D
A
LTO
N
FULL PAPER
DOI: 10.1039/a910260o J. Chem. Soc., Dalton Trans., 2000, 1649–1654 1649
Complexation and metallation of the multifunctional
[Ph
2P(o-C
6H
4)CH
᎐᎐N(CH
2)
2(o-C
5H
4N)] ligand on
triosmium carbonyl clusters
Wen-Yann Yeh,*
aChing-Chao Yang,
aShie-Ming Peng *
band Gene-Hsiang Lee
b aDepartment of Chemistry, National Sun Yat-Sen University, Kaohsiung, Taiwan 804
b
Department of Chemistry, National Taiwan University, Taipei, Taiwan 106
Received 22nd December 1999, Accepted 15th March 2000
Published on the Web 27th April 2000
Reaction of the phosphine–imine–pyridine containing ligand Ph2P(o-C6H4)CH᎐᎐N(CH2)2(o-C5H4N) (abbreviated to PNN) with [Os3(CO)10(NCMe)2] at ambient temperature affords {Os3(CO)10[η2-Ph2P(o-C6H4)CH᎐᎐N(CH2)2 -(o-C5H4N)]} 1. Thermolysis of 1 in refluxing toluene leads to CO dissociation to give {Os3(CO)9[µ-η3-Ph2P(o-C6H4 )-CH᎐᎐N(CH2)2(o-C5H4N)]} 2, and C–H activation of the PNN ligand to give {(µ-H)Os3(CO)8[µ3-η4-Ph2P(o-C6H4 )-CH᎐᎐NCH2CH(o-C5H4N)]} 3 and {(µ-H)Os3(CO)8[µ-η4-Ph2P(o-C6H4)C᎐᎐N(CH2)2(o-C5H4N)]} 4. In contrast, co-pyrolysis of [Os3(CO)12] with PNN in a sealed tube results in 2, 3 and the diosmium phosphido complex {Os2(CO)5 -(µ-PPh2)[µ-η3-Ph2P(o-C6H4)C᎐᎐N(CH2)2(o-C5H4N)]} 5. The structures of 1, 3, 4 and 5 have been determined by an X-ray diffraction study. Compounds 3 and 4 are isomers with the methylene and the imine C–H bonds of the PNN ligand being activated by the Os3 cluster, respectively.
Introduction
The ability of a metal atom (or ion) to organize a flexible multifunctional ligand around its coordination sphere has led to the design of several catalytic systems and intramolecularly organized recognition sites.1 The Ph
2P(o-C6H4)CH᎐᎐N(CH2)2 -(o-C5H4N) molecule (abbreviated to PNN), which contains a phosphine, an imine and a pyridyl electron-donating group, was previously prepared by Lavery and Nelson from the Schiff’s base condensation of Ph2P(o-C6H4)C(᎐᎐O)H and H2N(CH2)2 -(o-C5H4N).2 Due to its flexible structure, PNN can act either as a monodentate phosphine, a bidentate phosphine–imine or a tridentate P–N–N ligand. A few PNN complexes of Pd(0), Pd(), Pt() and Group VI metals are known.3 However, the coordination chemistry of PNN with metal clusters has received little attention, though the related poly(pyridyl) cluster complexes were recently described by Wang and co-workers.4 Arising from our interest in the systematic chemistry of organic substrates bound to transition-metal clusters,5 this paper shows the results of an investigation of PNN reacting with [Os3 -(CO)12] and [Os3(CO)10(NCMe)2].
Experimental
General methods
All manipulations were carried out under an atmosphere of purified dinitrogen with standard Schlenk techniques.6 [Os3(CO)12]7 and [Os3(CO)10(NCMe)2]8 were prepared by liter-ature methods. Ph2P(o-C6H4)CH᎐᎐N(CH2)2(o-C5H4N) was syn-thesized from condensation of Ph2P(o-C6H4)C(᎐᎐O)H and NH2(CH2)2(o-C5H4N) (Aldrich) as described in the literature.2 Solvents were dried over appropriate reagents under dinitrogen and distilled immediately before use.9 Preparative thin-layer chromatographic (TLC) plates were prepared from silica gel (Merck). Infrared spectra were recorded with a 0.1 mm path CaF2 solution cell on a Hitachi I-2001 IR spectrometer. 1H and 31P NMR spectra were obtained on a Varian VXR-300 spec-trometer at 300 and 121.4 MHz, respectively and referenced to SiMe4 and 85% aqueous H3PO4. Fast-atom-bombardment (FAB) mass spectra were recorded by using a VG Blotch-5022
mass spectrometer with 3-nitrobenzyl alcohol as the matrix. Elemental analyses were performed at the National Science Council Regional Instrumentation Center at National Chen-Kung University, Tainan, Taiwan.
Reaction of [Os3(CO)10(NCMe)2] with PNN
An oven-dried, 50 cm3 Schlenk flask, equipped with a magnetic stir bar, was evacuated and refilled with dinitrogen three times. The stopper was briefly removed and [Os3(CO)10(NCMe)2] (205 mg, 0.22 mmol) and PNN (96 mg, 0.24 mmol) were added against a dinitrogen flow. Freshly distilled dichloromethane (20 cm3) was then introduced by means of a cannula through the septum stopper. The mixture was stirred at ambient temper-ature for 1 h, resulting in a color change from pale yellow to orange-red. The volatile materials were removed under vacuum, and the residue was subjected to TLC with dichloromethane–n-hexane (1 : 1, v/v) as eluant. The major orange band afforded {Os3(CO)10[η 2-Ph 2P(o-C6H4)CH᎐᎐N(CH2)2(o-C5H4N)]} 1 (137 mg, 0.11 mmol, 50%) (Found: C, 34.65; H, 1.98; N, 2.18. C36H23N2O10POs3 requires C, 34.72; H, 1.86; N, 2.25%): IR (CH2Cl2, cm⫺1) ν(CO) 2092m, 2040s, 2000vs, 1980s, 1960 (sh) and 1912w; mass spectrum (FAB) m/z 1250 (M⫹, 192Os) and 1250⫺ 28n (n = 1–10); 1H NMR (C 6D6, 20⬚C) δ 8.34 (d, JP–H= 5 Hz, CH᎐᎐N), 7.87–6.50 (m, 18H, C6H4, C5H4N), 4.96 (m, 1H), 4.54 (m, 1H), 2.62 (m, 1H) and 2.23 (m, 1H, CH2). 31P{1H} NMR (C 6D6, 20⬚C) δ 15.62 (s). Thermolysis of 1
An oven-dried, 50 cm3 two-necked Schlenk flask was equipped with a magnetic stir bar. One neck was stopped and the other neck had a reflux condenser connected to an oil bubbler. A solution of 1 (123 mg, 0.099 mmol) in dry toluene (6 cm3) was introduced into the flask, heated to reflux under dinitrogen for 2 h and cooled to ambient temperature, forming an orange-yellow solution and a brown-red precipitate. The solid was filtered off and recrystallized from dichloromethane–n-hexane to afford {(µ-H)Os3(CO)8[µ3-η
4-Ph
2P(o-C6H4)CH᎐᎐NCH2 CH-(o-C5H4N)]} 3 (61 mg, 0.051 mmol, 52%). The filtrate was dried under vacuum and the residue was subjected to TLC with
Table 1 Crystallographic data for compounds 1, 3–5 1 3 4 5 Formula Crystal solvent M Crystal system Space group a/Å b/Å c/Å α/⬚ β/⬚ γ/⬚ U/Å3 T/K Z µ/mm–1 R1, wR2 C36H23N2O10Os3P 1245.13 Triclinic P1¯ 10.979(1) 12.592(2) 14.214(3) 80.23(2) 72.02(2) 79.29(1) 1823.2(5) 293(2) 2 10.531 0.0353, 0.0929 C34H23N2O8Os3P 1188.11 Triclinic P1¯ 10.0659(1) 13.4957(1) 13.6217(1) 66.709(1) 84.669(1) 85.686(1) 1690.81(2) 295(2) 2 11.345 0.0313, 0.0544 C34H23N2O8Os3P 0.5 n-hexane 1231.19 Triclinic P1¯ 9.5720(1) 10.6385(2) 20.2534(2) 79.228(1) 88.699(1) 68.027(1) 1876.39(4) 295(2) 2 10.227 0.0310, 0.0729 C43H32N2O5Os2P2 1099.05 Orthorhombic Pbcn 18.8768(3) 21.023(2) 20.026(4) 7901(2) 295(2) 8 6.555 0.0358, 0.0570
dichloromethane–n-hexane (1 : 1, v/v) as eluant. Isolation of the material forming the fourth yellow band gave {(µ-H)Os3(CO)8 -[µ-η4-Ph
2P(o-C6H4)C᎐᎐N(CH2)2(o-C5H4N)]} 4 (5 mg, 4%). Isolation of the material forming the seventh pale yellow band afforded {Os3(CO)9[µ-η3-Ph2P(o-C6H4)CH᎐᎐N(CH2)2 -(o-C5H4N)]} 2 (5 mg, 4%). The remaining several minor bands (<1%) were not characterized.
Compound 2 (Found: C, 34.80; H, 1.87; N, 2.22. C35H23 -N2O9POs3 requires C, 34.54; H, 1.90; N, 2.30%): IR (CH2Cl2, cm⫺1) ν(CO) 2088s, 2048vs, 2000vs, 1990 (sh), 1972 (sh), 1964 (sh) and 1942m; mass spectrum (FAB) m/z 1222 (M⫹, 192Os) and 1250⫺ 28n (n = 1–9); 1H NMR (CD 2Cl2, 20⬚C) δ 8.44 (d, JP–H= 5 Hz, CH᎐᎐N), 7.95–7.10 (m, 18H, C6H4, C5H4N), 4.23 (m, 1H), 3.46 (m, 1H), 2.95 (m, 1H) and 1.29 (m, 1H, CH2); 31P{1H} NMR (CD 2Cl2, 20⬚C) δ 35.25 (s). Compound 3 (Found: C, 34.35; H, 2.03; N, 2.16. C34H23 -N2O8POs3 requires C, 34.34; H, 1.95; N, 2.36%): IR (CH2Cl2, cm⫺1) ν(CO) 2060s, 2008vs, 1988s, 1970w, 1954m, 1944s and 1916w; mass spectrum (FAB) m/z 1194 (M⫹, 192Os) and 1194⫺ 28n (n = 1–8); 1H NMR (CD 2Cl2, 20⬚C) δ 8.54 (d, JP–H= 5 Hz, CH᎐᎐N), 7.61–6.45 (m, 18H, C6H4, C5H4N), 5.08 (m, 1H), 4.09 (m, 1H), 3.56 (m, 1H, CH2) and ⫺10.35 (d, JP–H= 15 Hz, Os–H); 31P{1H} NMR (CD2Cl2, 20⬚C) δ 3.56 (s). Compound 4 (Found: C, 34.25; H, 2.34; N, 2.58. C34H23 -N2O8POs3 requires C, 34.34; H, 1.95; N, 2.36%): IR (CH2Cl2, cm⫺1) ν(CO) 2068s, 1986vs, 1974 (sh), 1956w, 1932w and 1912m; mass spectrum (FAB) m/z 1194 (M⫹, 192Os) and 1194⫺ 28n (n = 1–8); 1H NMR (C 6D6, 20⬚C) δ 7.61–5.60 (m, 18H, C6H4, C5H4N), 3.47 (m, 1H), 3.16 (m, 1H), 3.05 (m, 1H), 1.72 (m, 1H, CH2) and ⫺13.23 (d, JP–H= 7 Hz, Os–H); 31P{1H} NMR (C6D6, 20⬚C) δ 31.15 (s).
Pyrolysis of [Os3(CO)12] with PNN
[Os3(CO)12] (100 mg, 0.11 mmol) and PNN (100 mg, 0.25 mmol) were ground, mixed and sealed in Pyrex glass tubing under vacuum (0.01 Torr). The tube was placed in a silicon oil bath at 120⬚C for 40 min and then at 160 ⬚C for 30 min. The tube was removed from the bath, cooled to room temper-ature and opened in air. The brown residue was dissolved in dichloromethane, applied to a preparative TLC plate and eluted with dichloromethane–n-hexane (1 : 1, v/v). The unreacted [Os3(CO)12] (35 mg, 35%) was recovered from the first yellow band. Compound 3 (30 mg, 23%) was obtained from the ninth band. Compound 2 (5 mg, 4%) was obtained from the tenth band. Isolation of the material forming the eleventh pale yellow band gave {Os2(CO)5(µ-PPh2)[µ-η3-Ph2P(o-C6H4)C ᎐᎐N-(CH2)2(o-C5H4N)]} 5 (21 mg, 0.019 mmol, 17%). The remaining minor bands were not characterized.
Compound 5 (Found: C, 46.89; H, 3.17; N, 2.71. C43H32 -N2O5P2Os2 requires C, 46.99; H, 2.93; N, 2.55%): IR (CH2Cl2,
cm⫺1) ν(CO) 2048s, 1976s, 1952m and 1930m; mass spectrum (FAB) m/z 1102 (M⫹, 192Os) and 1102⫺ 28n (n = 1–5); 1H NMR (CD2Cl2, 20⬚C) δ 8.49–6.61 (m, 28H, C6H4, C5H4N), 4.07 (m, 1H), 3.32 (m, 1H), 2.90 (m, 1H) and 2.45 (m, 1H, CH2); 31P{1H} NMR (CD2Cl2, 20⬚C) δ 80.97 (d, JP–P= 15 Hz, µ-PPh2) and 31.60 (d, JP–P= 15 Hz, PNN).
Thermolysis of 2
A solution of 2 (15 mg) in toluene (5 cm3) was heated to reflux under dinitrogen for 12 h. The reaction was monitored inter-mittently by IR and TLC. Unreacted 2 (52%) and compound 4 (12%) were obtained after separation by TLC, while no evidence supported the formation of compound 3.
Thermolysis of 3 and 4
Typically, 10 mg of 3 (or 4) was dissolved in 5 cm3 of dry toluene and heated to reflux under dinitrogen for 5 h, showing no reaction as evidenced by IR and TLC. This indicates that compounds 3 and 4 are not interconvertible.
Structure determination for 1 and 5
A crystal of 1 (ca. 0.45 × 0.40 × 0.40 mm) and 5 (ca. 0.40 × 0.25 × 0.10 mm) were each mounted in a thin-walled glass capillary and aligned on the Nonius CAD-4 diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.7107 Å). All data were corrected for Lorentz and polarization effects and for the effects of absorption. The structure was solved by direct methods and refined by full-matrix least-squares cycles on F2 on the basis of 5180 observed reflections (I > 2σ(I)) for 1 and 4243 observed reflections for 5. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included but not refined. All calculations were performed using the SHELXTL package.10a A summary of relevant crystallographic data is provided in Table 1.
Structure determination for 3 and 4
A crystal of 3 (ca. 0.20 × 0.12 × 0.03 mm) and 4 (ca. 0.35 × 0.13 × 0.08 mm) were each mounted in a thin-walled glass capil-lary and aligned on the Siemens SMART-CCD diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). 20361 reflections were measured and 6888 reflections (Rint= 0.0461) were unique for 3, while 23505 reflections were measured and 7633 reflections (Rint= 0.0341) were unique for 4. Sadabs absorption corrections10b were made for 3 (T
min= 0.2931, Tmax= 0.4921) and 4 (Tmin= 0.1440, Tmax= 0.2627). The structures were solved by direct methods and refined by full-matrix least-squares on F2. The program used was the SHELXTL package.10a The data collection and refinement parameters are collected in Table 1.
Scheme 1 CCDC reference number 186/1901.
See http://www.rsc.org/suppdata/dt/a9/a910260o/ for crystal-lographic files in .cif format.
Results and discussion
Reactions
Treating [Os3(CO)10(NCMe)2] with equimolar amounts of PNN at room temperature results in the facile substitution of the labile acetonitrile ligands by the phosphine and imine groups of PNN to generate {Os3(CO)10[η2-Ph2P(o-C6H4 )-CH᎐᎐N(CH2)2(o-C5H4N)]} 1 in 50% yield. Compound 1 remains intact to 80⬚C, while in refluxing toluene (110 ⬚C) it decomposes to generate several products. After separation by TLC, three major complexes are purified for characteriz-ation, namely {Os3(CO)9[µ-η3-Ph2P(o-C6H4)CH᎐᎐N(CH2)2 -(o-C5H4N)]} 2 (4%), {(µ-H)Os3(CO)8[µ3-η
4-Ph
2P(o-C6H4)CH᎐᎐ NCH2CH(o-C5H4N)]} 3 (52%) and {(µ-H)Os3(CO)8[µ-η4 -Ph2P(o-C6H4)C᎐᎐N(CH2)2(o-C5H4N)]} 4 (4%). Thermolysis of 1 in the presence of PNN gives the same products. A mechanistic study was taken to trace the origins of 3 and 4, such that heating pure 2 in refluxing toluene led to 4 but not to 3, and thermolysis of 3 (or 4) at 110⬚C did not generate 4 (or 3). The results, summarized in Scheme 1, imply that compounds 3 and 4 are formed via separate routes, and compound 2 is the precursor of 4.
Heating PNN and [Os3(CO)12] in refluxing toluene for 6 h affords 2, 3 and 4 in low yields, while co-pyrolysis of PNN with [Os3(CO)12] in a sealed tube results in 2, 3 and a diosmium phosphido complex {Os2(CO)5(µ-PPh2)[µ-η3-Ph2P(o-C6H4 )-C᎐᎐N(CH2)2(o-C5H4N)]} 5 (17%) as the major products (eqn.
(1)). For the latter reaction, formation of 5 is likely to involve coordination and rearrangement of two PNN ligands accom-panied by cluster fragmentation from Os3 to Os2 under harsh conditions.
Characterization of new compounds
Compounds 1–5 form air-stable crystalline solids which have been characterized by elemental analyses (C, H, and N), mass, IR and NMR. The FAB mass spectra of these complexes dis-play their molecular ion peaks and fragments resulting from successive loss of CO ligands. The isotopic distribution of the envelope surrounding the molecular ion matches that calcu-lated for each compound.
The 1H NMR spectrum of free PNN in CDCl
3 presents a doublet resonance at δ 9.01 (JP–H= 5 Hz) for the CH᎐᎐N proton and two 2H triplets at δ 3.93 and 3.04 (JH–H= 7 Hz) for the (CH2)2 protons, while its 31P{1H} NMR spectrum shows a sing-let at δ ⫺13.08 for the phosphine group. Upon coordination to
the osmium carbonyl clusters, the phosphine 31P resonances are shifted downfield to δ 3.56–35.25, and the imine CH᎐᎐N proton resonances are shifted upfield to ca. δ 8.4. Furthermore, the (CH2)2 proton resonances are split into four 1H multiplets (except in 3) in the range δ 4.9–1.3, indicating asymmetric coordination of the PNN ligands on the clusters to result in diastereotopic methylene groups.
The molecular ion peaks of 1 and 2 at m/z 1250 and 1222 suggest an [Os3(CO)10(PNN)] and an [Os3(CO)9(PNN)] form-ula, respectively. On the basis of the 1H and 31P NMR data, the PNN ligand of 1 likely forms a phosphine–imine chelate and of 2 donates its P,N,N set to result in a total of 48 valence electrons for the clusters.11 The IR absorptions in the carbonyl region for 1 and 2 are shifted to lower energy compared with [Os3(CO)12], consistent with the stronger net donor capability of the PNN compared with CO.12
Compounds 3 and 4 form brown-red and yellow crystals, respectively. Their mass spectra present identical molecular ion peaks at m/z 1194, which is 28 less than that of 2 to imply a formal [Os3(CO)8(PNN)] formula. The
1H NMR spectrum of 3 shows a doublet at δ 8.54 for the CH᎐᎐N proton, three 1H multi-plets at δ 5.08, 4.09 and 3.56 for the methylene protons, and a doublet at δ ⫺10.35 for the hydride resonance. In contrast, the 1H NMR spectrum of 4 shows four 1H multiplets in the range
δ 3.47–1.72 for the methylene protons and a hydride resonance at δ ⫺13.23, while the CH᎐᎐N resonance is absent. It appears that compounds 3 and 4 are configurational isomers with the methylene and the imine C–H bonds of the PNN ligand being activated by the Os3 cluster. An X-ray diffraction study was thus conducted to elucidate the exact way in which the ligands are bound to the cluster in 3 and 4.
The mass spectrum of 5 shows the parent ion at m/z 1102 plus ions corresponding to the successive loss of 5 carbonyls. The isotopic distribution surrounding the molecular ion peak matches an Os2 composition. The 1H NMR spectrum indicates the presence of PNN but without the CH᎐᎐N proton resonance. The 31P{1H} NMR spectrum reveals two doublet signals at
δ 31.60 and 80.97 (JP–P= 15 Hz) in about equal intensities. The former 31P resonance, assigned to a phosphine group, is com-patible with that of 4, while the latter resonance falls in the range measured for a bridging phosphido (µ-PPh2) moiety.13 The spectroscopic data suggest an [Os2(CO)5(PNN–H)(PPh2)] formula for 5. Owing to the absence of diagnostic spectral features to reveal the structure of 5, an single-crystal X-ray diffraction study was performed.
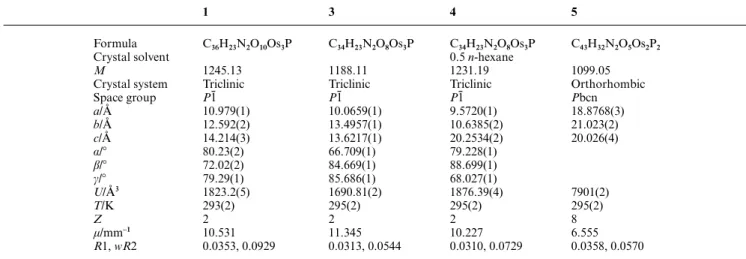
Crystal structure of 1
An ORTEP diagram of 1 is shown in Fig. 1. Selected bond distances and bond angles are collected in Table 2. There are no abnormally short intermolecular contacts. The molecule is formally derived from [Os3(CO)12], but with two carbonyls being replaced by the phosphine and imine groups of PNN. The osmium–osmium bonds show substantial variation, with Os(1)–Os(2)= 2.9232(8) Å, Os(1)–Os(3) = 2.8829(6) Å and Os(2)–Os(3)= 2.8933(7) Å, which are longer than the values found in the parent compound [Os3(CO)12] (Os–Os(av)= 2.877(3) Å).14
The carbonyl ligands are terminally bonded to the Os3 cluster with the Os–C–O angles in the range 172.7(8)–179.5(9)⬚. The Os(2) and Os(3) atoms are each linked to four carbonyl ligands with the Os–CO lengths ranging from 1.88(1) Å to 1.95(1) Å, while two carbonyls are associated with the Os(1) atom showing a shorter Os–CO bond length of 1.85(1) Å. This can be attrib-uted to enhancement of the Os(1)←CO back-donation by the stronger net electron-donating phosphine and imine groups.
The PNN ligand chelates the Os(1) atom with the bite angle N(1)–Os(1)–P(1)= 81.3(2)⬚. The pyridyl group is pendent and pointed away from the cluster. The bulky phosphine group is in the more opened equatorial position with Os(1)–P(1)= 2.308(2)
Å, while the imine group takes up an axial position with Os(1)–N(1)= 2.204(7) Å. The P(1), C(11), C(16) and C(17) atoms are coplanar to within ±0.01 Å, and the dihedral angle between this plane and the P(1)–Os(1)–N(1) plane is 50.42⬚. The C(17)–N(1) distance of 1.28(1) Å retains a C᎐᎐N double bond character, while the C(18)–N(1) single bond distance is 1.49(1) Å.
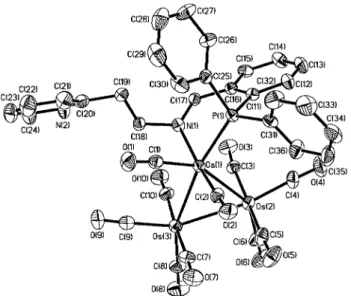
Crystal structure of 3
An ORTEP diagram of 3 is shown in Fig. 2. Selected bond distances and bond angles are collected in Table 3. The tri-metallic parts are based on a triangular array of osmium atoms in which the Os(1)–Os(2) distance of 3.0229(3) Å is significantly longer than the other two intermetallic distances, i.e. Os(1)– Os(3)= 2.8085(4) Å and Os(2)–Os(3) = 2.8535(4) Å. The Os(2), Os(1) and Os(3) atoms are each associated with two, three and three terminal carbonyl ligands. The Os–CO distances range from 1.863(7) Å through 1.924(8) Å, and the Os–C–O angles are in the range 175.1(7)–178.4(8)⬚. The equatorial carbonyl C(1), C(2), C(5) and C(7) atoms lie approximately on the tri-osmium plane and the C(8) atom is bent towards the pyridyl group with a Os(1)–Os(2)–Os(3)–C(8) torsion angle of 34.22⬚. The hydride atom was not located, but the distribution of carbonyl groups indicates that it probably binds the Os(3) atom (or bridges the Os(3)–Os(2) edge) to provide an 18-electron configuration for each Os atom.
The PNN ligand bridges the three Os atoms. The phosphine and imine groups chelate the Os(2) atom with the distances P(1)–Os(2)= 2.324(2) Å, N(2)–Os(2) = 2.159(5) Å and a bite angle P(1)–Os(2)–N(2)= 88.4(1)⬚. The pyridyl group replaces an axial carbonyl of Os(1) with the distance N(1)–Os(1)= 2.195(5) Å and the angles N(1)–Os(1)–C(3)= 176.0(3)⬚, N(1)– Os(1)–Os(2)= 95.5(1)⬚ and N(1)–Os(1)–Os(3) = 85.5(1)⬚. The methylene C(14) atom is metallated on the Os(3) atom. The Os(3)–C(14) length of 2.204(6) Å is characteristic of an Os– C(sp3) σ bond.
Fig. 1 ORTEP17 drawing of 1. Thermal ellipsoids are drawn at the 30% probability level.
Table 2 Selected bond distances (Å) and bond angles (⬚) for 1 Os(1)–Os(2) Os(2)–Os(3) Os(1)–P(1) N(1)–C(18) Os(1)–Os(2)–Os(3) Os(2)–Os(1)–Os(3) C(17)–N(1)–C(18) 2.9232(8) 2.8933(7) 2.308(2) 1.491(10) 59.42(2) 59.77(2) 112.8(7) Os(1)–Os(3) Os(1)–N(1) N(1)–C(17) Os(1)–Os(3)–Os(2) N(1)–Os(1)–P(1) C(16)–C(17)–N(1) 2.8829(6) 2.204(7) 1.283(10) 60.80(2) 81.3(2) 128.4(8)
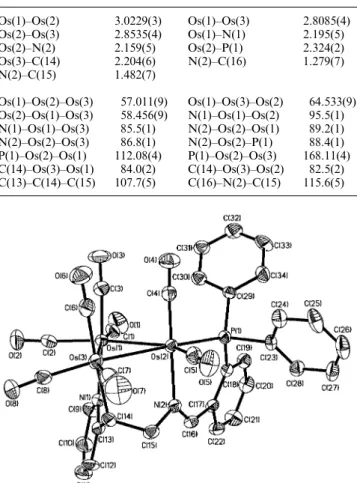
Crystal structure of 4
An ORTEP diagram of 4 is shown in Fig. 3. Selected bond distances and bond angles are collected in Table 4. The mole-cule consists of a triangular array of osmium atoms in which the individual bond lengths are Os(1)–Os(2)= 2.9772(3) Å, Os(1)–Os(3)= 2.8319(4) Å and Os(2)–Os(3) = 2.8701(4) Å. The hydride ligand was not located directly, but the lengthened Os(1)–Os(2) distance and the orientation of carbonyls indicates that it likely bridges the Os(1)–Os(2) edge. The Os(1) and Os(2) atoms are each linked to two terminal carbonyl ligands, and Os(3) has four. The Os–C–O angles are in the range 175.1(7)– 179.4(7)⬚. The Os–CO distances are diverse, ranging from 1.858(7) Å to 1.956(8) Å.
The imine C(16)–H bond of the PNN ligand has been acti-vated by the Os(2) atom. In contrast to 1 and 3, the imine and pyridyl groups chelate the Os(1) atom with a bite angle N(1)– Os(1)–N(2)= 80.1(2)⬚. The C(16)–N(2) bond is parallel to the Os(1)–Os(2) edge, forming a σ bond to Os(2) (2.100(6) Å) and a dative bond to Os(1) (2.130(5) Å). The dihedral angle between the triosmium plane and the Os(1)–Os(2)–C(16)–N(2) plane is 71.73⬚. The C(16)–Os(2) distance of 2.100(6) Å is between those measured for Os–C σ bonds (such as C(14)–Os(3) = 2.204(6) Å in 3) and Os᎐᎐C double bonds (2.04–2.07 Å) in some carbene systems,15 suggesting a partial double bond feature. The phosphine P(1) and pyridine N(1) atoms are in the equatorial positions to bind Os(2) (2.317(2) Å) and Os(1) (2.166(5) Å), respectively. However, the torsional angles P(1)–Os(2)–Os(1)– Os(3)= 166.01⬚, N(1)–Os(1)–Os(2)–Os(3) = 178.40⬚ and N(1)– Os(1)–Os(2)–P(1)= 12.39⬚ indicate that the P(1) atom is bent away from the Os3 plane.
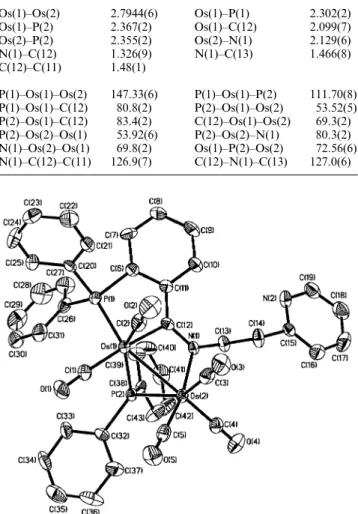
Crystal structure of 5
An ORTEP diagram of 5 is shown in Fig. 4. Selected bond Fig. 2 ORTEP drawing of 3. Thermal ellipsoids are drawn at the 30% probability level.
Table 3 Selected bond distances (Å) and bond angles (⬚) for 3 Os(1)–Os(2) Os(2)–Os(3) Os(2)–N(2) Os(3)–C(14) N(2)–C(15) Os(1)–Os(2)–Os(3) Os(2)–Os(1)–Os(3) N(1)–Os(1)–Os(3) N(2)–Os(2)–Os(3) P(1)–Os(2)–Os(1) C(14)–Os(3)–Os(1) C(13)–C(14)–C(15) 3.0229(3) 2.8535(4) 2.159(5) 2.204(6) 1.482(7) 57.011(9) 58.456(9) 85.5(1) 86.8(1) 112.08(4) 84.0(2) 107.7(5) Os(1)–Os(3) Os(1)–N(1) Os(2)–P(1) N(2)–C(16) Os(1)–Os(3)–Os(2) N(1)–Os(1)–Os(2) N(2)–Os(2)–Os(1) N(2)–Os(2)–P(1) P(1)–Os(2)–Os(3) C(14)–Os(3)–Os(2) C(16)–N(2)–C(15) 2.8085(4) 2.195(5) 2.324(2) 1.279(7) 64.533(9) 95.5(1) 89.2(1) 88.4(1) 168.11(4) 82.5(2) 115.6(5)
distances and bond angles are collected in Table 5. This com-plex consists of Os(CO)2 and Os(CO)3 units connected by a three-electron-donating diphenylphosphido group and a five-electron-donating metallated PNN ligand, resulting in a total of 34 valence electrons. This electron count requires one metal– metal bond to provide an 18-electron configuration on each osmium atom, in agreement with the observed Os(1)–Os(2) distance (2.7944(6) Å). In addition, on the basis of the angles subtended, the coordination about both Os(1) and Os(2) atoms can be described as distorted octahedral.
The phosphido group bridges the two osmium atoms about equally, being 2.367(2) Å to Os(1) and 2.355(2) Å to Os(2). The Os(1) and Os(2) atoms are each associated with two and three terminal carbonyl ligands, with the Os–C distances ranging from 1.87(1) to 1.92(1) Å and the Os–CO angles in the range 176.2(8)–179.3(8)⬚. The PNN ligand is connected to Os(1) through the phosphine P(1) atom (2.302(2) Å) and the imine C(12) atom (2.099(7) Å), and to Os(2) through the imine N(1) atom (2.129(6) Å). The Os(1), Os(2), N(1) and C(12) atoms are planar to within 0.05 Å, and the Os(1), P(1), C(6), C(11) and C(12) atoms are to within 0.1 Å, giving a dihedral angle of 13.11⬚ between the two planes. Presumably, this coplanar feature results in delocalization of the C᎐᎐N π electrons, such that the C(12)–N(1) length (1.326(9) Å) is 0.03 Å longer than the value measured for 4.
Probable reaction mechanism
It is apparent that compound 2 is formed from substitution of a CO ligand by the pendent pyridyl group of 1, and compounds 3 and 4 arise from C–H activation of the PNN ligand on the Os3 cluster together with CO loss.
Compound 1 is thermally stable to 80⬚C in either solid or solution state. At higher temperatures, however, decarbonyl-Fig. 3 ORTEP drawing of 4. Thermal ellipsoids are drawn at the 30% probability level.
Table 4 Selected bond distances (Å) and bond angles (⬚) for 4 Os(1)–Os(2) Os(2)–Os(3) Os(1)–N(1) Os(2)–C(16) N(2)–C(15) Os(1)–Os(2)–Os(3) Os(2)–Os(1)–Os(3) N(1)–Os(1)–Os(3) N(2)–Os(1)–Os(3) P(1)–Os(2)–Os(1) C(16)–N(2)–C(15) 2.9772(3) 2.8701(4) 2.166(5) 2.100(6) 1.475(9) 57.900(9) 59.154(9) 168.5(1) 93.1(1) 116.90(4) 123.6(5) Os(1)–Os(3) Os(1)–N(2) Os(2)–P(1) N(2)–C(16) C(16)–C(17) Os(1)–Os(3)–Os(2) N(1)–Os(1)–Os(2) N(2)–Os(1)–Os(2) N(2)–Os(1)–N(1) P(1)–Os(2)–Os(3) N(2)–C(16)–C(17) 2.8319(4) 2.130(5) 2.317(2) 1.294(8) 1.485(9) 62.947(9) 109.4(1) 67.3(1) 80.1(2) 166.77(4) 123.8(6)
ation might occur to generate an unsaturated species [Os3(CO)9 -(η2-PNN)], which is followed by coordination of the pendent pyridyl group to generate [Os3(CO)9(η3-PNN)]. Since heating 1 in the presence of free PNN gives the same results, the reaction is presumably governed by the chelate effect. Two configur-ational isomers 2 and 2⬘ can be drawn to account for [Os3 -(CO)9(η3-PNN)]. The structure of 2⬘ is related to 1 with a P–N chelate, while of 2 the imine group has moved to the adjacent Os atom to form a N–N chelate. Compound 2⬘ is likely to be the kinetic product, which then undergoes ligand migration to give 2 or C–H activation to lead to 3. It is probable the different arrangements of PNN ligands constrain the pyridyl α-CH2 moiety of 2⬘ and the imine H–C bond of 2 close to the cluster framework, and the subsequent C–H activation reactions of which afford 3 and 4, respectively. Apparently, compound 3 is more stable (less strained) than 4 and should be dominant, in agreement with the observed product distribution. However, since compound 2⬘ is not isolated, an alternative route via initial C–H activation of 1 followed by CO/pyridyl substitution to generate 3 cannot be excluded.
On the other hand, two PNN molecules might be added into the cluster in the co-pyrolysis of Os3(CO)12 and PNN, and sub-sequent cluster fragmentation from Os3 to Os2 accompanied by elimination of a PhCH᎐᎐N(CH2)2(C5H4N) species under harsh conditions could lead to 5, though no mechanistic information is available at this stage.
Fig. 4 ORTEP drawing of 5. Thermal ellipsoids are drawn at the 30% probability level.
Table 5 Selected bond distances (Å) and bond angles (⬚) for 5 Os(1)–Os(2) Os(1)–P(2) Os(2)–P(2) N(1)–C(12) C(12)–C(11) P(1)–Os(1)–Os(2) P(1)–Os(1)–C(12) P(2)–Os(1)–C(12) P(2)–Os(2)–Os(1) N(1)–Os(2)–Os(1) N(1)–C(12)–C(11) 2.7944(6) 2.367(2) 2.355(2) 1.326(9) 1.48(1) 147.33(6) 80.8(2) 83.4(2) 53.92(6) 69.8(2) 126.9(7) Os(1)–P(1) Os(1)–C(12) Os(2)–N(1) N(1)–C(13) P(1)–Os(1)–P(2) P(2)–Os(1)–Os(2) C(12)–Os(1)–Os(2) P(2)–Os(2)–N(1) Os(1)–P(2)–Os(2) C(12)–N(1)–C(13) 2.302(2) 2.099(7) 2.129(6) 1.466(8) 111.70(8) 53.52(5) 69.3(2) 80.3(2) 72.56(6) 127.0(6)
In summary, activation of the methylene and imine C–H bonds presented herein is unique because it is orthometallation of the phenyl and pyridyl groups which commonly occurs in related cluster reactions.16 Our observations illustrate that pri-marily geometrical factors control the structure of the products formed rather than the electron donor ability of the various donor groups in this potentially tetradentate ligand.
Acknowledgements
We are grateful for support of this work by the National Science Council of Taiwan.
References
1 B. Linton and A. D. Hamilton, Chem. Rev., 1997, 97, 1669; Y. Kobuke and Y. Satoh, J. Am. Chem. Soc., 1992, 114, 789; H.-J. Schneider and D. Ruf, Angew. Chem., Int. Ed. Engl., 1990, 29, 1159; N. Fujita, Acc. Chem. Res., 1999, 32, 53; P. D. Beer, Acc.
Chem. Res. 1998, 31, 71; J. Sprinz, M. Kiefer, G. Helmchen,
M. Reggelin, G. Huttner, O. Walter and L. Zsolnai, Tetrahedron
Lett., 1994, 35, 1523; P. von Matt and A. Pfaltz, Angew. Chem., Int. Ed. Engl., 1993, 32, 566.
2 A. Lavery and S. M. Nelson, J. Chem. Soc., Dalton Trans., 1984, 615. 3 P. Wehman, R. E. Rülke, V. E. Kaasjager, P. C. J. Kamer, H. Kooijman, A. L. Spek, C. J. Elsevier, K. Vrieze and P. W. N. M. van Leeuwen, J. Chem. Soc., Chem. Commun., 1995, 331; R. E. Rülke, V. E. Kaasjager, P. Wehman, C. J. Elsevier, P. W. N. M. van Leeuwen, K. Vrieze, J. Fraanje, K. Goubitz and A. L. Spek,
Organometallics, 1996, 15, 3022; C.-C. Yang, W.-Y. Yeh, G.-H. Lee
and S.-M. Peng, J. Organomet. Chem., 2000, 598, 353.
4 Y.-Y. Choi and W.-T. Wang, J. Chem. Soc., Dalton Trans., 1999, 331; Y.-Y. Choi and W.-T. Wang, J. Organomet. Chem., 1996, 542, 121.
5 W.-Y. Yeh, M.-A. Hsu, S.-M. Peng and G.-H. Lee, Organometallics, 1999, 18, 880; W.-Y. Yeh, C.-Y. Wu and L.-W. Chiou,
Organo-metallics, 1999, 18, 3547; W.-Y. Yeh, S. C. N. Hsu, S.-M. Peng and
G.-H. Lee, Organometallics, 1998, 17, 2477; M.-A. Hsu, W.-Y. Yeh, G.-H. Lee and S.-M. Peng, J. Organomet. Chem., 1999, 588, 32; M.-A. Hsu, W.-Y. Yeh, G.-H. Lee and S.-M. Peng, Inorg. Chim.
Acta, 1999, 294, 232.
6 D. F. Shriver and M. A. Drezdzon, The Manipulation of
Air-Sensitive Compounds, Wiley, New York, 2nd edn., 1986.
7 B. F. G. Johnson and J. Lewis, Inorg. Synth., 1972, 13, 92.
8 D. Braga, F. Grepioni, E. Parisini, B. F. G. Johnson, C. M. Martin, J. G. M. Nairn, J. Lewis and M. Martinelli, J. Chem. Soc., Dalton
Trans., 1993, 1891.
9 D. D. Perrin and W. L. F. Armarego, Purification of Laboratory
Chemicals, Pergamon, Oxford, 4th edn., 1996.
10 (a) G. M. Sheldrick, SHELXTL, University of Göttingen, 1993; (b) R. H. Blessing, Acta Crystallogr., Sect. A, 1995, 51, 33.
11 D. M. P. Mingos and A. May, in The Chemistry of Metal Cluster
Complexes, VCH, New York, 1990; S. M. Owen, Polyhedron, 1988,
7, 253.
12 J. P. Collman, L. S. Hegedus, J. R. Norton and R. G. Finke,
Principles and Applications of Organotransition Metal Chemistry,
University Science Books, Mill Valley, CA, 1987.
13 R. J. Haines, in Comprehensive Organometallic Chemistry II, Pergamon, Oxford, 1995, vol. 7, p. 625.
14 M. R. Churchill and B. G. DeBoer, Inorg. Chem., 1977, 16, 878. 15 J. R. Shapley, W.-Y. Yeh, M. R. Churchill and Y.-J. Li,
Organometallics, 1985, 4, 1898; W.-Y. Yeh, J. R. Shapley, J. W.
Ziller and M. R. Churchill, Organometallics, 1987, 6, 1; C. M. Jensen, C. B. Knobler and H. D. Kaesz, J. Am. Chem. Soc., 1984,
106, 5926.
16 K. A. Azam, C. C. Yin and A. J. Deeming, J. Chem. Soc., Dalton
Trans., 1978, 1201; G. A. Foulds, B. F. G. Johnson and J. Lewis, J. Organomet. Chem., 1985, 296, 147; W.-Y. Yeh, S.-B. Chen,
S.-M. Peng and G.-H. Lee, J. Organomet. Chem., 1994, 481, 183; A. E. Shilov and G. B. Shul’pin, Chem. Rev., 1997, 97, 2879. 17 M. N. Burnett and C. K. Johnson, ORTEP 3, Report ORNL-6895,