as Mechanism-Based Inactivators of the
Severe Acute Respiratory Syndrome 3CL Protease
Chung-Yi Wu,
1,2Ke-Yung King,
1Chih-Jung Kuo,
1,3Jim-Min Fang,
1,4Ying-Ta Wu,
1Ming-Yi Ho,
1Chung-Lin Liao,
1Jiun-Jie Shie,
1Po-Huang Liang,
1,*
and Chi-Huey Wong
1,2,4,*
1The Genomics Research Center and
Institute of Biological Chemistry
Academia Sinica No. 128
Academia Road Section 2
Nan-Kang
Taipei, 115
Taiwan
2Department of Chemistry and
Skaggs Institute for Chemical Biology
The Scripps Research Institute
10550 North Torrey Pines Road
La Jolla, California 92037
3
Taiwan International Graduate Program
Academia Sinica
Nan-Kang
Taipei, 115
Taiwan
4Department of Chemistry
National Taiwan University
Taipei, 106
Taiwan
Summary
Severe acute respiratory syndrome (SARS) is caused
by a newly emerged coronavirus that infected more
than 8000 individuals and resulted in more than 800
fa-talities in 2003. Currently, there is no effective
treat-ment for this epidemic. SARS-3CL
prohas been shown
to be essential for replication and is thus a target for
drug discovery. Here, a class of stable benzotriazole
esters was reported as mechanism-based inactivators
of 3CL
pro, and the most potent inactivator exhibited
a k
inactof 0.0011 s
21and a K
iof 7.5 nM. Mechanistic
in-vestigation with kinetic and mass spectrometry
analy-ses indicates that the active site Cys145 is acylated,
and that no irreversible inactivation was observed
with the use of the C145A mutant. In addition, a
non-covalent, competitive inhibition became apparent by
using benzotriazole ester surrogates in which the
bridged ester-oxygen group is replaced with carbon.
Introduction
Severe acute respiratory syndrome (SARS), a newly
emerged infectious disease, first occurred in Guandong
(China) in November of 2002 and spread through many
countries in 2003; it reemerged in China in December
2003 and in the spring of 2004. This disease is caused
by infection with a novel human coronavirus
(SARS-CoV)
[1–3]
, which affected more than 8000 individuals
across 32 countries and resulted in more than 800
fatal-ities in 2003
[4–6]
. The origin of SARS-CoV is unclear,
though studies on the molecular evolution of
SARS-CoV indicate that the virus may have emerged from
non-human species
[7]
. At present, no efficacious therapy for
SARS is available. Therefore, a search for effective
anti-virals for the SARS-CoV is of current interest.
SARS coronavirus is a positive-strand RNA virus that
encodes two polyproteins, pp1a and pp1ab
[8–10]
, for
further proteolytic processing to provide the functional
proteins for viral propagation. These processes are
me-diated primarily by the main protease (M
pro), known as
dimeric chymotrypsin-like protease (3CL
pro)
[11–13]
.
The active site of 3CL
procontains Cys145 and His41,
constituting a catalytic dyad in which the cysteine thiol
functions as the nucleophile in the proteolytic process
[11–13]
. Due to its essential role in viral replication, the
protease is an attractive target for the development of
therapeutics against SARS.
So far, only a few inhibitors of 3CL
prohave been
re-ported, including the HIV protease inhibitor TL-3
[14]
,
zinc-conjugated compounds
[15]
, aryl boronic acids
analogs
[16]
, a quinolinecarboxylate derivative
[17]
, a
thiophenecarboxylate
[18]
, phthalhydrazide-substituted
keto-glutamine analogs
[19]
, and anilides
[20]
.
How-ever, none of these inhibitors exhibits activities in the
low nanomolar range, and no preclinical studies were
reported.
As a part of our efforts directed toward the
develop-ment of potent anti-SARS agents, we report here the
dis-covery of a new class of mechanism-based irreversible
inactivators with inhibition constants in the nanomolar
range, by using the strategy of combinatorial reaction
in microtiter plates followed by screening in situ
[21–
23]
. This approach relies on the use of high-yield organic
reactions that can be carried out in water or
water-mis-cible, nontoxic solvents on microscales without
protect-ing groups, allowprotect-ing the product to be assayed directly
in situ without isolation and purification. Using this
approach, one can quickly modify a lead compound
with a small set of building blocks to identify an optimal
inhibitor.
Results and Discussion
Previously
[14]
, we have reported that the HIV protease
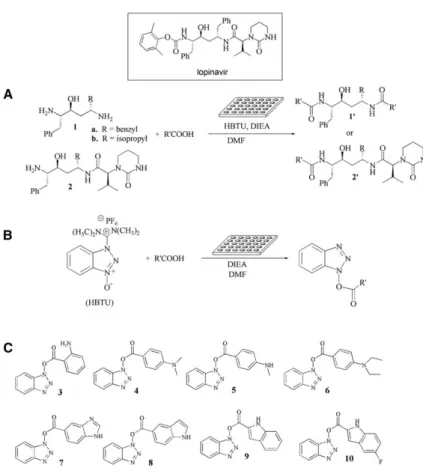
inhibitor Lopinavir (
Figure 1
) also inhibits 3CL
prowith an
IC
50of w50 mM. In order to find more potent 3CL
proin-hibitors, a library of Lopinavir-like compounds was
as-sembled by using either diamine
1 or amine 2 as the
core structure
[24]
for reaction with various acids in
mi-crotiter plates followed by screening in situ (
Figure 1
). In
a typical procedure
[21–23]
, a library of 90 carboxylic
acids (see the
Supplemental Data
available with this
ar-ticle online) (10 mmol each) in a microtiter plate was used
to couple with amine
1 or 2 (10 mmol) in the presence
of N,N-diisopropylethylamine (22 mmol) and
2-(1H-ben-zotriazole-1-yl)-1,1,3,3-tetramethyluronium
hexafluoro-phosphate (HBTU, 11 mmol) in DMF (100 ml) for 4 hr. An
aliquot of the product, based on a putative 100%
*Correspondence: phliang@gate.sinica.edu.tw (P.-H.L.); wong@ scripps.edu(C.-H.W.)
conversion of starting materials, was taken and
sub-jected to the 3CL
proinhibition assay in a 20 mM
Bis-Tris buffer (pH 7.0) at 25ºC. To obtain the IC
50values,
the initial velocities of the inhibited reactions with 50
nM of the protease and 6 mM of a fluorogenic substrate
were plotted against different inhibitor concentrations.
In this study, we noticed that the products derived
from 2-aminobenzoic acid (well C9, IC
50= 0.2 mM),
4-(methylamino)benzoic acid (well D5, IC
50= 0.3 mM),
4-(dimethylamino)benzoic acid (well D6, IC
50= 0.5 mM),
and 4-(diethylamino)benzoic acid (well D7, IC
50= 0.5
mM) showed the best inhibition and that the degree of
in-hibition was independent of the amine used.
In order to characterize the inhibitors, we attempted to
separately prepare the pure amide derivatives, but we
found that the amide formation was very slow, and
that the intermediates benzotriazole esters
326 were
isolated as major products from silica gel columns (the
X-ray ORTEP structure of compound
4 is shown in the
Supplemental Data
). To our surprise, all of the
Lopina-vir-like compounds showed only modest inhibitory
ac-tivities toward 3CL
pro(IC
50
R
10 mM), whereas
benzo-triazole esters
326 showed high inhibition activities.
To our knowledge, benzotriazole esters have been
used as acylating agents and have never been found
to be enzyme inhibitors. We then prepared a series of
benzotriazole esters by condensation of HBTU with
var-ious carboxylic acids (
Figure 1
B), and we found that the
benzotriazole esters derived from benzoic acid
contain-ing electron-withdrawcontain-ing substituents, e.g., NO
2, CN,
and CF
3, were susceptible to hydrolysis, whereas
ben-zotriazole esters
3–6 and those with electron-donating
groups were relatively stable in pH 5.0–8.0 over 24 hr
at room temperature. The relative stability of each
deriv-ative depends on the pKa of the corresponding benzoic
acid and basically follows the Hammett equation
[25–
27]
. These stable benzotriazole esters, esters
3–10
(
Figure 1
C), were assayed, and their inhibition results
against 3CL
proare shown in
Table 1
.
Further study of the inhibition of benzotriazole esters
3–10 showed that there was a time-dependent decrease
in enzyme activity as a function of the inhibitor
concen-tration (e.g., the inhibition of compound
4) (
Figure 2
A). In
the presence of 1 mM DTT (dithiothreitol), the
preincu-bation of enzyme with inhibitor did not affect the enzyme
activity, indicating that DTT can protect the enzyme from
inactivation (
Figure 2
B). These experiments indicate an
irreversible mode of action and point to the active site
Cys being involved in the inactivation process. The
ki-netic results for k
inactand K
ideterminations of
com-pound
4 are shown in
Figures 2
C and 2D (the detailed
Figure 1. Microtiter Plate-Based Reactions and In Situ Screening
(A) Reactions of1 or 2 with 90 acids in micro-titer plates, followed by in situ screening for inhibitors of SARS-CoV 3CLpro
.
(B) Reactions of HBTU and 90 acids in mi-crotiter plates, followed by in situ screening for inhibitors of SARS-CoV 3CLpro. (C) Molecular structures of inhibitors3–10 against SARS-CoV 3CLpro
.
Table 1. IC50, Ki, kinact, and CC50of Benzotriazole Esters
Compounds Ki (nM) kinact (s21 ) 3 103 kinact/Ki (M21 s21 ) 3 1023 CC50 (mM) 3 19.5 1.6 82.0 >100 4 17.4 1.3 74.7 >100 5 12.1 0.9 74.4 >100 6 11.1 0.8 72.1 >100 7 22.9 1.1 48.0 >100 8 7.5 1.1 146.7 >100 9 12.3 0.9 73.2 >100 10 13.8 1.2 86.9 >100
procedures are shown in the
Supplemental Data
). We
used the initial rate of inactivation (0–10 min) showing
pseudo first-order kinetics to determine the inhibition
constants. The enzyme activity is, however, not reduced
to zero over the time period, perhaps because of the
de-composition of the thioester intermediate or the
exis-tence of two populations of the enzymes. As shown in
Table 1
, benzotriazole esters
3–10 are strong inhibitors,
and, among these,
8 is the most potent, with an
inactiva-tion constant of 1.1 3 10
23s
21and an inhibition
con-stant of 7.5 nM. In addition, these esters are not toxic
to Vero E6 cells, which are often used in the cell-based
assay for SARS-CoV
[14]
, at a concentration of
100 mM. Compound
8 represents the most potent
mech-anism-based 3CL
proinhibitor reported to date.
We also investigated the nature of the inhibited
en-zyme by mass spectrometry. Electrospray ionization
mass spectra of wild-type 3CL
proand 3CL
protreated
with 4-(dimethylamino)benzoyl ester
4 (2 hr of
incuba-tion and 18 hr of dialysis) and their deconvoluted mass
spectra were determined (see the
Supplemental Data
).
The mass difference of 148.6 Da between the peaks of
33847.35 ([M + H]
+) and 33995.93 ([M + H]
+) implies the
acylation of 3CL
prowith a 4-(dimethylamino)benzoyl
moiety (mass 147.12). To further investigate the
acyla-tion site, MALDI-TOF mass spectrometric analysis of
the trypsin digest of 3CL
proand 3CL
protreated with
4-(dimethylamino)benzoyl ester
4 was performed. From
the MALDI spectra of the tryptic 3CL
proand the tryptic
acylated 3CL
proof G138-K180 peptide fragments, a
mass shift of 147 Da between T15 (4594.11 Da) and
ac-ylated T15 (4741.31 Da) indicates that this peptide
frag-ment contains an acylated residue (
Figures 3
A and 3B).
In order to determine the acylation site on the acylated
T15 peptide, we performed a sequence analysis by
MALDI MS/MS for peptides T15 and acylated T15 as
de-picted in
Figure 3
C. There is no mass difference among y
series fragment ions up to y
27, but a mass shift of 147.2
Da (4149.6 Da versus 4296.8 Da) on b
39clearly shows
that Cys145 is the only acylation site. This is consistent
with the observation of no mass shift of mutant C145A
3CL
protreated with 4-(dimethylamino)benzoyl ester
4.
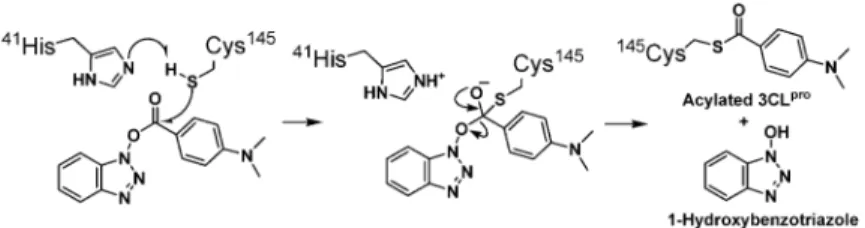
All of these results support the mechanism of
irrevers-ible inhibition of 3CL
proby 4-(dimethylamino)benzoyl
es-ter
4 via acylation of Cys145 (
Figure 4
).
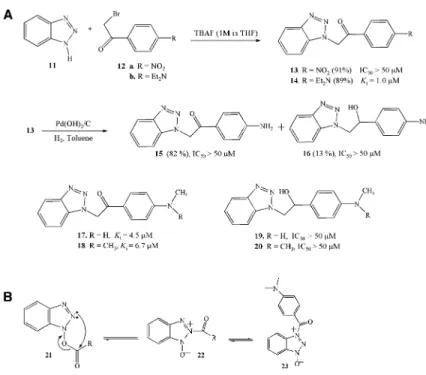
In order to develop stable, noncovalent inhibitors
based on the benzotriazole esters discovered in this
study, we synthesized compounds
13 and 14 by using
TBAF-assisted N-alkylation of 1H-benzotriazole
11
[28,
29]
. In addition, compound
13 was hydrogenated with
Pd(OH)
2/C as a catalyst at room temperature to obtain
compounds
15 (82%) and 16 (13%). Compounds 15
and
16 were methylated or dimethylated to compounds
17–20 by using TBAF as a reagent (
Figure 5
A).
Com-pounds
13–20 have all the features of benzotriazole
es-ter inhibitors, except that the eses-ter oxygen is replaced
by a carbon. Inhibition analysis shows that compounds
13–20 are noncovalent competitive inhibitors, albeit
rel-atively weak ones compared to the corresponding
es-ters; of these, compound
14 is most potent, with a K
iof 1.0 mM. Reduction of the carbonyl group, however,
re-sults in a significant loss of activity (
Figure 5
A). It is noted
that the benzotriazole compounds contain three
equilib-rium structures in solution (
Figure 5
B)
[25, 26]
, and that
compounds
13–20 may mimic the ester forms instead
of the three equilibrium forms found in solution.
To gain further insight into the mode of inhibition,
a docking experiment based on computer modeling
(Au-todock version 3.0.5)
[30]
for the binding of compounds
3, 4, 8, and 9 with 3CL
pro(1uk4)
[12]
was carried out, and
the result indicated that the benzotriazole moiety was
disposed in the pocket formed by Cys145, Ser144, and
Gly143 in the active site (
Figure 6
). The g-S atom of
Cys145 was close enough, within 3.5 A˚ in a rigid model,
to the carbonyl group of the benzotriazole ester to
ren-der a nucleophilic attack. The aminophenyl group (for
3 and 4) and the indole moiety (for 8 and 9) were in the
region surrounded by Thr25, Thr26, His41, Thr45,
Ala46, and Met49. The NH group of the indole moiety
of
8 was hydrogen bonded with the side chain OH of
Thr25. In comparison, the indole group of
9 lacks such
hydrogen bonding and thus shows a weaker affinity
Figure 2. Kinetic Studies of Inhibitor4 and SARS-CoV 3CLpro
(A) The progress curves in the presence of 0.3–3.0 mM inhibitor for reactions initiated by adding enzyme (final concentration of 0.05 mM) into a mixture of substrate (6 mM) and inhibitor4. Over the entire 5 min time window, the uninhibited enzyme displayed a linear progress curve, whereas the inhibited enzyme with a different concentration of in-hibitor showed a time-dependent reduction of activity.
(B) The same experiments as performed in (A), but with 1 mM DTT in the preincubation mixture.
(C) Preincubation time dependence of the fractional velocity of the protease-catalyzed reaction in the presence of 0.02–0.2 mM time-dependent inhibitor4.
(D) Kitz and Wilson replot of the half-life (t1/2) of enzyme inactivation as a function of the reciprocal of the slow inactivator concentra-tion. The kinactis 0.0013 s
21
and Kiis 17.4 nM for the time-dependent inactivator4 based on the kinetic data.
toward 3CL
prothan does isomer
8. Compounds 13–20
do not fit into the pocket well enough to interact with
the residues mentioned above, and the calculated
min-imal energies of binding are w0.5–1.0 kcal/mol higher
than those of the corresponding esters, consistent
with the inhibition assay result (see the
Supplemental
Data
).
Significance
Severe acute respiratory syndrome (SARS) is a newly
emerged disease caused by a novel human
corona-virus. Currently, no effective antiviral agents exist
against this deadly epidemic. The main protease of
SARS-CoV, 3CL
pro, is an attractive target for drug
dis-covery due to its essential role in viral replication. We
have discovered several stable benzotriazole esters as
a new class of irreversible enzyme inhibitors, and, to
our knowledge, these compounds are the most potent
mechanism-based 3CL
proinhibitors known to date.
The mode of action has been studied and has been
shown to proceed through acylation of the active site
Cys145 assisted by the catalytic dyad.
Experimental Procedures
Materials and Methods
SARS-CoV 3CL protease was prepared according to the previously described procedure[31]. Reactions requiring dry conditions were carried out under an inert atmosphere by using standard tech-niques. All of the reagents and solvents were reagent grade and were used without further purification unless otherwise specified. THF was distilled from sodium benzophenone ketyl under N2. HRMS values were obtained by using the EI as the ionization source.
General Procedure for the Preparation of Benzotriazole Esters To a solution of acid compound (1.0 equiv.), 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU, 1.1 equiv) in DMF (1.0 ml), as well as N,N-diisopropylethylamine (DIEA, 1.1 equiv.), was added. After the solution was stirred at 25ºC for 3.0 hr, the reaction mixture was added to 1 M NaHCO3(10 ml) and extracted with ethyl acetate (EA) (15 ml 3 3), and the organic layer was collected and concentrated under reduced pressure. The resi-due was purified by use of column chromatography on silica gel to provide the desired 1-hydroxybenzotriazole esters.
2-Aminobenzoic Acid Benzotriazol-1-yl Ester, 3
The standard procedure was followed by use of 2-aminobenzoic acid (C9, 34.8 mg, 0.2537 mmol, 1.0 equiv.), HBTU (105.9 mg, 0.2791 mmol, 1.1 equiv.), and DIEA (49 ml, 0.2791 mmol, 1.1 equiv.). After the reaction mixture was worked up, the residue was purified by use of column chromatography (5% MeOH in chloroform as
eluant). Benzotriazole ester3 (57.4 mg, 0.2258 mmol) was obtained in 89% yield as a yellow solid: TLC Rf= 0.81 (5% MeOH in CHCl3as eluant);1 H NMR (CDCl3, 400 MHz) d = 5.31 (brs, 2 H, NH2), 6.73–6.78 (m, 2 H, 2 3 ArH), 7.41 (t, 2 H, J = 6.0 Hz, 2 3 ArH), 7.46 (d, 1 H, J = 6.6 Hz, ArH), 7.52 (t, 1 H, J = 6.0 Hz, ArH), 8.06 (d, 2 H, J = 6.7 Hz, ArH), 8.14 (d, 1 H, J = 6.7 Hz, ArH);13 C NMR (CDCl3, 100 MHz) d = 104.26, 108.45, 116.82, 116.98, 120.42, 124.78, 128.66, 129.00, 130.84, 136.71, 143.50, 152.23, 163.65; IR (KBr) 3456 (m, NH), 3322 (m, NH), 1736 (s, C=O), 1632 (s), 1566 (m), 1485 (s), 1372 (m), 1228 (s), 1153 (s), 977 (s), 740 (s) cm21 ; HRMS [M + 1] calcd for C13H11N4O2: 255.0875, found 255.0882.
4-Dimethylamino-Benzoic Acid Benzotriazol-1-yl Ester, 4 The standard procedure was followed by use of (dimethylamino)-benzoic acid (D6, 120.3 mg, 0.7283 mmol, 1.0 equiv.), HBTU (303.8 mg, 0.8011 mmol, 1.1 equiv.), and DIEA (140 ml, 0.8011 mmol, 1.1 equiv.). After the reaction mixture was worked up, the residue was purified by use of column chromatography (chloroform as eluant). Benzotriazole ester 4 (191.2 mg, 0.6773 mmol) was obtained in 93% yield as a light-yellow solid: TLC Rf= 0.50 (CHCl3as eluant); 1 H NMR (CDCl3, 400 MHz) d = 3.13 (s, 6 H, 2 3 CH3), 6.74 (d, J = 8.9 Hz, 2 H, 2 3 ArH), 7.42 (t, J = 6.7 Hz, 1 H, ArH), 7.47–7.55 (m, 2 H, 2 3 ArH), 8.07–8.12 (m, 3 H, 3 3 ArH);13 C NMR (CDCl3, 100 MHz) d = 40.01, 108.64, 110.02, 111.06, 120.35, 124.54, 128.36, 129.11, 132.73, 143.57, 154.38, 163.51; IR (KBr) 2914 (w), 1768 (s, C=O), 1609 (s), 1537 (s), 1441 (s), 1386 (s), 1263 (s), 1183 (s), 957 (s), 770 (s) cm21 ; HRMS [M + 1] calcd for C15H15N4O2: 283.1195, found 283.1199.
4-Methylamino-Benzoic Acid Benzotriazol-1-yl Ester, 5
The standard procedure was followed by use of (4-methylamino)-benzoic acid (D5, 53.5 mg, 0.3539 mmol, 1.0 equiv.), HBTU (147.7 mg, 0.3894 mmol, 1.1 equiv.), and DIEA (68 ml, 0.3894 mmol, 1.1 equiv.). After the reaction mixture was worked up, the residue was purified by use of column chromatography (chloroform as eluant). Benzotriazole ester5 (88.3 mg, 0.3291 mmol) was obtained in 93% yield as a white solid: TLC Rf= 0.28 (CHCl3as eluant);1H NMR (CDCl3, 400 MHz) d = 2.92 (d, 3 H, J = 1.5 Hz, CH3N), 6.63 (d, 2 H, J = 8.7 Hz, 2 3 ArH), 7.41 (t, 1 H, J = 7.6 Hz, ArH), 7.46–7.53 (m, 2 H, 2 3 ArH), 8.04–8.07 (m, 3 H, 3 3 ArH);13C NMR (CDCl3, 100 MHz) d = 29.91, 108.61, 111.05, 111.52, 120.29, 124.62, 128.43, 129.05, 133.00, 143.52, 154.70, 162.70; IR (KBr) 3346 (m, NH), 3010 (m), 2907 (w), 1764 (s, C=O), 1602 (s), 1545 (s), 1497 (s), 1446 (s), 1362 (s), 1238 (s), 1068 (s), 957 (s), 791 (s) cm21 ; HRMS [M + 1] calcd for C14H13N4O2: 269.1039, found 269.1032.
4-Diethylamino-Benzoic Acid Benzotriazol-1-yl Ester, 6 The standard procedure was followed by use of (diethylamino)ben-zoic acid (D7, 48.4 mg, 0.2505 mmol, 1.0 equiv.), HBTU (104.5 mg, 0.2755 mmol, 1.1 equiv.), and DIEA (48 ml, 0.2755 mmol, 1.1 equiv.). After the reaction mixture was worked up, the residue was purified by use of column chromatography (chloroform as eluant). Benzotria-zole ester6 (71.5 mg, 0.2304 mmol) was obtained in 92% yield as a light-yellow solid: TLC Rf = 0.50 (CHCl3 as eluant); 1H NMR (CDCl3, 400 MHz) d = 1.17–1.25 (m, 6 H, 2 3 CH3), 3.38–3.49 (m, 4 H, 2 3 CH2), 6.70 (d, 2 H, J = 8.8 Hz, 2 3 ArH), 7.40 (t, J = 7.4 Hz,
Figure 3. MALDI Spectrum of Inhibitor4 and SARS 3CLpro (A) MALDI spectrum of tryptic 3CLpro
. (B) MALDI spectrum of tryptic acylated 3CLpro
.
(C) MALDI MS/MS spectra of T15 and acylated T15 peptides (GSFLNGSC*GSVGFNIDYDCVSFCYMHHMELPTGVHAGTDLEGK) showed a mass shift of 147.2 Da (4149.6 Da versus 4296.8 Da) on b39, indicating that Cys145 (C*) is the acylation site.
Figure 4. Proposed Mechanism for Inhibition of SARS-CoV 3CLpro
by Acylation with Ben-zotriazole Esters
1 H, ArH), 7.42–7.53 (m, 2 H, 2 3 ArH), 6.70 (d, 1 H, J = 8.8 Hz, ArH), 8.06 (d, J = 8.8 Hz, 2 H, 2 3 ArH);13 C NMR (CDCl3, 100 MHz) d = 12.39, 44.66, 108.63, 110.64, 120.29, 124.53, 128.33, 128.89, 132.23, 133.01, 143.53, 152.61, 162.69; IR (KBr) 2980 (m), 1774 (s, C=O), 1602 (s), 1529 (s), 1445 (s), 1380 (s), 1260 (s), 1155 (s), 963 (s), 766 (s) cm21 ; HRMS [M + 1] calcd for C17H19N4O2: 311.1508, found 311.1503.
1H-Benzoimidazole-5-Carboxylic Acid Benzotriazol-1-yl Ester, 7 The standard procedure was followed by use of 5-benzimidazole-carboxylic acid (G5, 39.1 mg, 0.2411 mmol, 1.0 equiv.), HBTU (100.6 mg, 0.2652 mmol, 1.1 equiv.), and DIEA (46 ml, 0.2652 mmol, 1.1 equiv.). After the reaction mixture was worked up, the residue was purified by use of column chromatography (chloroform as elu-ant). Benzotriazole ester7 (48.5 mg, 0.1737 mmol) was obtained in 72% yield as a light-brown solid: TLC Rf = 0.21 (10% MeOH in CHCl3 as eluant); 1 H NMR (d4-methanol + CDCl3, 400 MHz) d = 7.47–7.53 (m, 2 H, 2 3 ArH), 7.79–7.84 (m, 2 H, 2 3 ArH), 8.04 (d, 1 H, J = 8.5 Hz, ArH), 8.17 (dd, 1 H, J = 8.5, 1.6 Hz, ArH), 8.49 (s, 1 H, N=CH), 8.61 (d, 1 H, J = 1.6 Hz, ArH);13 C NMR (d4-methanol + CDCl3, 100 MHz) d = 108.53, 110.19, 115.19, 117.00, 118.35, 119.38, 124.66, 125.11, 125.96, 126.82, 128.88, 143.11, 144.83, 163.12; IR (KBr) 3099 (m, NH), 2901 (w), 1778 (s, C=O), 1620 (s), 1576 (m), 1421 (s), 1362 (s), 1281 (s), 1155 (s), 993 (s), 739 (s) cm21 ; HRMS [M + 1] calcd for C14H10N5O2: 280.0834, found 280.0834.
1H-Indole-5-Carboxylic Acid Benzotriazol-1-yl Ester, 8
The standard procedure was followed by use of indole-5-carboxylic acid (70.6 mg, 0.4381 mmol, 1.0 equiv.), HBTU (182.8 mg, 0.4819 mmol, 1.1 equiv.), and DIEA (84 ml, 0.4819 mmol, 1.1 equiv.). After the reaction mixture was worked up, the residue was purified by use of column chromatography (chloroform as eluant). Benzotria-zole ester8 (107.3 mg, 0.3855 mmol) was obtained in 88% yield as a brown solid:1
H NMR (d6-acetone, 400 MHz) d = 6.78 (d, 1 H, J = 3.08 Hz,), 7.52 (t, 1 H, J = 7.1 Hz, ArH), 7.59 (t, 1 H, J = 2.3 Hz, ArH), Figure 5. Synthesis of Noncovalent Inhibi-tors
(A) Synthesis of compounds13–20 and their IC50s or Kis for SARS-CoV 3CLpro. (B) The equilibrium structures of the benzo-triazole compounds in solution.
Figure 6. A Modeling Complex of SARS 3CLpro
and Benzotriazole Esters
Binding modes of compounds3 (yellow), 4 (red), 8 (blue), and 9 (green) in the active site of SARS-CoV 3CLpro
(PDBluk4). Models were gen-erated by Autodock and displayed by MGLTOOLS (MGL, Scripps).
7.65 (t, 1 H, J = 8.4 Hz, ArH), 7.68 (d, 1 H, J = 8.6 Hz, =CH), 7.79 (d, 1 H, J = 8.4 Hz, ArH), 8.01 (d, 1 H, J = 8.4 Hz, ArH), 8.10 (d, 1 H, J = 8.6 Hz, =CH), 8.68 (s, 1 H, ArH);13 C NMR (d6-acetone, 100 MHz) d = 104.06, 109.58, 112.82, 115.65, 120.64, 123.78, 125.56, 125.79, 128.53, 128.90, 129.50, 129.78, 141.00, 144.27, 164.87; IR (KBr) 3213 (m, NH), 2907 (w), 1767 (s, C=O), 1614 (s), 1582 (m), 1446 (s), 1359 (s), 1266 (s), 1157 (s), 990 (s), 789 (s) cm21 ; HRMS [M + 1] calcd for C15H11N4O2: 279.0882, found 279.0878.
1H-Indole-2-Carboxylic Acid Benzotriazol-1-yl Ester, 9
The standard procedure was followed by use of indole-2-carboxylic acid (F1, 74.9 mg, 0.4648 mmol, 1.0 equiv.), HBTU (193.9 mg, 0.5113 mmol, 1.1 equiv.), and DIEA (89 ml, 0.5113 mmol, 1.1 equiv.). After the reaction mixture was worked up, the residue was purified by use of column chromatography (chloroform as eluant). Benzotriazole ester 9 (115.1 mg, 0.4136 mmol) was obtained in 89% yield as a light-yel-low solid: TLC Rf= 0.82 (5% MeOH in CHCl3as eluant);1H NMR (CDCl3, 400 MHz) d = 7.24 (t, 1 H, J = 8.0 Hz, ArH), 7.41–7.46 (m, 2 H, 2 3 ArH), 7.49–7.58 (m, 3 H, 3 3 ArH), 7.69 (s, 1 H, =CH), 7.78 (d, 1 H, J = 8.2 Hz, ArH), 8.10 (1 H, J = 8.4 Hz, ArH), 9.97 (br, 1 H, NH); 13 C NMR (CDCl3, 100 MHz) d = 38.58, 108.37, 112.48, 113.28, 120.45, 120.69, 121.69, 123.11, 124.90, 127.06, 127.29, 128.83, 138.60, 143.42, 157.99; IR (KBr) 3282 (m, NH), 1773 (s, C=O), 1670 (s), 1517 (s), 1459 (s), 1389 (s), 1340 (s), 1161 (s), 1052 (s), 778 (s) cm21
; HRMS [M + 1] calcd for C15H11N4O2: 279.0882, found 279.0878.
1H-Benzoimidazole-5-Carboxylic Acid Benzotriazol-1-yl Ester, 10
The standard procedure was followed by use of 5-fluoroindole-2-carboxylic acid (F4, 26.1 mg, 0.1457 mmol, 1.0 equiv.), HBTU (60.8 mg, 0.1603 mmol, 1.1 equiv.), and DIEA (28 ml, 0.1603 mmol, 1.1 equiv.). After the reaction mixture was worked up, the residue was purified by use of column chromatography (chloroform as eluant). Benzotriazole ester 10 (39.3 mg, 0.1327 mmol) was obtained in 91% yield as a light-yellow solid: TLC Rf = 0.83 (5% MeOH in CHCl3as eluant);1H NMR (d6-DMSO and CDCl3, 400 MHz) d = 7.15 (dd, 1 H, J = 7.4, 2.4 Hz, ArH), 7.41–7.52 (m, 3 H, 3 3 ArH), 7.58– 7.65 (m, 2 H, ArH + =CH), 7.71 (d, 1 H, J = 8.4 Hz, ArH), 8.07 (1 H, J = 8.4 Hz, ArH); 13 C NMR (d6-DMSO and CDCl3, 100 MHz) d = 106.15 (d, J = 24.0 Hz), 108.60, 111.92 (d, J = 6.0 Hz), 114.14 (d, J = 10.0 Hz), 115.61 (d, J = 27.0 Hz), 119.66, 121.80, 124.79, 126.35 (d, J = 11.0 Hz), 128.26, 128.77, 135.58, 142.65, 156.32 (d, J = 235.0 Hz), 157.34; IR (KBr) 3231 (m, NH), 2981 (w), 1782 (s, C=O), 1681 (s), 1521 (m), 1434 (s), 1342 (s), 1222 (s), 1186 (s), 921 (s), 757 (s) cm21
; HRMS [M + 1] calcd for C15H10FN4O2: 297.0779, found 297.0788.
General Procedure for the TBAF-Assisted Benzotriazole N-Alkylation, 14
Benzotriazole (100 mg, 0.84 mmol) and a-bromo-4-(diethylamino) acetophenone (272 mg, 1.00 mmol, 1.2 equiv.) were placed in a 5 ml flask with a stirring bar, followed by the addition of TBAF (1.00 ml, 0.84 mmol, 1.2 equiv., 1 M in THF) at room temperature. After being stirred for 2 hr at room temperature, the reaction was di-rectly loaded into the column, and the product was eluted with a so-lution of 6:1 hexane:ethyl acetate to yield 217.4 mg (84%) of product as a pale-yellow solid.1 H NMR (500 MHz, CDCl3) d 8.05 (d, J = 8.5 Hz, 1 H), 7.96 (d, J = 9.2 Hz, 2 H), 7.50–7.30 (m, 3 H), 6.68 (d, J = 9.2 Hz, 2 H), 5.98 (s, 2 H), 3.42 (q, J = 7.0 Hz, 4 H), 1.19 (t, J = 7.0 Hz, 6 H); 13 C NMR (125 MHz, CDCl3) d 187.92, 152.46, 146.42, 134.38, 131.37, 128.00, 124.21, 121.41, 120.25, 110.91, 110.46, 53.76, 45.06, 12.87; ESI-MS calculated for C18H20N4O 308.16, found 308.12.
Inhibition Assay against the SARS-CoV 3CL Protease
A fluorometric assay[31]was utilized to determine the inhibition constants of the prepared samples. Briefly, a fluorogenic peptide, Dabcyl-KTSAVLQSGFRKME-Edans, was used as the substrate[31], and the enhanced fluorescence due to cleavage of this substrate catalyzed by the protease was monitored at 538 nm with excitation at 355 nm. The IC50value of individual inhibitors was measured in a reaction mixture containing 50 nM SARS 3CL protease and 6 mM fluorogenic substrate in 20 mM Bis-Tris (pH 7.0). The enzyme stock solution was kept in 12 mM Tris-HCl (pH 7.5) containing 120 mM
NaCl, 0.1 mM EDTA, plus 7.5 mM b-ME before being added to the as-say solution. The Kimeasurements (for the noncovalent inhibitors case) were performed at two fixed inhibitor concentrations and var-ious substrate concentrations. In the mechanism-based inactivator cases, we used t1/2versus 1/[inactivator] for Ki measurements (for more detailed procedures for the behavior of inhibitors, please see theSupplemental Data).
Expression and Purification of SARS-CoV 3CLpro
Wild-type and the C145A mutant of the SARS protease were cloned in pET 28 with N-terminal Trx, His tag, and FXa site. The tags were removed by FXa protease after the proteins were purified with NiNTA column chromatography. For more detailed experimental proce-dures, please see[32].
Mass Spectrometric Analysis
The ESI-MS experiments were conducted on a Bruker Daltonics BioTOF III high-resolution mass spectrometer, equipped with a home-built nanoESI source. Mass resolution was better than 20,000 on one-pass mode with a mass range of w100–3,000. The samples were diluted to 1.0 mM with bidistilled aqueous solution containing 40% methanol and 0.1% formic acid (v/v/v) and were in-fused with a syringe pump at a flow rate of 300 nl/min. The actual amount of samples consumed was less than 300 fmole.
The MALDI-MS measurements were performed on a MALDI-TOF/ TOF mass spectrometer (Applied Biosystems 4700 Proteomics An-alyzer). Tryptic digest solution was mixed 1:1 with matrix solution (CHCA [a-cyano-4-hydroxycinnamic acid] 10 mg/ml in solvents 49.9/50/0.1 H2O/CH3CN/TFA), and 1.0 ml was then spotted in each well. Each MALDI spectrum was accumulated from up to 4,000 laser shots from a random sampling of 40 positions per well.
Computer Modeling of SARS-CoV 3CL Protease Inhibition Docking was performed by using Autodock, version 3.05[30]. Pre-computed energy grid maps with grid point spacing of 0.375 A˚ and 50 3 50 3 50 grid points centered at the active site were used (autogrid tool in Autodock, version 3.05). During a docking experi-ment, each compound was kept flexible (except their rings and am-ide bonds), and the built-in LGA method was adopted. In each com-pound structure, 1.5 3 106
energy was evaluated, and 40 poses were selected from 2.7 3 105
generations per run.
The crystal structure of SARS-CoV 3CL protease in complex with a substrate-analog inhibitor (coded1uk4) was obtained from The Protein Data Bank (PDB;http://www.rcsb.org/pdb/) (for more de-tailed procedures, please see theSupplemental Data).
Supplemental Data
Supplemental data including 90 different acids, the X-ray data for compound4, synthesis and characterization of compounds 15–20, a supplemental enzyme kinetic study, and the detailed molecular docking data are available at http://www.chembiol.com/cgi/ content/full/13/3/261/DC1/.
Acknowledgments
This work is supported by the National Science Council, Taiwan and Genomics Research Center, Academia Sinica. We also thank Ms. Hsien-Hua Hsu for her help with the cytotoxicity assay.
Received: July 12, 2005 Revised: December 16, 2005 Accepted: December 27, 2005 Published online: March 24, 2006
References
1. Ksiazek, T.G., Erdman, D., Goldsmith, C.S., Zaki, S.R., Peret, T., Emery, S., Tong, S., Urbani, C., Comer, J.A., Lim, W., et al. (2003). A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966.
2. Drosten, C., Gu¨nther, S., Preiser, W., van der Werf, S., Brodt, H.-R., Becker, S., Rabenau, H., Panning, M., Kolesnikova, L., Fouchier, R.A.M., et al. (2003). Identification of a novel
coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1967–1976.
3. Peiris, J.S.M., Lai, S.T., Poon, L.L.M., Guan, Y., Yam, L.Y.C., Lim, W., Nicholls, J., Yee, W.K.S., Yan, W.W., Cheung, M.T., et al. (2003). Coronavirus as a possible cause of severe acute respira-tory syndrome. Lancet 361, 1319–1325.
4. Lee, N., Hui, D., Wu, A., Chan, P., Cameron, P., Joynt, G.M., Ahuja, A., Yung, M.Y., Leung, C.B., To, K.F., et al. (2003). A major outbreak of severe acute respiration syndrome in Hong Kong. N. Engl. J. Med. 348, 1986–1994.
5. Poutanen, S.M., Low, D.E., Henry, B., Finkelstein, S., Rose, D., Green, K., Tellier, R., Draker, R., Adachi, D., Ayers, M.N., et al. (2003). Canadian severe acute respiratory syndrome study, identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 348, 1995–2005.
6. Tsang, K.W., Ho, P.L., Ooi, G.C., Yee, W.K., Wang, T., Chan-Yeung, M., Lam, W.K., Seto, W.H., Yam, L.Y., Cheung, T.M., et al. (2003). A cluster of cases of severe acute respiratory syn-drome in Hong Kong. N. Engl. J. Med. 348, 1977–1985. 7. He, J.-F., Peng, G.-W., Min, J., Yu, D.-W., Liang, W.-J., Zhang,
S.-Y., Xu, R.-H., Zheng, H.-Y., Wu, X.-W., Xu, J., et al. (2004). Mo-lecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303, 1666–1669. 8. Rota, P.A., Oberste, M.S., Monroe, S.S., Nix, W.A., Campagnoli,
R., Icenogle, J.P., Penaranda, S., Bankamp, B., Maher, K., Chen, M.-H., et al. (2003). Characterization of a novel coronavirus asso-ciated with severe acute respiratory syndrome. Science 300, 1394–1399.
9. Marra, M.A., Jones, S.J.M., Astell, C.R., Holt, R.A., Brooks-Wil-son, A., Butterfield, Y.S.N., Khattra, J., Asano, J.K., Barber, S.A., Chan, S.Y., et al. (2003). The genome sequence of the SARS-associate coronovirus. Science 300, 1399–1404. 10. Ruan, Y., Wei, C.L., Lin, A.E., Vega, V.B., Thoreau, H., Se-Thoe,
S.Y., Chia, J.M., Ng, P., Chiu, K.P., Lim, L., et al. (2003). Compar-ative full-length genome sequence analysis of 14 SARS corono-virus isolates and common mutations associated with putative origins of infection. Lancet 361, 1779–1785.
11. Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J.R., and Hilgen-feld, R. (2003). Coronavirus main protease (3CLpro
) structure: ba-sis for design of anti-SARS drugs. Science 300, 1763–1767. 12. Yang, H., Yang, M., Ding, Y., Liu, Y., Lou, Z., Zhou, Z., Sun, L.,
Mo, L., Ye, S., Pang, H., et al. (2003). The crystal structure of se-vere acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U.S.A. 100, 13190–13195.
13. Chou, K., Wei, D., and Zhong, W. (2003). Binding mechanism of coronavirus main protease with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 308, 148–151.
14. Wu, C.-Y., Jan, J.-T., Ma, H.-H., Kuo, C.-J., Juan, H.-F., Cheng, Y.-S.E., Hsu, H.-H., Huang, H.-C., Wu, D., Brik, A., et al. (2004). Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. USA 101, 10012– 10017.
15. Hsu, J.T.-A., Kuo, C.-J., Hsieh, H.-P., Wang, Y.-C., Huang, K.-K., Lina, C.P.-C., Huang, P.-F., Chen, X., and Liang, P.-H. (2004). Evaluation of metal-conjugated compounds as inhibitors of 3CL protease of SARS-CoV. FEBS Lett. 574, 116–120. 16. Bacha, U., Barrila, J., Velazquez-Campoy, A., Leavitt, S.A., and
Freire, E. (2004). Identification of novel inhibitors of the SARS co-ronavirus main protease 3CLpro
. Biochemistry 43, 4906–4912. 17. Kao, R.Y., Tsui, W.H.W., Lee, T.S.W., Tanner, J.A., Watt, R.M.,
Huang, J.-D., Hu, L., Chen, G., Chen, Z., Zhang, L., et al. (2004). Identification of novel small-molecular inhibitors of se-vere acute respiratory syndrome-associated coronavirus by chemical genetics. Chem. Biol. 11, 1293–1299.
18. Blanchard, J.E., Elowe, N.H., Huitema, C., Fortin, P.D., Cechetto, J.D., Eltis, L.D., and Brown, E.D. (2004). High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem. Biol. 11, 1445–1453.
19. Jain, R.P., Pettersson, H.I., Zhang, J., Aull, K.D., Fortin, P.D., Hui-tema, C., Eltis, L.D., Parrish, J.C., James, M.N.G., Wishart, D.S., et al. (2004). Synthesis and evaluation of keto-glutamine
ana-logues as potent inhibitors of severe acute respiratory syndrome 3CLpro
. J. Med. Chem. 47, 6113–6116.
20. Shie, J.-J., Fang, J.-M., Kuo, C.-J., Kuo, T.-H., Liang, P.-H., Huang, H.-J., Yang, W.-B., Lin, C.-H., Chen, J.-L., Wu, Y.-T., et al. (2005). Discovery of potent anilide inhibitors against the se-vere acute respiratory syndrome 3CL protease. J. Med. Chem. 48, 4469–4473.
21. Brik, A., Lin, Y.-C., Elder, J., and Wong, C.-H. (2002). A quick di-versity-oriented amide-forming reaction to optimize p-subsite residues of HIV protease inhibitors. Chem. Biol. 9, 891–896. 22. Wu, Y., Chang, F., Chen, J.S.-Y., Wong, H., and Lin,
C.-H. (2003). Rapid diversity-oriented synthesis in microtiter plates for in situ screening: discovery of potent and selective a-fucosi-dase inhibitors. Angew. Chem. Int. Ed. 42, 4661–4664. 23. Chang, C.-F., Ho, C.-W., Wu, C.-Y., Chao, T.-A., Wong, C.-H.,
and Lin, C.-H. (2004). Discovery of picomolar slow tight-binding inhibitors of a-fucosidase. Chem. Biol. 11, 1301–1306. 24. Stoner, E.J., Cooper, A.J., Dickman, D.A., Kolaczkowski, L.,
Lal-laman, J.E., Liu, J.-H., Oliver-Shaffer, P.A., Patel, K.M., Paterson, J.B., Jr., Plata, D.J., et al. (2000). Synthesis of HIV protease inhib-itor ABT-378 (Lopinavir). Org. Proc. Res. Dev. 4, 264–269. 25. McCarthy, D.G., Hegarty, A.F., and Hathaway, B.J. (1977).
N-hy-droxy-compounds as acyl transfer agents. Part 1. kinetics and mechanism of nucleophilic displacements on 1-hydroxybenzo-triazole esters and crystal and molecular structure of 1-benzoyl-oxybenzotriazole. J. Chem. Soc. Perkin II 2, 224–231. 26. Barlos, K., Papaioannou, D., and Voliotis, S. (1985). Crystal
structure of 3-(Na
-tritylmethionyl)-benzotriazole 1-oxide, a syn-thon in peptide synthesis. J. Org. Chem. 50, 696–697. 27. Hansh, C., Leo, A., and Taft, R.W. (1991). A survey of Hammett
substituent constants and resonance and field parameters. Chem. Rev. 91, 165–195.
28. Brik, A., Wu, C.-Y., Best, M.D., and Wong, C.-H. (2005). Tetrabu-tylammonium fluoride-assisted rapid N9-alkylation on purine ring: application to combinatorial reactions in microtiter plates for the discovery of potent sulfotransferase inhibitors in situ. Bioorg. Med. Chem. 13, 4622–4626.
29. Wu, C.-Y., Brik, A., Wang, S.-K., Chen, Y.-S., and Wong, C.-H. (2005). Tetrabutylammonium fluoride-mediated rapid alkylation reaction in microtiter plates for discovery of enzyme inhibitors in situ. Chembiochem. 6, 2176–2180.
30. Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., and Olsen, A.J. (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662.
31. Kuo, C.-J., Chi, Y.-H., Hsu, T.-A., and Liang, P.-H. (2004). Char-acterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biochem. Biophys. Res. Commun. 318, 862–867.
32. Hsu, M.-F., Kuo, C.-J., Chang, K.-T., Chang, H.-C., Chou, C.-C., Ko, T.-P., Shr, H.-L., Chang, G.-G., Wang, A.H.-J., and Liang, P.-H. (2005). Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 280, 31257–31266.