Open Access
Research article
Molecular and clinical analyses of 84 patients with tuberous sclerosis
complex
Chia-Cheng Hung
†1, Yi-Ning Su
†2,10, Shu-Chin Chien
3, Horng-Huei Liou
4,5,
Chih-Chuan Chen
4, Pau-Chung Chen
6, Chia-Jung Hsieh
6, Chih-Ping Chen
7,
Wang-Tso Lee
8, Win-Li Lin
1and Chien-Nan Lee*
9Address: 1Institute of Biomedical Engineering, College of Medicine and College of Engineering, National Taiwan University, Taipei, Taiwan, 2Department of Medical Genetics, National Taiwan University Hospital, Taipei, Taiwan, 3Departments of Medical Genetics and Obstetrics and Gynecology, China Medical University Hospital, Taichung, Taiwan, 4Department of Neurology, National Taiwan University Hospital, Taipei, Taiwan, 5Department of Pharmacology, College of Medicine, National Taiwan University, Taipei, Taiwan, 6Institute of Occupational Medicine and Industrial Hygiene, National Taiwan University College of Public Health, Taipei, Taiwan, 7Department of Obstetrics and Gynecology, Mackay Memorial Hospital, Taipei, Taiwan, 8Department of Pediatrics, National Taiwan University Hospital, Taipei, Taiwan, 9Department of Obstetrics and Gynecology, National Taiwan University Hospital, Taipei, Taiwan and 10Graduate Institute of Clinical Medicine, College of Medicine, National Taiwan University, Taipei, Taiwan
Email: Chia-Cheng Hung - r91548020@ntu.edu.tw; Yi-Ning Su - ynsu@ntumc.org; Shu-Chin Chien - chien-sc@yahoo.com.tw; Horng-Huei Liou - liou@ha.mc.ntu.edu.tw; Chih-Chuan Chen - chihch@ha.mc.ntu.edu.tw; Pau-Chung Chen - pchen@ntu.edu.tw; Chia-Jung Hsieh - r92841014@ntu.edu.tw; Chih-Ping Chen - cpc_mmh@yahoo.com; Wang-Tso Lee - leeped@hotmail.com; Win-Li Win-Lin - winli@ntu.edu.tw; Chien-Nan Lee* - leecn@ntumc.org
* Corresponding author †Equal contributors
Abstract
Background: Tuberous sclerosis complex (TSC) is an autosomal dominant disease characterized
by the development of multiple hamartomas in many internal organs. Mutations in either one of 2 genes, TSC1 and TSC2, have been attributed to the development of TSC. More than two-thirds of TSC patients are sporadic cases, and a wide variety of mutations in the coding region of the TSC1 and TSC2 genes have been reported.
Methods: Mutational analysis of TSC1 and TSC2 genes was performed in 84 Taiwanese TSC
families using denaturing high-performance liquid chromatography (DHPLC) and direct sequencing.
Results: Mutations were identified in a total of 64 (76 %) cases, including 9 TSC1 mutations (7
sporadic and 2 familial cases) and 55 TSC2 mutations (47 sporadic and 8 familial cases). Thirty-one of the 64 mutations found have not been described previously. The phenotype association is consistent with findings from other large studies, showing that disease resulting from mutations to
TSC1 is less severe than disease due to TSC2 mutation.
Conclusion: This study provides a representative picture of the distribution of mutations of the
TSC1 and TSC2 genes in clinically ascertained TSC cases in the Taiwanese population. Although
nearly half of the mutations identified were novel, the kinds and distribution of mutation were not different in this population compared to that seen in larger European and American studies.
Published: 18 September 2006
BMC Medical Genetics 2006, 7:72 doi:10.1186/1471-2350-7-72
Received: 28 February 2006 Accepted: 18 September 2006 This article is available from: http://www.biomedcentral.com/1471-2350/7/72
© 2006 Hung et al; licensee BioMed Central Ltd.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Background
Tuberous sclerosis complex (TSC) is an autosomal domi-nant disorder having an incidence of 1 in 6,000 to 1 in 10,000 live births [1]. The severity of TSC and its impact on the quality of life are extremely variable among patients [2]. Common clinical manifestations of this dis-ease include intellectual handicap, autistic disorders, and epilepsy due to the frequent, widespread occurrence of cortical tubers, which are focal disruptions of the cortical architecture due to undifferentiated giant cells. Hamarto-mas are also found in multiple other organ systems, including the heart, lungs, kidneys, and skin [3].
Patients often seek medical attention for dermal lesions or frequent seizures. The clinical diagnostic guidelines on TSC were prepared based on clinical features, radio-graphic findings, and histopathological findings [3]. Accurate clinical diagnoses are relatively easy in patients with classic multisystem involvement, but are often diffi-cult due to the diversity of clinical findings in TSC patients.
The genetic basis of TSC has been determined to be due to mutation in either one of two unlinked genes, TSC1 and
TSC2 [4]. The human TSC1 gene on chromosome 9q34
consists of 23 exons giving an 8.6-kb mRNA transcript, which has a coding region of 3.5-kb and encodes a 130-kDa protein spanning 1164 amino acids [5]. The TSC2 gene, which is located on chromosome 16p13.3, contains 41 exons and encodes a 200-kDa protein with 1807 amino acid [4,6]. Both TSC1 and TSC2 are tumor suppres-sor genes and their protein products, hamartin and tuberin, respectively, form a complex that regulates the mammalian target of rapamycin (mTOR) in the phosph-oinositide 3-kinases (PI3-kinase)/AKT pathway to control cellular proliferation, adhesion, growth, differentiation or migration [7,8]. Furthermore, both genes play a role in cortical differentiation and growth control.
The mutation spectra of the TSC genes are very heteroge-neous and no hotspots for mutations have been reported. There are many mutations in each gene that are seen recurrently, but no single mutation accounts for more than about 1% of all TSC patients. TSC2 mutations are about five times more common than TSC1 mutations [9] and new mutations are typically found in the two-thirds of TSC cases that are sporadic [10]. Despite complete pen-etrance of the disease in TSC patients, phenotypic variabil-ity can make the determination of disease status difficult among family members of affected individuals.
In this study, we analyzed both TSC1 and TSC2 genes in 84 independent Taiwanese TSC probands for whom detailed information on clinical manifestations and phe-notype were available. Furthermore, we also assessed the
mutational distribution and possible genotype-pheno-type correlations between and within the two genes.
Methods
Patient Population
This study was approved by the Ethics Committee of the Division of Obstetrics and Gynecology, National Taiwan University Hospital. Eighty-four unrelated patients with confirmed clinical diagnoses of TSC and their family members were tested for mutations in TSC1 and TSC2 genes.
The general clinical features of TSC patients were deter-mined by clinicians in accordance with the TSC diagnosis criteria set forth by the Tuberous Sclerosis Consensus Conference [3]. All patients' symptoms were investigated by a person blind to mutational status. High-resolution brain magnetic-resonance imaging (MRI) or computed tomography (CT) was performed on most patients. The extent of facial angiofibroma or forehead plaques, non-traumatic ungal or periungal fibromas, hypomelan-otic macules, shagreen patches, multiple retinal nodular hamartomas, cortical tubers, subependymal nodules, sub-ependymal giant cell astrocytomas, cardiac rhabdomyo-mas, lymphangiomyomatoses, renal angiomyolipomas and confetti-like lesions were all assessed. Moreover, most patients' medical histories of mental development were assessed by a certified psychologist.
Sample Preparation
After genetic counseling and obtaining informed consent, 5–10 mL of peripheral blood were collected from the par-ticipants. Genomic DNA was isolated from peripheral whole blood using the Puregene DNA Isolation Kit (Gen-tra Systems, Inc., Minneapolis, MN, USA).
Mutational Analysis of TSC Genes
PCR primers and running conditions for each exon were available from previous studies [11-13]. The PCR reaction was run on each exon with a total sample volume of 25 µL containing 100 ng of genomic DNA, 0.12 µM of each respective primer, 100 µM dNTPs, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 2 mM MgCl2, and 0.5 units of AmpliTaq Gold enzyme (PE Applied Biosystems, Foster City, CA, USA). Amplification was performed in a multiblock sys-tem thermocycler (ThermoHybaid, Ashford, UK). The PCR amplification started with a denaturing step at 95°C for 5 minutes, followed by 35 cycles of denaturing at 94°C for 30 seconds, annealing at melting temperature (Tm) for 30 seconds, extension at 72°C for 45 seconds, and ends with a final extension step at 72°C for 10 min-utes.
The screening of mutations was performed using the Transgenomic Wave Nucleic Acid Fragment Analysis Sys-tem (Transgenomic Inc, San Jose, CA) with a C18 reversed-phase column containing 2-µm nonporous poly (styrene/ divinylbenzene) particles (DNASep Column, Transge-nomic Inc). PCR products were analyzed using linear ace-tonitrile gradients and triethylammonium acetate acting as mobile phases with the provision of buffer A (0.1 M TEAA) and buffer B (0.1 M TEAA with 25% acetonitrile) (WAVE Optimized, Transgenomic Inc). Heteroduplex analyses were performed according to the manufacturer's protocol and of previous studies [14,15].
Statistical method
The χ2 and Fisher exact tests were used to examine the dif-ferences in clinical manifestations, phenotypes, and mutation distributions in independent Taiwanese probands between patients with TSC1 and TSC2 genes.
Direct Sequence Analysis
PCR products were purified by solid-phase extraction and bidirectionally sequenced using Applied Biosystems' Taq DyeDeoxy terminator cycle sequencing kit (Applied Bio-systems). Sequencing reactions were separated on a PE Biosystems 373A/3100 sequencer.
Results and Discussion
Identification and Characterization of Mutations
In the current study, we performed mutational analysis on the coding exons and the exon/intron junctions of both
TSC1 and TSC2 in a total of 84 individuals with TSC and
their family members. The determination of mutation vs. polymorphism was done by: 1) checking the mutation tables at the Chromium site (http://chromium.liacs.nl/); 2) comparison of findings to those of 100 healthy Taiwan-ese controls; and 3) checking the families similarly.
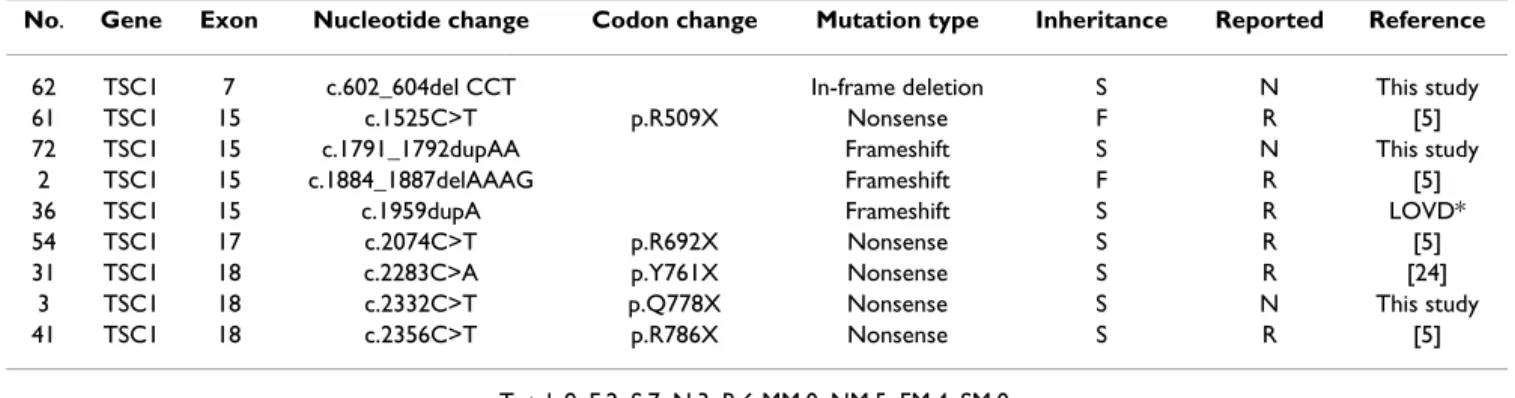
Nine mutations were identified in the TSC1 gene while 55 were identified in the TSC2 gene. Mutations in the TSC1 gene included five nonsense mutations with early termi-nation codons and four insertions/deletions which caused frameshifts and resulted in premature truncation of the protein. Three of these mutations were novel, while six were previously reported (Table 1).
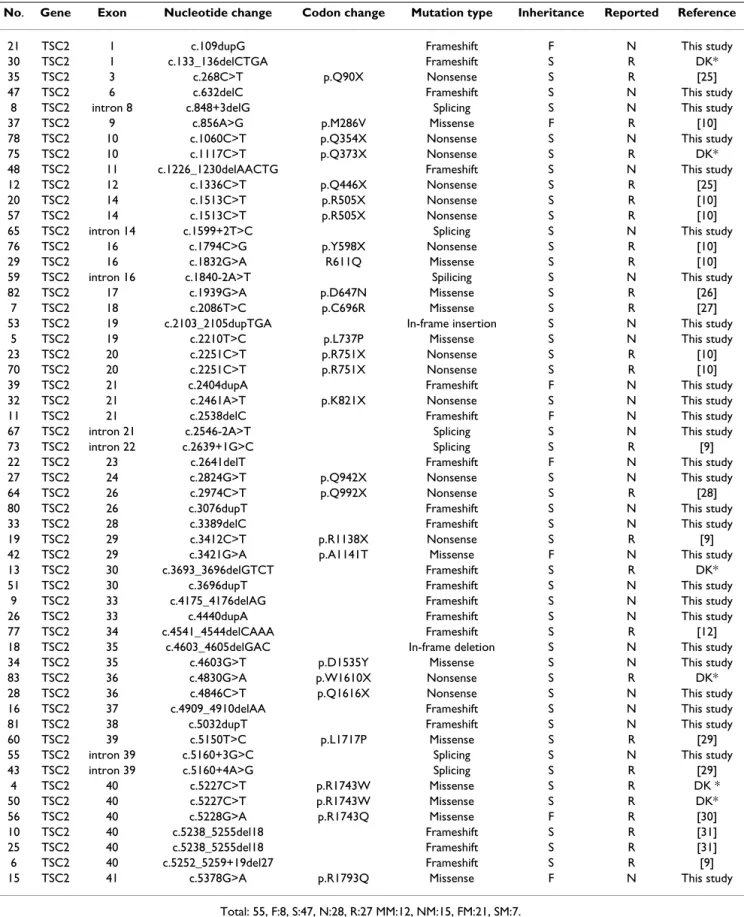
The 55 mutations in the TSC2 gene included 12 missense, 15 nonsense, 21 frameshifts due to insertions and dele-tions and 7 putative splice-site mutadele-tions. Twenty-seven of these mutations were previously reported while 28 were novel (Table 2). Of the familial TSC2 missense muta-tions, A1141T and R1793Q may be rare polymorphic var-iants co-segregating with TSC. There was no direct evidence that these familial TSC2 missense mutational changes were pathogenic.
For both genes, sequence variants that were possible mutations were tested in all other family members, including the parents and both the affected and the unaf-fected family members. In total, 31 of the 64 mutations (48%) had not been reported elsewhere. Moreover, no mutational hotspots were identified in either gene, with only four different mutations being found twice in TSC2. Compared with those of European and American counter-parts [9,10,16], the distribution of the TSC1 and TSC2 mutations among Taiwanese population is similar. There-fore, the spectrum of mutations seen among the Taiwan-ese is no different in comparison to those already reported thus far for these two genes, based on the genetic analyses of European and American TSC patients using the Fisher exact test (P = 0.85, 0.46, and 0.14, respectively).
Table 1: Status of TSC1 mutations in Taiwanese patients with TSC
No. Gene Exon Nucleotide change Codon change Mutation type Inheritance Reported Reference
62 TSC1 7 c.602_604del CCT In-frame deletion S N This study
61 TSC1 15 c.1525C>T p.R509X Nonsense F R [5]
72 TSC1 15 c.1791_1792dupAA Frameshift S N This study
2 TSC1 15 c.1884_1887delAAAG Frameshift F R [5]
36 TSC1 15 c.1959dupA Frameshift S R LOVD*
54 TSC1 17 c.2074C>T p.R692X Nonsense S R [5]
31 TSC1 18 c.2283C>A p.Y761X Nonsense S R [24]
3 TSC1 18 c.2332C>T p.Q778X Nonsense S N This study
41 TSC1 18 c.2356C>T p.R786X Nonsense S R [5]
Total: 9, F:2, S:7, N:3, R:6 MM:0, NM:5, FM:4, SM:0. F: familial case, S:sporadic case.
N: non-reported, R: reported.
MM: missense mutations, NM: nonsense mutations, FM: frameshift/in-frame mutations, SM: splicing site mutations. * The the Leiden Open (source) Variation Database which was available at http://chromium.liacs.nl/lovd/.
Table 2: Status of TSC2 mutations in Taiwanese patients with TSC
No. Gene Exon Nucleotide change Codon change Mutation type Inheritance Reported Reference
21 TSC2 1 c.109dupG Frameshift F N This study
30 TSC2 1 c.133_136delCTGA Frameshift S R DK*
35 TSC2 3 c.268C>T p.Q90X Nonsense S R [25]
47 TSC2 6 c.632delC Frameshift S N This study
8 TSC2 intron 8 c.848+3delG Splicing S N This study
37 TSC2 9 c.856A>G p.M286V Missense F R [10]
78 TSC2 10 c.1060C>T p.Q354X Nonsense S N This study
75 TSC2 10 c.1117C>T p.Q373X Nonsense S R DK*
48 TSC2 11 c.1226_1230delAACTG Frameshift S N This study
12 TSC2 12 c.1336C>T p.Q446X Nonsense S R [25]
20 TSC2 14 c.1513C>T p.R505X Nonsense S R [10]
57 TSC2 14 c.1513C>T p.R505X Nonsense S R [10]
65 TSC2 intron 14 c.1599+2T>C Splicing S N This study
76 TSC2 16 c.1794C>G p.Y598X Nonsense S R [10]
29 TSC2 16 c.1832G>A R611Q Missense S R [10]
59 TSC2 intron 16 c.1840-2A>T Spilicing S N This study
82 TSC2 17 c.1939G>A p.D647N Missense S R [26]
7 TSC2 18 c.2086T>C p.C696R Missense S R [27]
53 TSC2 19 c.2103_2105dupTGA In-frame insertion S N This study
5 TSC2 19 c.2210T>C p.L737P Missense S N This study
23 TSC2 20 c.2251C>T p.R751X Nonsense S R [10]
70 TSC2 20 c.2251C>T p.R751X Nonsense S R [10]
39 TSC2 21 c.2404dupA Frameshift F N This study
32 TSC2 21 c.2461A>T p.K821X Nonsense S N This study
11 TSC2 21 c.2538delC Frameshift F N This study
67 TSC2 intron 21 c.2546-2A>T Splicing S N This study
73 TSC2 intron 22 c.2639+1G>C Splicing S R [9]
22 TSC2 23 c.2641delT Frameshift F N This study
27 TSC2 24 c.2824G>T p.Q942X Nonsense S N This study
64 TSC2 26 c.2974C>T p.Q992X Nonsense S R [28]
80 TSC2 26 c.3076dupT Frameshift S N This study
33 TSC2 28 c.3389delC Frameshift S N This study
19 TSC2 29 c.3412C>T p.R1138X Nonsense S R [9]
42 TSC2 29 c.3421G>A p.A1141T Missense F N This study
13 TSC2 30 c.3693_3696delGTCT Frameshift S R DK*
51 TSC2 30 c.3696dupT Frameshift S N This study
9 TSC2 33 c.4175_4176delAG Frameshift S N This study
26 TSC2 33 c.4440dupA Frameshift S N This study
77 TSC2 34 c.4541_4544delCAAA Frameshift S R [12]
18 TSC2 35 c.4603_4605delGAC In-frame deletion S N This study
34 TSC2 35 c.4603G>T p.D1535Y Missense S N This study
83 TSC2 36 c.4830G>A p.W1610X Nonsense S R DK*
28 TSC2 36 c.4846C>T p.Q1616X Nonsense S N This study
16 TSC2 37 c.4909_4910delAA Frameshift S N This study
81 TSC2 38 c.5032dupT Frameshift S N This study
60 TSC2 39 c.5150T>C p.L1717P Missense S R [29]
55 TSC2 intron 39 c.5160+3G>C Splicing S N This study
43 TSC2 intron 39 c.5160+4A>G Splicing S R [29]
4 TSC2 40 c.5227C>T p.R1743W Missense S R DK * 50 TSC2 40 c.5227C>T p.R1743W Missense S R DK* 56 TSC2 40 c.5228G>A p.R1743Q Missense F R [30] 10 TSC2 40 c.5238_5255del18 Frameshift S R [31] 25 TSC2 40 c.5238_5255del18 Frameshift S R [31] 6 TSC2 40 c.5252_5259+19del27 Frameshift S R [9]
15 TSC2 41 c.5378G>A p.R1793Q Missense F N This study
Total: 55, F:8, S:47, N:28, R:27 MM:12, NM:15, FM:21, SM:7. F: familial case, S:sporadic case.
N: non-reported, R: reported.
MM: missense mutations, NM: nonsense mutations, FM: frameshift/in-frame mutations, SM: splicing site mutations. * The database of Dr David Kwiatkowski which was available at http://tsc-project.partners.org/.
Identification and Characterization of Polymorphism
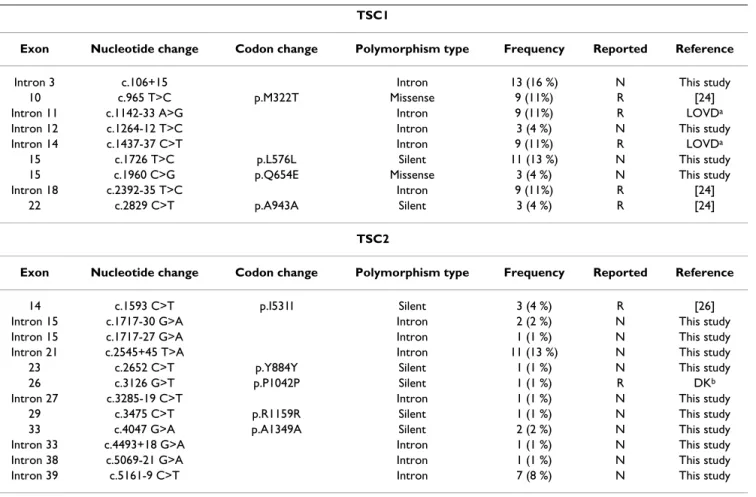
In order to identify whether the observed changes were mutations or polymorphisms, samples from 100 normal individuals serving as controls were analyzed. Changes that were not found in more than 200 control alleles were considered pathogenic. Therefore, unique or less frequent changes such as missense and splicing site mutations (Table 2) were considered likely pathogenic mutations. The nonpathogenic TSC1 and TSC2 mutations identified in the Taiwanese TSC patients are described in Table 3. We identified nine nonpathogenic polymorphisms in the
TSC1 gene and 12 in the TSC2 gene. The nonpathogenic
sequence variants were identified in both the TSC patients and the normal controls. Fourteen of these polymor-phisms had not been reported previously (4 at the TSC1 locus and 10 at the TSC2 locus) that included one mis-sense variant within the TSC1 coding region.
Genotype-Phenotype Correlation: Familial or Sporadic TSC mutations
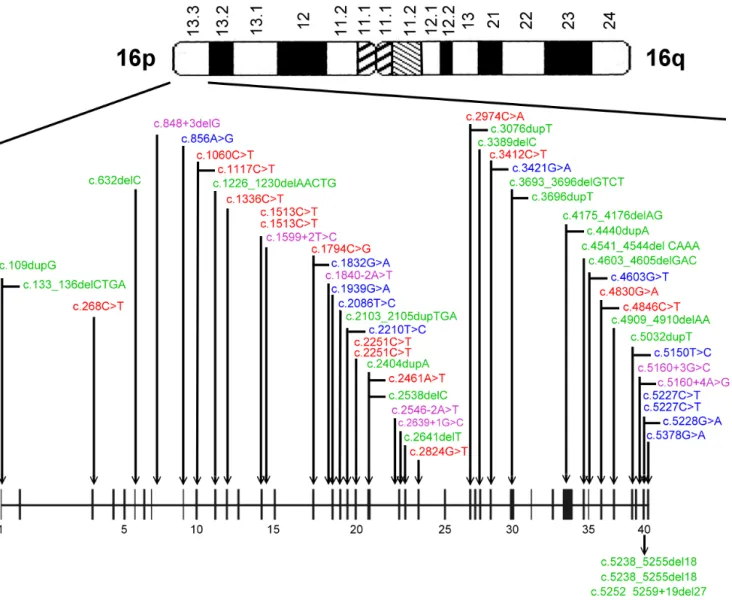
Mutations were identified and located in exons of both
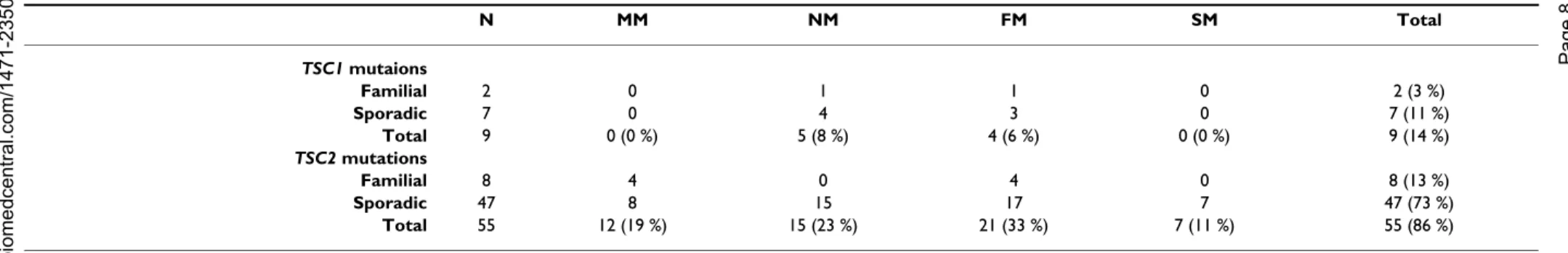
TSC1 and TSC2 genes (see Figure 1 and 2). Of the 64
mutations found, nine and 55 were associated with TSC1 (14%) and TSC2 (86%), respectively, as shown in Table 4. Of the 10 familial cases, 2 (20%) and 8 (80%) were
TSC1and TSC2 mutations, respectively. Among the 54
sporadic cases, 7 TSC1 (13%) and 47 TSC2 (87%) muta-tions were found. Accordingly, there was no significant difference between sporadic and familial TSC cases with respect to the frequency of TSC1 vs TSC2 mutation (P = 0.62).
Genotype-Phenotype Correlation: Clinical Manifestations
The clinical characteristics associated with each mutation in the proband are shown in Tables 5 (eight TSC1 muta-tions) and Table 6 (43 TSC2 mutamuta-tions). Most patients with TSC1 and TSC2 mutations had seizures, brain lesions (subependymal nodules and/or cortical tubers detected by MRI), and dermal manifestations. Our criteria for intellectual disability included any degree of mental retardation and learning disorder. The incidence of intel-lectual disability appeared lower in patients with TSC1 mutations (3/8 = 38%) compared to that of patients with
TSC2 mutations (27/43 = 63%). However, this difference Table 3: Polymorphisms identified for TSC1 and TSC2 in Taiwanese TSC population.
TSC1
Exon Nucleotide change Codon change Polymorphism type Frequency Reported Reference
Intron 3 c.106+15 Intron 13 (16 %) N This study
10 c.965 T>C p.M322T Missense 9 (11%) R [24]
Intron 11 c.1142-33 A>G Intron 9 (11%) R LOVDa
Intron 12 c.1264-12 T>C Intron 3 (4 %) N This study
Intron 14 c.1437-37 C>T Intron 9 (11%) R LOVDa
15 c.1726 T>C p.L576L Silent 11 (13 %) N This study
15 c.1960 C>G p.Q654E Missense 3 (4 %) N This study
Intron 18 c.2392-35 T>C Intron 9 (11%) R [24]
22 c.2829 C>T p.A943A Silent 3 (4 %) R [24]
TSC2
Exon Nucleotide change Codon change Polymorphism type Frequency Reported Reference
14 c.1593 C>T p.I531I Silent 3 (4 %) R [26]
Intron 15 c.1717-30 G>A Intron 2 (2 %) N This study
Intron 15 c.1717-27 G>A Intron 1 (1 %) N This study
Intron 21 c.2545+45 T>A Intron 11 (13 %) N This study
23 c.2652 C>T p.Y884Y Silent 1 (1 %) N This study
26 c.3126 G>T p.P1042P Silent 1 (1 %) R DKb
Intron 27 c.3285-19 C>T Intron 1 (1 %) N This study
29 c.3475 C>T p.R1159R Silent 1 (1 %) N This study
33 c.4047 G>A p.A1349A Silent 2 (2 %) N This study
Intron 33 c.4493+18 G>A Intron 1 (1 %) N This study
Intron 38 c.5069-21 G>A Intron 1 (1 %) N This study
Intron 39 c.5161-9 C>T Intron 7 (8 %) N This study
* Frequence means the number of cases in 84 Taiwanese TSC patients.
a The the Leiden Open (source) Variation Database which was available at http://chromium.liacs.nl/lovd/. b The database of Dr David Kwiatkowski which was available at http://tsc-project.partners.org/.
was not statistically significant (P = 0.25), but this would be expected because of such small sample sizes. Similarly, the incidence of mental retardation in patients with TSC1 mutations (1/8 = 13%) appeared to be less than that of patients with TSC2 mutations (17/43 = 40%), but this dif-ference was not statistically significant (P = 0.23). Simi-larly, the frequencies of renal findings, cortical tubers, subependymal giant cell astrocytomas, liver tumors, car-diac tumors, or skin manifestations, including hypomelanotic macules, facial angiofibromas, shagreen patches, and ungual fibromas did not significantly differ between the patients with TSC1 and TSC2 mutations. However, all of these comparisons are under-powered due to the relatively small number of patients with TSC1 mutations that were studied. For nearly all of the clinical
features studied, the frequencies were less for those bear-ing TSC1 mutations than for those bearbear-ing TSC2 muta-tions. This is consistent with findings from other large studies, showing that TSC1 disease is less severe than
TSC2 disease [9,10,16].
Conclusion
This study is the first analysis of TSC1 and TSC2 genes in the Taiwanese population. We identified 64 mutations among a total of 84 patients (76%); 9 were TSC1 muta-tions (14%) and 55 were TSC2 mutamuta-tions (86%). These numbers are similar to other studies with larger cohorts [9,10,16-18] and would be expected if the germ line mutation rate at the TSC2 locus were higher than that at the TSC1 locus. The failure to detect mutations in the
Diagram depicting the locations of mutations in the TSC1 gene
Figure 1
Diagram depicting the locations of mutations in the TSC1 gene. Nonsense (red), missense (blue), frameshift/in-frame (green) and splicing site (purple) mutations were identified.
remaining 24% of the patients may be due to a combina-tion of lack of screening for large genomic delecombina-tion and rearrangement mutations in either TSC1 or TSC2. The occurrence of mosaic mutations [19,20] in some of these patients that may be difficult to detect. Another reason is mutation detection failure.
According to previous reports, somatic and general mosa-icism are seen in 6%-10% of all TSC patients [20,21]. In addition, large deletions have been identified in about 2%-4% of TSC2 mutations [6] and less commonly in the
TSC1 gene [22,23]. Thus, both of these situations likely
contributed to patients in which mutations were not iden-tified.
In summary, sixty-four different mutations were identi-fied and characterized for the Taiwanese population. Of those, 31 were not previously described. The diverse mutation spectrum of TSC was also seen in different fam-ilies and different populations.
Abbreviations
DHPLC: Denaturing high performance liquid chromatog-raphy
Diagram depicting the locations of mutations in the TSC2 gene
Figure 2
Diagram depicting the locations of mutations in the TSC2 gene. Nonsense (red), missense (blue), frameshift/in-frame (green) and splicing site (purple) mutations were identified.
BMC Med ical Ge netics 200 6, 7:72 http ://www.bio m e dcent ral.com/147 1-235 0/7/7 Pa ge 8 of (page nu mber not for cit a tion pur poses) N MM NM FM SM Total TSC1 mutaions Familial 2 0 1 1 0 2 (3 %) Sporadic 7 0 4 3 0 7 (11 %) Total 9 0 (0 %) 5 (8 %) 4 (6 %) 0 (0 %) 9 (14 %) TSC2 mutations Familial 8 4 0 4 0 8 (13 %) Sporadic 47 8 15 17 7 47 (73 %) Total 55 12 (19 %) 15 (23 %) 21 (33 %) 7 (11 %) 55 (86 %) N: screening numbers. MM: missense mutations. NM: nonsense mutations. FM: frameshift/in-frame mutations. SM: splicing site mutations.
Table 5: Clinical data of patients with TSC1 mutations
Family no. Familial/
Sporadic
Mutation type
Sex Onset age
of seizure Intellectual performance Brain tubers Renal tumors Hepatic tumors Cardiac rhabdomyoma Hypomela notic macules Facial angiofibro ma Shagreen patch Ungual fibroma 2 F FS F 2 y N + 0 NA NA 0 + 0 + 3 S NM M 8 y 3 m N + 0 0 0 + + + 0 41 S NM F 1 y N + 0 0 + + 0 0 0 31 S NM F 6 m LD + + 0 + + 0 0 0 36 S FS M 2 y N + 0 0 0 + + 0 0 61 F NM M 3 y 6 m LD + 0 0 0 + + 0 0 62 S FS M 1 m LD + + 0 0 + + + + 72 S FS M 3 y N + 0 0 + + 0 + 0
BMC Med ical Ge netics 200 6, 7:72 http ://www.bio m e dcent ral.com/147 1-235 Page 9 of (page nu mber not for cit a tion pur 4 S MM M 10 m N + + 0 0 + + + 0 5 S MM F 1 y N + NA NA 0 + + 0 0 6 S FS M 1 m LD + 0 0 + + 0 0 0 7 S MM M 6 m MR NA NA NA NA + + 0 0 8 S S F 3 m LD + + 0 0 + + + 0 9 S FS M 5 y LD + 0 0 0 + + + 0 10 S FS F 6 m LD + 0 0 + + + 0 0 11 F FS F 4 m LD + + 0 + 0 + + 0 12 S NM M 10 m N + + 0 NA + + + 0 13 S FS F 4 m MR + NA NA + 0 0 0 0 15 F MM F 7 m MR + 0 0 0 + 0 0 0 16 S FS M 1 y N + + 3 NA + + + + 18 S FS F 5 m LD + NA NA NA + + + 0 19 S NM M 3 m MR + + 0 NA + 0 0 0 20 S NM M 1 y 6 m N + + 0 0 + + 0 0 21 F FS F 9 y N + + 1 0 + + + + 22 F FS M 1 y LD NA NA NA NA + + + 0 23 S NM F 7 m MR + NA NA NA + 0 0 0 25 S FS M 6 m LD + 0 NA 0 + + + 0 26 S FS F 8 m LD + + 0 0 + + + 0 27 S NM M 1 y MR + + 0 0 + + 0 0 28 S NM M 3 m LD + NA NA NA + + + 0 29 S MM F 6 m MR + 0 NA 0 + 0 + 0 30 S FS F 1 y MR + + 0 0 + + 0 0 32 S NM M 3 m N + + + 0 + + 0 0 33 S FS F 1 m N + + + 0 + + + + 34 S MM F 7 y N + + 0 0 + 0 + 0 35 S NM M 1 y MR + + 0 + + + + 0 37 F MM M 9 m MR + 0 0 + + + + 0 39 F FS F 3 m MR + NA NA NA + + + 0 42 F MM M 7 y N 0 + + NA + + + + 47 S FS F 2 m N + 0 0 + + 0 0 0 48 S FS F 3 m MR + + 0 0 + + + 0 50 S MM M 3 m MR + NA NA NA + + + 0 53 S FS M 3 y MR + + 0 0 + + + 0 56 F MM F 2 y N + + + 0 + + + + 57 S NM F 6 m MR + 0 0 0 + + + 0 59 S S F 1 m MR + + 0 0 + + 0 0 64 S NM M 2 m N + 0 0 0 + 0 0 0 67 S S F 3 m MR + 0 0 + + + 0 0 73 S S F 21 y N + 0 0 0 + + 0 + 75 S NM F 1 y N + + 0 0 + + 0 0 82 S MM M 1 m N 0 0 0 + 0 0 0 0
TSC: Tuberous sclerosis complex CT: Computed tomography MRI: Magnetic-resonance imaging PCR: Polymerase chain reaction Tm: Melting temperature
Competing interests
We received financial support in the form of a grant from the National Science Council of Taiwan (NSC 92-2314-B-002-319). We have no other competing interests to declare.
Authors' contributions
CCH and YNS performed the molecular genetics studies
and drafted the manuscript. SCC participated in the molecular genetics studies. HHL and CCC performed the clinical characterization of the patients. PCC and CJH per-formed the statistical analyses. CPC, WTL and WLL partic-ipated in the design of the study. CNL conceived the study, participated in its design and coordination, and helped draft the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors gratefully extend their gratitude to the families and patients affected by TSC for their participation and cooperation in this study. We thank Prof. David Kwiatkowski, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School for deeply review and rewrite the manuscript. We thank Dr. Fon-Jou Hsieh for his expertise and assist-ance. This work was supported by a grant from the National Science Coun-cil of Taiwan (NSC 92-2314-B-002-319).
References
1. Osborne JP, Fryer A, Webb D: Epidemiology of tuberous sclero-sis. Ann N Y Acad Sci 1991, 615:125-127.
2. Gomez MR SJRHWV: Tuberous sclerosis complex. 3rd edn New
York: Oxford University Press 1999.
3. Roach ES, Gomez MR, Northrup H: Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J
Child Neurol 1998, 13(12):624-628.
4. Povey S, Burley MW, Attwood J, Benham F, Hunt D, Jeremiah SJ, Franklin D, Gillett G, Malas S, Robson EB, et al.: Two loci for tuber-ous sclerosis: one on 9q34 and one on 16p13. Ann Hum Genet 1994, 58 (Pt 2):107-127.
5. van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A, Halley D, Young J, Bur-ley M, Jeremiah S, Woodward K, Nahmias J, Fox M, Ekong R, Osborne J, Wolfe J, Povey S, Snell RG, Cheadle JP, Jones AC, Tachataki M, Ravine D, Sampson JR, Reeve MP, Richardson P, Wilmer F, Munro C, Hawkins TL, Sepp T, Ali JB, Ward S, Green AJ, Yates JR, Kwiatkowska J, Henske EP, Short MP, Haines JH, Jozwiak S, Kwiatkowski DJ: Iden-tification of the tuberous sclerosis gene TSC1 on chromo-some 9q34. Science 1997, 277(5327):805-808.
6. The European Chromosome 16 Tuberous Sclerosis Consortium.: Identification and characterization of the tuberous sclerosis gene on chromosome 16. The European Chromosome 16 Tuberous Sclerosis Consortium. Cell 1993, 75(7):1305-1315. 7. Astrinidis A, Henske EP: Tuberous sclerosis complex: linking
growth and energy signaling pathways with human disease.
Oncogene 2005, 24(50):7475-7481.
8. Kwiatkowski DJ, Manning BD: Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet 2005, 14 Spec No. 2:R251-8.
9. Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, Choy YS, Reeve MP, Thiele E, Egelhoff JC, Kasprzyk-Obara J, Domanska-Pakiela D, Kwiatkowski DJ: Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs.
Am J Hum Genet 2001, 68(1):64-80.
10. Jones AC, Shyamsundar MM, Thomas MW, Maynard J, Idziaszczyk S, Tomkins S, Sampson JR, Cheadle JP: Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet 1999, 64(5):1305-1315.
11. Jones AC, Daniells CE, Snell RG, Tachataki M, Idziaszczyk SA, Krawc-zak M, Sampson JR, Cheadle JP: Molecular genetic and pheno-typic analysis reveals differences between TSC1 and TSC2 associated familial and sporadic tuberous sclerosis. Hum Mol
Genet 1997, 6(12):2155-2161.
12. Maheshwar MM, Cheadle JP, Jones AC, Myring J, Fryer AE, Harris PC, Sampson JR: The GAP-related domain of tuberin, the product of the TSC2 gene, is a target for missense mutations in tuberous sclerosis. Hum Mol Genet 1997, 6(11):1991-1996. 13. Maheshwar MM, Sandford R, Nellist M, Cheadle JP, Sgotto B, Vaudin
M, Sampson JR: Comparative analysis and genomic structure of the tuberous sclerosis 2 (TSC2) gene in human and puff-erfish. Hum Mol Genet 1996, 5(1):131-137.
14. Su YN, Lee CN, Hung CC, Chen CA, Cheng WF, Tsao PN, Yu CL, Hsieh FJ: Rapid detection of beta-globin gene (HBB) muta-tions coupling heteroduplex and primer-extension analysis by DHPLC. Hum Mutat 2003, 22(4):326-336.
15. Xiao W, Oefner PJ: Denaturing high-performance liquid chro-matography: A review. Hum Mutat 2001, 17(6):439-474. 16. Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C,
Maat-Kievit A, Zonnenberg B, Verhoef S, Halley D, van den Ouweland A: Mutational analysis of the TSC1 and TSC2 genes in a diag-nostic setting: genotype--phenotype correlations and com-parison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet 2005, 13(6):731-741.
17. Lewis JC, Thomas HV, Murphy KC, Sampson JR: Genotype and psy-chological phenotype in tuberous sclerosis. J Med Genet 2004, 41(3):203-207.
18. Jones AC, Sampson JR, Hoogendoorn B, Cohen D, Cheadle JP: Appli-cation and evaluation of denaturing HPLC for molecular genetic analysis in tuberous sclerosis. Hum Genet 2000, 106(6):663-668.
19. Kwiatkowska J, Wigowska-Sowinska J, Napierala D, Slomski R, Kwiatkowski DJ: Mosaicism in tuberous sclerosis as a potential cause of the failure of molecular diagnosis. N Engl J Med 1999, 340(9):703-707.
20. Sampson JR, Maheshwar MM, Aspinwall R, Thompson P, Cheadle JP, Ravine D, Roy S, Haan E, Bernstein J, Harris PC: Renal cystic ease in tuberous sclerosis: role of the polycystic kidney dis-ease 1 gene. Am J Hum Genet 1997, 61(4):843-851.
21. Verhoef S, Bakker L, Tempelaars AM, Hesseling-Janssen AL, Mazurc-zak T, Jozwiak S, Fois A, Bartalini G, Zonnenberg BA, van Essen AJ, Lindhout D, Halley DJ, van den Ouweland AM: High rate of mosa-icism in tuberous sclerosis complex. Am J Hum Genet 1999, 64(6):1632-1637.
22. Nellist M, Sancak O, Goedbloed MA, van Veghel-Plandsoen M, Maat-Kievit A, Lindhout D, Eussen BH, de Klein A, Halley DJ, van den Ouweland AM: Large deletion at the TSC1 locus in a family with tuberous sclerosis complex. Genet Test 2005, 9(3):226-230. 23. Longa L, Saluto A, Brusco A, Polidoro S, Padovan S, Allavena A, Car-bonara C, Grosso E, Migone N: TSC1 and TSC2 deletions differ in size, preference for recombinatorial sequences, and loca-tion within the gene. Hum Genet 2001, 108(2):156-166. 24. Dabora SL, Sigalas I, Hall F, Eng C, Vijg J, Kwiatkowski DJ:
Compre-hensive mutation analysis of TSC1 using two-dimensional DNA electrophoresis with DGGE. Ann Hum Genet 1998, 62 ( Pt 6):491-504.
25. Choy YS, Dabora SL, Hall F, Ramesh V, Niida Y, Franz D, Kasprzyk-Obara J, Reeve MP, Kwiatkowski DJ: Superiority of denaturing high performance liquid chromatography over single-stranded conformation and conformation-sensitive gel
elec-Publish with BioMed Central and every scientist can read your work free of charge
"BioMed Central will be the most significant development for disseminating the results of biomedical researc h in our lifetime."
Sir Paul Nurse, Cancer Research UK Your research papers will be:
available free of charge to the entire biomedical community peer reviewed and published immediately upon acceptance cited in PubMed and archived on PubMed Central yours — you keep the copyright
Submit your manuscript here:
http://www.biomedcentral.com/info/publishing_adv.asp
BioMedcentral trophoresis for mutation detection in TSC2. Ann Hum Genet
1999, 63 ( Pt 5):383-391.
26. Zhang H, Nanba E, Yamamoto T, Ninomiya H, Ohno K, Mizuguchi M, Takeshita K: Mutational analysis of TSC1 and TSC2 genes in Japanese patients with tuberous sclerosis complex. J Hum
Genet 1999, 44(6):391-396.
27. Roberts PS, Ramesh V, Dabora S, Kwiatkowski DJ: A 34 bp deletion within TSC2 is a rare polymorphism, not a pathogenic muta-tion. Ann Hum Genet 2003, 67(Pt 6):495-503.
28. Beauchamp RL, Banwell A, McNamara P, Jacobsen M, Higgins E, Northrup H, Short P, Sims K, Ozelius L, Ramesh V: Exon scanning of the entire TSC2 gene for germline mutations in 40 unre-lated patients with tuberous sclerosis. Hum Mutat 1998, 12(6):408-416.
29. Chen CP, Su YN, Hung CC, Lee CN, Hsieh FJ, Chang TY, Chen MR, Wang W: Molecular genetic analysis of the TSC genes in two families with prenatally diagnosed rhabdomyomas. Prenat
Diagn 2005, 25(2):176-178.
30. Ichikawa T, Wakisaka A, Daido S, Takao S, Tamiya T, Date I, Koizumi S, Niida Y: A case of solitary subependymal giant cell astrocy-toma: two somatic hits of TSC2 in the tumor, without evi-dence of somatic mosaicism. J Mol Diagn 2005, 7(4):544-549. 31. Niida Y, Lawrence-Smith N, Banwell A, Hammer E, Lewis J,
Beau-champ RL, Sims K, Ramesh V, Ozelius L: Analysis of both TSC1 and TSC2 for germline mutations in 126 unrelated patients with tuberous sclerosis. Hum Mutat 1999, 14(5):412-422.
Pre-publication history
The pre-publication history for this paper can be accessed here: