilic substitution products such as 11 or unexpected reductive-dehalogenation to regenerate complexes 3 and 5, respectively, with the apparently external anions being oxidized into alkyl or aryl disul-fides (Scheme 3). Various spectral data and five X-ray crystal structures (Figures 1-5) help to establish that the two thiolato bridges in 3-11 may adopt a syn form or an anti form, or they may involve an equilibrium mixture of both forms with one form or the other favored in solution. An unprecedented structure type with the two thiolato bridges located cis to one of two phosphine ligands in the compounds but trans to the other was (13) The coupling constant is compatible with the reported values.14
(14) Gill, D. F.; Mann, B. E.; Shaw, B. L. J. Chem. Soc., Dalton Trans. 1973, 311.
(15) Astruc, D., Ed. Electron Transfer and Radical Processes in Transition-Metal Chemistry; VCH: New York, 1995.
Scheme 4 Scheme 5
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
observed in 6 (Figure 4) and retained in the iodination product 10 (Figure 5), in spite of the anti geometry in 6 being changed into syn in 10.
Acknowledgment. Financial support for this work by the National Science Council of Republic of China (Contract NSC87-2113-M006-007) and skillful assis-tance by Ms. Fang-Chung Chou are gratefully acknowl-edged.
Supporting Information Available: Tables of
non-hydrogen atomic coordinates and equivalent isotropic displace-ment coefficients, complete bond lengths and angles, aniso-tropic displacement coefficients, and hydrogen coordinates for
3, 4A, 5B, 6, and 10 (30 pages). Ordering information is given
on any current masthead page.
OM9710024
Downloaded by NATIONAL TAIWAN UNIV on August 14, 2009
![Figure 1. ORTEP plot of [Ru 2 (CO) 4 (µ-S t Bu) 2 (PPh 3 ) 2 ] (3).](https://thumb-ap.123doks.com/thumbv2/9libinfo/8874599.249686/4.918.485.834.74.260/figure-ortep-plot-ru-µ-s-bu-pph.webp)
![Figure 2. ORTEP plot of [Ru 2 (CO) 4 (µ-S i Pr) 2 (PPh 3 ) 2 ] (4A).](https://thumb-ap.123doks.com/thumbv2/9libinfo/8874599.249686/5.918.100.423.72.287/figure-ortep-plot-ru-µ-s-pr-pph.webp)
![Figure 5. ORTEP plot of [Ru 2 (CO) 4 (µ-SPh) 2 I 2 (PMe 3 ) 2 ] (10).](https://thumb-ap.123doks.com/thumbv2/9libinfo/8874599.249686/6.918.503.814.73.310/figure-ortep-plot-ru-µ-sph-i-pme.webp)
