-bulge in the folding of the -hairpin
structure in ubiquitin
PEI-YEH CHEN,
1,2B.G. GOPALACUSHINA,
1,3CHIEN-CHIH YANG,
1,4SUNNEY I.CHAN,
2 ANDPHILIP A.EVANS
11
Cambridge Center for Molecular Recognition and Department of Biochemistry, University of Cambridge, Cambridge CB2 1QW, UK
2
Institute of Chemistry, Academia Sinica, Taipei, Taiwan, R.O.C.
(RECEIVEDFebruary 15, 2001; FINALREVISIONJuly 6, 2001; ACCEPTEDJuly 12, 2001)
Abstract
It is known that the peptide corresponding to the N-terminal-hairpin of ubiquitin, U(1–17), can populate the monomeric -hairpin conformation in aqueous solution.In this study, we show that the Gly-10 that forms the bulge of the-turn in this hairpin is very important to the stability of the hairpin.The deletion of this residue to desG10(1–16) unfolds the structure of the peptide in water.Even under denaturing conditions, this bulge appears to be important in maintaining the residual structure of ubiquitin, which involves tertiary interactions within the sequence 1 to 34 in the denatured state.We surmise that this residual structure functions as one of the nucleation centers in the folding process and is important in stabilizing the transition state.In accordance with this idea, deleting Gly-10 slows down the refolding and unfolding rate by about one half.
Keywords: Ubiquitin; hairpin; bulge; turn; peptide; structure; folding; stability; kinetics
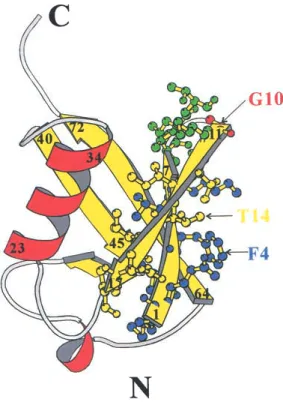
Ubiquitin is an excellent paradigm for protein folding.It is tightly hydrogen-bonded: 87% of the polypeptide chain is involved in a hydrogen-bonded secondary structure that consists of a 5-strand mixed -sheet (1–7, 11–17, 40–45, 48–50, 64–72), 3.5 turns of an␣-helix (23–34), a short piece of 310helix (56–59), and seven reverse turns (7–11, 18–21, 37–40, 45–48, 51–54, 57–60, 62–65) connecting the sec-ondary elements (Vijay-kumar et al.1987) (Fig.1).The N-terminal -hairpin of ubiquitin is one of the few cases where a short peptide can populate the monomeric-hairpin conformation in aqueous solution (Cox et al.1993).
The structure of the N-terminal-hairpin of ubiquitin is shown in Fig.1.There are six residues on each strand and
a 3:5 type “5-residue turn” in between.This turn is a type I
-turn plus a G1-type -bulge. -Bulges are a common
feature of-sheets.The -bulge is caused by an extra resi-due on the bulged strand, which increases the backbone length, disrupts the hydrogen bonding, and causes the strand to bulge out of the plane of the sheet.They thus provide a mechanism to change the direction of the-strand and the orientation of the sidechains.These features suggest that bulges might have a special role in the folding, structural stability, and function of the protein.For example, Axe et al. highlighted the importance of a-bulge in the stability and function of barnase (Axe et al.1999).
-Bulges are classified into five types based on protein
backbones and hydrogen-bonding diagrams: classic, G1, wide, bent, and special (Chan et al.1993).The distortions produced by the special, bent, and G1 bulges are the most pronounced.About 50% of the bulges found so far belong to the classic type.The next most common class is the G1 bulge, which is found only at the edges of antiparallel
-sheets and is usually associated with -hairpin turns
(Mil-ner-white and Poet 1987).The first of the two residues forming a G1 bulge is typically in a␥Lor␣Lconformation; Reprint requests to: Sunney I.Chan, Institute of Chemistry, Academia
Sinica, Taipei, 11529, Taiwan, R.O.C.; e-mail: chans@chem.sinica.edu.tw; fax: 886-2-2783-1237.
3Present address: National Institute of Pharmaceutical Education and Research, Punjab, India.
4Present address: Department of Agricultural Chemistry, National Tai-wan University, Taipei, TaiTai-wan, R.O.C.
Article and publication are at http://www.proteinscience.org/cgi/doi/ 10.1101/ps.07101.
accordingly, there is a strong preference for this position to be occupied by residues tolerant of this conformation.Gly is by far the predominant residue, hence the name of the class. The second position of the G1 bulge seems to be favored by charged groups, including Arg, Lys, and Asp.It is a Lys that is occupying this position in the G1 -bulge of the N-ter-minal hairpin of ubiquitin.
The N-terminal sequence of ubiquitin has many residues, for example, Val, Thr, Ile, and Phe, with high propensity toward-sheet formation.However, the -hairpin stability of the N-terminal peptide here is significantly higher than that of other peptides with similar-structure-forming pro-pensities [cf.sequence 1–20 of Streptococcal protein-G B1 domain (Blanco and Serrano 1995) and sequence 1–20 of ferredoxin (Searle et al.1996)].This raises the question as to whether or not the structural stability of ubiquitin peptide (1–17), U(1–17), is also augmented by the residues in the special bulge turn.Addressing this issue, Searle et al.re-placed the native turn-sequence (TLTGK) in the-hairpin with the four-residue sequence NPDG, with the intent of maximizing the stability of the -turn.Unexpectedly, this one amino-acid frameshift in the alignment of the peptide main chain led to a strand flip in the stem of the hairpin, and a new bulge turn-sequence (NPDGT) was established
(Searle et al.1995).This result suggests that the nativelike register of the side chains is not critical for hairpin stability of the peptide fragment.It also highlights the importance of the G1-bulge in maintaining the hairpin structure.
In addition to the work of Searle and coworkers, de Alba et al.have designed a peptide (sequence IYSNPDGTWT) that forms a hairpin with a type I-turn plus a G1 -bulge in the sequence NPDGT as well (de Alba et al.1996).Taken together, these data suggest that this turn type might be especially stable and favors the formation of a hairpin.Not surprisingly, it has been suggested that the hairpin of ubiq-uitin is stabilized by the TLTGK turn.
The type I turn with the G1 -bulge is actually quite common.Interestingly, in addition to ubiquitin, it is also found in hairpins very close to the N-termini of a few pro-teins; for example, the sequence SLQDKTGFHFC in
␥-chymotrypsin A (32–42) (PDB entry name 2GCH), the
sequence FRKAADDTWEP in prealbumin (33–43) (PDB entry name 2PAB), the sequence IYKDTEGYYTI in T4 lysozyme (17–27) (PDB entry name 2LZM), and the se-quence VGRGVLGDQKN in thermolysin (9–19) (PDB en-try name 8TLN) (Sibanda and Thornton 1991).In light of these observations, it is possible that this special turn type has a role in the folding of these proteins.
In the present study, we sought to establish the impor-tance of the-bulge for stabilizing the hairpin in ubiquitin, and by implication, in the other proteins alluded to above. Gly-10 in position 1 of the bulge was deleted from the turn sequence in ubiquitin, and the effects of the mutation on the whole protein and the N-terminal peptide fragments were examined.The structural differences between the wild-type ubiquitin and the Gly-10 deletion mutant, in the whole pro-tein and in the peptide fragment, were studied using two-dimensional nuclear magnetic resonance (2D-NMR).Equi-librium studies of GdnHCl denaturation were also used to compare the stability of the proteins.NMR magnetization transfer and GdnHCl titration experiments were employed to detect the existence of any residual structure in the pres-ence of high denaturant concentrations.Finally, the role of this residual structure in the folding and unfolding kinetics was investigated.
Results
Peptides
Initially, an E18D mutation was introduced in ubiquitin to generate an N-terminal fragment E18D(1–18), MQIFVK TLTGKTITLEVD, from the expressed protein by mild-acid cleavage of the introduced Asp-Pro linkage between se-quences 18 and 19.Our strategy was to engineer the G10 deletion in this protein expression system and to obtain the N-terminal mutant peptide desG10E18D(1–17) in exactly the same manner.Unfortunately, deleting Gly-10
signifi-Fig. 1. Ribbon diagram of ubiquitin structure produced using the program MOLSCRIPT (Kraulis 1991).-Strands and ␣-helices are displayed in yellow and red, respectively.The residues in the N-terminal-hairpin are displayed by ball-and-stick figures.The first strand, the turn region, and the second strand are shown in blue, green, and yellow, respectively.The G1
cantly decreased the solubility of the N-terminal mutant peptide desG10E18D(1–17).Following its cleavage from the expressed desG10/E18D protein, the yield of the peptide after purification was also insufficient for NMR experi-ments.Accordingly, we resorted to solid-phase peptide syn-thesis to prepare the desG10(1–16) peptide for our NMR structural studies.
Structure of desG10(1–16)
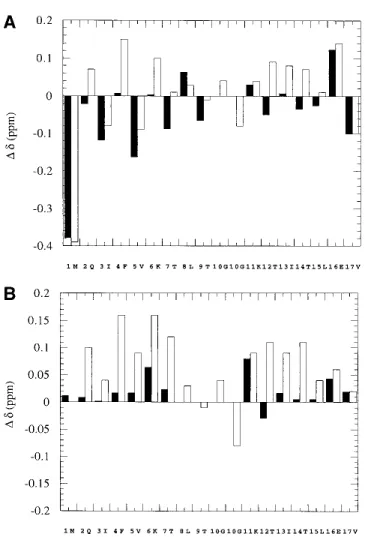
The solubility of the desG10(1–16) peptide was found to be much lower than that of the wild-type peptide, U(1–17).The crosspeaks in the NMR spectra were also much less dis-persed.We compared the C␣-proton chemical shift devia-tions of desG10(1–16) from their estimated unfolded state values (Bundi and Wu¨thrich 1979; Wishart et al.1991) with those of the synthetic wild-type peptide U(1–17) (Zerella et al.1999) (Fig.2A).It is evident from this comparison that the C␣-proton chemical shifts of residues 2–7 and 12–15 in desG10(1–16) are very different from those in U(1–17). Except in a few cases such as Ile-3 and Val-5, where the values are upfield shifted, the C␣-proton chemical shifts in desG10(1–16) are close to the literature random coil values. Zerella et al.compared these C␣-proton chemical shifts of U(1–17) with the synthetic peptides U(1–7) and U(11–17) instead of referring them to the random coil values in the literature, and showed that these upfield shifts in Ile-3 and Val-5 are actually due to sequence and conformational ef-fects (Zerella et al.1999).In the same manner, if the C␣ -proton shifts observed for desG10(1–16) and U(1–17) are referred to the corresponding values in the peptides U(1–7) and U(11–17), U(1–17) shows generally downfield shifts in the strand regions, whereas those observed for desG10(1– 16) are close to the random coil values (Fig.2B).Based on these spectral results, we conclude that the desG10(1–16) peptide does not adopt a nativelike conformation with well defined secondary structure, but rather exist largely as an unfolded structure.
The ␥-methyl proton chemical shifts of Thr-7, -9, -12, and -14 are compared between desG10(1–16) and U(1–17) in Table 1.It is clear from these data that the threonines in the turn region generally have ␥-methyl proton chemical shifts that are close to the random coil value of 1.2 ppm. In contrast, the ␥-methyl proton chemical shifts of the thre-onines in the strand region are upfield shifted.The␥-methyl protons of Thr-12 and -14, for example, appear significantly upfield in the spectra of U(1–17).The most likely origin of these upfield shifts would be the ring current effect of Phe-4, as mutating Phe-4 to Trp yielded an even more upfield shift of the␥-methyl protons of Thr-14 (data not shown).It is known that when Thr-9 is mutated to Asp, the peptide forms an even more stable hairpin, and concomitantly, the
␥-methyl protons of Thr-12 and Thr-14 become further
up-field shifted (Zerella et al.2000).Thus, the threonine
␥-methyl proton chemical shifts provide a good indicator
for the formation of the hairpin structure.When these indi-cators are applied to the desG10(1–16) peptide, we find that the corresponding upfield shifts of the␥-methyl protons of Thr-12 and Thr-14 in desG10(1–16) are grossly attenuated, consistent with unfolding of the hairpin when Gly-10 is excised from the peptide.
Structure of desG10/E18D
To compare the structures of the various mutant proteins with the wild-type ubiquitin, we recorded a two-dimen-sional total correlation spectroscopy (2D-TOCSY)
spec-Fig. 2. (A) Deviations of the C␣H chemical shifts of desG10(1–16) (black bar) and U(1–17) (clear bar) from literature random coil values.(B) De-viations of the C␣H chemical shifts of desG10(1–16) (black bar) and U(1– 17) (clear bar) from the chemical shifts of U(1–7) and U(1–17) for the sequence 1–7 and 11–17 and literature random coil values for residues 8, 9, and 10.The sequence of U(1–17) was adopted in the x-axis.The two
␣-protons of Gly-10 in U(1–17) are shown separately.There is no Gly-10
in desG10(1–16).⌬␦ > 0 for downfield shifts.The pH of the solutions were 3.8 for U(1–17), U(1–7), and U(11–17), and 1.9 for desG10(1–16) to take advantage of improved solubility at this pH.The chemical shifts observed are independent of pH over the pH of interest in this study.
trum, and at least one two-dimensional nuclear Overhauser effect spectroscopy (2D-NOESY) spectrum for the wild-type ubiquitin and the E18D and desG10/E18D mutant pro-teins.
From these data, we found that the deviations of the C␣-proton chemical shifts for the E18D mutant protein from those for the wild-type ubiquitin are generally within 0.1 ppm (data not shown).The largest deviation, about 0.1 ppm, was observed for the␣-proton of Ser-20.Thus we conclude that the E18D mutation affects only the local structure near the mutation site and does not substantially affect other parts of the protein structure.
The chemical shifts of the C␣-protons for desG10/E18D are compared with those for the wild-type ubiquitin in Fig-ure 3.These results indicate that desG10/E18D maintains essentially a nativelike overall structure.Deleting Gly-10 changes the chemical shifts of the C␣-protons significantly only for residues Lys-6, Thr-7, Thr-9, Lys-11, and Val-70 (numbering according to the wild-type protein sequence). Although Val-70 is in the C-terminal strand, it lies close to
the turn of the N-terminal -hairpin in the three-dimen-sional fold of the wild-type protein.The remaining affected residues are also close to the turn.Thus, it appears that any tertiary structural perturbations arising from deletion of Gly-10 are largely localized in this mutant.
Equilibrium unfoldingstudies by spectrofluorometry
We now proceed to compare the structural stability of the three ubiquitins under study.In earlier work, it was dem-onstrated that the folding of ubiquitin was well accounted for by a two-state system based on tryptophan fluorescence of the F45W mutant protein (Khorasanizadeh et al.1993). In the present study, we exploited the fluorescence of Tyr-59 to monitor the conformational behavior of the wild-type, E18D, and desG10/E18D ubiquitins under study.Although the fluorescence of Tyr-59 is quenched by a deprotonated carboxyl group, probably that of Asp-52 in the native pro-tein, as the aspartate becomes protonated, a marked increase in the tyrosine fluorescence was observed (Jenson et al. 1980; Ibarra-Molero et al.1999).Accordingly, we carried out the equilibrium denaturation studies at pH 3 to take advantage of the tyrosine fluorescence.No pH-induced transitions have been noted in ubiquitin between pH 8.48 and 1.18 (Lenkinski et al. 1977).
The denaturation curves of the proteins are shown in Figure 4, where the data are normalized to the maximum intensity in each case.From these data, the⌬GH2O, m value, and Cm were calculated and are given in Table 2.Neither mutant protein shows any significant differences from the wild-type protein in⌬GH2O, m value, or Cm.These results are consistent with the data obtained from the NMR struc-tural studies (see below).
Table 1. Comparison of the C␥H chemical shifts of threonine 7, 9, 12 and 14 between the wild-type peptide and the Gly-10 deletion peptide Peptide C␥H chemical shift (ppm) Thr-7 Thr-9 Thr-12 Thr-14 U(1–17)a 1.20 1.20 1.16 1.12 desG10(1–16)b 1.217 1.204 1.183 1.184
aThe assignments of U(1–17) were adopted from Zerella et al.(1999). bThe NMR spectra of desG10(1–16) were recorded in aqueous solution (pH 1.9) at 280 K to take advantage of the enhanced solubility of the peptide at the lower pH.These C␥H chemical shifts are independent of pH over the pH range 1.18–8.48 examined.
Fig. 3. Deviations of the C␣H chemical shifts of desG10/E18D from those of ubiquitin.Proteins were dissolved in unbuffered H2O/D2O (9/1), and the pH value was adjusted to 7.2. Residues Val-5, Pro-19, Glu-24, Pro-37, Pro-38, and Gly-53 are not included.
Fig. 4. Fluorescence-detected GdnHCl denaturation of ubiquitin:䡺,
wild-type;䡬, E18D; 䉭, desG10/E18D.The protein samples were dissolved in 20 mM glycine buffer, pH3 with different concentrations of GdnHCl as described in Materials and Methods.
NMR titrations of the wild-type protein and desG10/E18D in the presence of denaturants
We also compared the structural stability of ubiquitin and its desG10/E18D mutant at pH 3 by NMR titration in the pres-ence of denaturants.One-dimensional (1D) NMR spectra of the wild-type and the desG10/E18D mutant in the presence of different concentrations of GdnDCl in D2O at pH 3 were recorded at 290 K (Figs.5 and 6).In these experiments, His-68 provided a very good indicator of the solution
con-formation, as the histidine H2 ring proton resonances are well resolved in the 1D spectrum in D2O.In the spectrum of the wild-type protein with GdnDCl close to Cm, the H2 protons of His-68 appear at 8.87 ppm in the native state, but in the denatured state, they become upfield shifted to 8.74 ppm.The chemical exchange between the native and dena-tured forms is slow on the NMR timescale.Thus, the change in these peak intensities around the midpoint of denaturation can be used to monitor the denaturation process.
The titration of the desG10/E18D and wild-type proteins revealed that the midpoint of the denaturation of the desG10/E18D protein is very close to 3.5 M, compared to near 3.9 M for the midpoint of the wild-type protein. This observation confirms the relatively small change in the pro-tein stability resulting from the G10 deletion, as deduced from the fluorescence studies.In contrast, the stability of the peptide segment is grossly affected by the same deletion (see above).
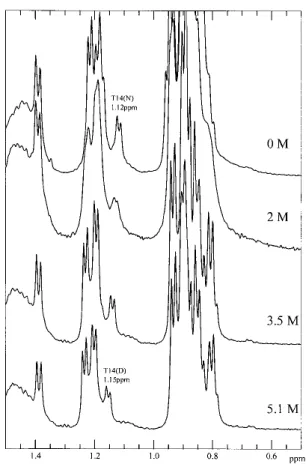
A striking result from these spectral studies is that the wild-type ubiquitin retains some structural features in the denatured state even at high denaturant concentrations.In the spectrum of the denatured ubiquitin, there remains con-siderable dispersion of the Thr␥-methyl peaks, even at 5.1 M GdnDCl.The ␥-methyl protons of the Thr residues are expected to resonate close to 1.2 ppm in a random coil peptide.However, one of the Thr␥-methyl peaks resonates at the unexpected high field position of∼1.15 ppm. It there-fore seems that there is some persistence of the hairpin
Table 2. Effects of different mutations on the protein stability of
ubiquitin as measured by GdnHCl denaturation
Proteina ⌬G H2O (kcal mol−1) m value (kcal mol−1M−1) Cm (M) wild-type −5.31 ± 0.46 1.42 ± 0.12 3.74 ± 0.32 E18D −5.39 ± 0.46 1.51 ± 0.13 3.57 ± 0.30 desG10/E18D −5.53 ± 0.43 1.58 ± 0.12 3.50 ± 0.27 aAll measurements were performed at room temperature in H
2O with different concentrations of GdnHCl (pH 3).The samples were excited at 277 nm and the tyrosine fluorescence emission was recorded at 303 nm. The free energy changes and m values were obtained by a nonlinear least-squares fit with the fitting errors noted.
Fig. 5. 1D-NMR spectra of wild-type ubiquitin as a function of GdnDCl
concentration, pH3 at 290 K.The GdnDCl concentrations are highlighted in the middle of the figure.
Fig. 6. 1D-NMR spectra of desG10/E18D as a function of GdnDCl
con-centration, pH3 at 290 K.The GdnDCl concentrations are highlighted in the middle of the figure.
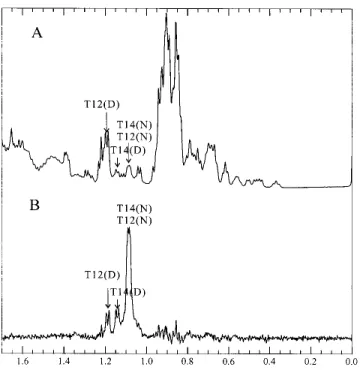
structure in the wild-type ubiquitin, even at high denaturant concentrations.In order to verify this, it was necessary to assign the shifted Thr␥-methyl peaks in the spectrum of the denatured state.We thus performed magnetization transfer experiments to try to obtain a cross-assignment from the native state spectrum (Evans et al.1991).
In the presence of 3.1 M GdnDCl at pH 3 in D2O, the wild-type ubiquitin is almost half denatured at 320 K.Under these conditions, the peaks arising from the␥-methyl pro-tons of Thr-12 and Thr-14 in the native state merge to a combined resonance at around 1.085 ppm. A 2D-NOESY of the wild-type protein under the same conditions revealed the expected crosspeaks between the C␣-proton and ␥-methyl protons and between the C-proton and␥-methyl protons of Thr-12 and Thr-14 in the native state, confirming the as-signment of the 1.085 ppm resonance to the Thr ␥-methyl protons (Fig.7).If the exchange between the native and denatured states is sufficiently rapid, presaturation of this peak will reduce the intensity of the peaks originating from the Thr-12 and Thr-14 ␥-methyl protons of the denatured protein.Figure 8 depicts the difference spectrum obtained from subtraction of the spectrum recorded upon presatura-tion of the Thr-12 and -14␥-methyl protons at 1.085 ppm from a control spectrum obtained upon presaturation in a blank region.In this manner, the peaks of the Thr-12 and Thr-14 ␥-methyl protons were located at 1.185 ppm and 1.14 ppm, respectively (Fig.8).We assigned the peak at 1.14 ppm to the Thr-14␥-methyl protons, because the 2D-spectrum of U(1–17) indicates that the␥-methyl protons of Thr-14 are more upfield shifted than those of Thr-12.Using diagonal subtraction, it was also possible to detect the chemical exchange crosspeaks between the “native” and the “denatured” ␥-methyl protons of Thr-12 and Thr-14, con-firming the 1D saturation transfer results above and cor-roborating that the peak at 1.14 ppm originates from the
␥-methyl protons of Thr-14 in the denatured state (Fig.7).
In contrast, the Thr␥-methyl protons are all shifted back close to 1.2 ppm in the 1D NMR spectrum of desG10/E18D in the denatured state, indicating that the residual structure observed for the “denatured” wild-type protein is essentially lost with the Gly-10 deletion (Fig.6).Similarly, we found that the desG10(1–16) peptide is essentially unfolded in aqueous solution.Taken together, these findings implicate a prominent role for Gly-10 in the-hairpin-forming propen-sity of the N-terminal hairpin segment of the wild-type pro-tein, in both its native and denatured states.Given that the stability of a protein at a given temperature is determined by the free energy difference between the native and denatured states, the rather marginal change in the stability of the protein derived from the desG10/E18D mutations, as in-ferred from the equilibrium NMR titration studies in this section, most likely arises from compensating effects of the hairpin destabilization on the free energy of both the folded and denatured states.
NMR titrations of peptides
To ascertain whether or not the residual structure in the denatured state of wild-type ubiquitin involves only local interactions, or both local interactions as well as longer range interactions, we recorded 1D NMR spectra of the two peptides U(1–17) and U(1–34), which correspond to the
-hairpin and the -hairpin plus the ␣-helix, respectively, in
D2O at pH 3 in the presence of different concentrations of GdnDCl at 290 K.At the denaturing GdnDCl of 5.1 M, the
␥-methyl protons of Thr-14 in the spectra of U(1–34) (Fig.
9) exhibit the same chemical shift (1.15 ppm) as they do in the spectrum of the full-length denatured protein (Fig.5). This observation suggests that residues from the C-terminal
Fig. 7. 2D-NOESY of ubiquitin in the vicinity of the midpoint of
dena-turation in the presence of 3.1 M GdnDCl, pH 3 at 320 K. The diagonal was suppressed by subtraction of two NOESY data sets acquired with different mixing times.
part of ubiquitin are not important in stabilizing these re-sidual interactions.In the spectra of U(1–17) (Fig.10), how-ever, the␥-methyl protons of Thr-14 are less upfield shifted, and appeared around 1.17 ppm even in the presence of only 4.4 M GdnDCl. Thus, the residues from the-hairpin alone are not enough to stabilize this residual structure in the presence of high concentrations of GdnHCl; in other words, this structural feature appears to require some interactions with the adjacent␣-helix for stabilization.It is remarkable that this embryonic tertiary interaction persists at the high denaturant concentrations studied.However, the deletion in the bulge of the hairpin destroys this embryonic structure.
Foldingand unfoldingkinetics
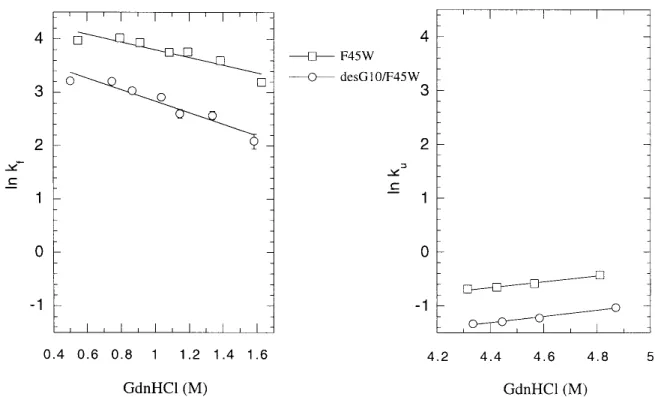
The F45W mutation was originally introduced to provide a better probe for kinetic studies of ubiquitin by Roder and his colleagues (Khorasanizadeh et al.1993).It turns out that this mutation does not significantly affect the structure and the stability of ubiquitin.In the present work, we adopted the same strategy to study the effect of deleting Gly-10 on the folding/unfolding kinetics of ubiquitin, taking advantage of the stronger emission intensity of tryptophan relative to that of tyrosine.The results on the refolding and the
unfold-ing kinetics are summarized in Figure 11.The refoldunfold-ing and unfolding rates are about one-half slower in desG10F45W than in F45W.We interpret this finding as supporting the notion that Gly-10 is important in stabilizing the transition state.Since Gly-10 is important in stabilizing the residual structure in the denatured state, we surmise that this residual structure is one of the nucleation sites in the folding path-way.If so, deleting Gly-10 should destabilize this residual structure, raise the free energy of the transition state, and retard the unfolding and refolding reactions.
The free energy diagrams for the unfolding and refolding of the wild-type (F45W) and the Gly-10 deletion mutant (desG10/F45W) are summarized in Figure 12.The kinetic data can be converted to the free energy scale using the following relationships: ⌬G‡-F(WT)=RT关ln共kBTⲐh兲–ln kU兴, ⌬G‡-F(M)=RT关ln共kBTⲐh兲–ln kU⬘兴, ⌬G‡-U(WT)=RT关ln共kBTⲐh兲–ln kF兴, ⌬G‡-U(M)=RT关ln共kBTⲐh兲–ln kF⬘兴, ⌬⌬G‡-F=⌬G ‡−⌬G F=⌬G‡-F共M兲 −⌬G‡-F共WT兲=RT ln共kUⲐkU⬘兲, ⌬⌬G‡-U=⌬G ‡−⌬G U=⌬G‡-U(M) −⌬G‡-U共WT兲=RT ln共kFⲐkF⬘兲, ⌬GU−⌬GF=⌬⌬G‡-F−⌬⌬G‡-U
where⌬G‡-Fand⌬G‡-Uare the activation free energies for unfolding and refolding, respectively; kU, kF, kU⬘, and kF⬘ are the rates of unfolding and refolding of the wild-type (F45W) and the mutant protein (desG10/F45W), respec-tively, obtained in the kinetic experiments; ⌬⌬G‡-F and
⌬⌬G‡-Udenote the free energy difference in the⌬G‡-Fand ⌬G‡-U between the mutant (M) and wild-type (F45W); ⌬GU,⌬GF, and ⌬G
‡denote the free energy difference be-tween mutant and wild-type (F45W) in the unfolded, folded, and transition states, respectively; and the remaining sym-bols take on their standard meanings.
In our equilibrium and NMR titration studies, we have shown that deleting Gly-10 raises the free energy of both the folded and unfolded states.When these⌬GFand⌬GUare combined with the present kinetic data using the above equations, we obtain⌬G‡-F⳱ 19.22±0.01 kcal/mol (F45W) and 19.70±0.01 kcal/mol (desG10/F45W); ⌬G‡-U ⳱ 14.76±0.07 kcal/mol (F45W) and 15.12±0.07 kcal/mol (desG10/F45W).The refolding reaction is much more facile than the unfolding reaction under the present circumstances. Therefore the kinetic traces are noisier, and larger errors appear in the fitting of the kinetic data.In any case, the deletion of Gly-10 retards the refolding and unfolding rates, indicating that the mutation has elevated the transition state more, although not dramatically, than the initial and final equilibrium states; ⌬⌬G‡-F ⳱ 0.48±0.02 kcal/mol and
⌬⌬G‡-U ⳱ 0.36±0.1 kcal/mol. The effect of destabilizing
Fig. 8. Saturation transfer assignment of the Thr-12 and Thr-14 resonances
in denatured ubiquitin.(A) 1D spectrum of ubiquitin in the vicinity of the midpoint of denaturation in the presence of 3.1 M GdnDCl, pH 3 at 320 K. (B) Difference spectrum obtained by subtraction from A of a comparable spectrum acquired with presaturation of the peak originating from the
␥-methyl protons of native Thr-12 and Thr-14 (1.085 ppm). Individual
assignments of Thr-12 and Thr-14 were made by analogy with the U(1–17) and U(1–34) peptides.
this hairpin on the transition state is about 0.4 kcal/mol (⌬G‡) plus the energy difference between the initial states of the wild-type and the mutant.Subtracting⌬⌬G‡-Ufrom
⌬⌬G‡-F, we obtain ⌬GU − ⌬GF⳱ 0.12±0.1 kcal/mol; or, deleting Gly-10 from the turn seems to have a slightly larger effect on destabilizing the denatured state than the native state, although the error is quite large compared with the free energy difference.
Discussion
It has been suggested that turns might play a role in direct-ing hairpin formation (Ramirez-Alvarado et al.1997; de Alba et al.1999; Griffiths-Jones et al.1999).It is known, for example, that only a one-residue change in the turn region can affect the populations of-hairpins with a dif-ferent register (Blanco et al.1993; Searle et al.1995; de Alba et al.1997).Moreover, it has been amply demon-strated that turns can exist without substantial populations of a-hairpin (Dyson et al.1985; Blanco et al.1991; Chan-drasekhar et al.1991; Shin et al.1993), and turn mutations can substantially affect a protein’s thermodynamic stability (Hynes et al.1989; Predki et al.1996) and folding kinetics
(Kim et al.1997).In light of these observations, it has been postulated that there exists in a protein the tendency to bend in predetermined regions of the polypeptide chain from the very beginning of the folding process, and these regions act as nucleation centers to drive the subsequent folding of the protein (Zimmerman and Scheraga 1977).In other words, the bends are intrinsically stable due to local forces rather than from the tertiary interactions acquired during the as-sembly of the protein.
The results of the present study show that Gly-10 in the bulge of the turn is an important determinant of the struc-tural stability of the N-terminal-hairpin of ubiquitin.De-leting Gly-10 unfolds the structure in the peptide but has only a very localized effect on the structure of the protein. Since the type I turn is not a favored turn type in-hairpins, the existence of a bulge might be a way to change the twist and the direction of the polypeptide chain to form a more stable turn.In the protein, the unfavored turn is apparently compensated for by structural changes and interactions in other parts of the protein.This compensation probably ac-counts, in part, for the rather small effect of the deletion on the stability of the native structure.
Fig. 9. 1D-NMR spectra of U(1–34) as a function of GdnDCl
concentra-tion, pH3 at 290 K.
Fig. 10. 1D-NMR spectra of U(1–17) as a function of GdnDCl
During the past ten years, largely due to improvements in NMR methods, scientists studying protein folding have compiled convincing evidence showing that the so-called “denatured” state is not always fully unfolded.For example, Fersht and coworkers have reported the persistence of some residual structures in urea-denatured barnase.These struc-tures are located in the center of the first helix and the tight turn between the third and fourth strands of the-sheet in the native protein (Arcus et al.1995; Freund et al.1996). Detection of any residual structures in unfolded proteins can
provide clues to the folding initiation sites.The embryonic initiation sites existing in protein denatured states could well develop into nucleation centers in the transition state. This is relevant to the Levinthal paradox (Levinthal 1968), which places great emphasis on the impossibility of random searches of a polypeptide chain in the folding process.A tendency to form nativelike structures reduces the space of the conformational search.
Our NMR titration results argue for the existence of some residual structure in the denatured state of wild-type ubiq-uitin.Under denaturing conditions, deleting the bulge de-stabilizes the residual structure in the wild-type and raises the free energy of the unfolded state.Although this structure has not been elucidated in detail, it is clear that Thr-14 is involved from the sidechain chemical shift perturbations observed.Phe-4 is also involved in this structure, because it is the only source of a ring-current effect.In addition, a turn must be formed to bring these two residues to a close jux-taposition.A comparison of the NMR spectra of the peptide fragments U(1–17) and U(1–34) also suggests that the
-hairpin at the N-terminus of ubiquitin is stabilized by
some sort of interaction with the segment that follows it in the sequence, probably the␣-helix.It seems that the entire
␣-helix is not required to stabilize this tertiary structure,
because NMR and circular dichrosim (CD) studies of the peptide U(1–35) suggest that the entire ␣-helix is not formed (B.Trotter, unpubl.).However, we do not know
Fig. 12. Free energy diagrams for the folding and unfolding of wild-type
ubiquitin and the desG10 mutant.The free energy axis is not to scale.
Fig. 11. Refolding/unfolding rates of ubiquitin F45W and desG10/F45W.䡺, F45W; 䡬, desG10/F45W.The refolding and unfolding
rates were determined in 0.1 M citrate buffer, pH3 with different concentrations of GdnHCl as described in Materials and Methods. Error bars are within the size of the data markers.
which residues in the sequence following the hairpin are involved.More titration experiments with peptides of dif-ferent lengths are required to clarify this issue.
Consistent with our conclusion, deleting Gly-10 affected the -hairpin-forming propensity in the N-terminal part of ubiquitin, and the characteristic spectral features of the re-sidual structure were not discerned in the spectrum of the denatured desG10/E18D.These results demonstrate the im-portance of the extruding bulge residue in stabilizing the
-hairpin.Deleting the bulge also retards both the refolding
and unfolding rates of the protein by raising the energy of the transition state.On the basis of these findings, we pro-pose that the residual structure might function as one of the nucleation centers during protein folding, with the bulge in the turn of the hairpin playing a prominent role in directing the formation of the hairpin and this nucleation center.
The transition state most likely involves a group of pep-tide conformations.In addition, the residual structure might not be the only precursor that leads to the transition state, because the deletion mutant can fold into its native confor-mation as well, albeit with slower kinetics.Accordingly, there must be other interactions, possibly weaker ones and hence difficult to detect, that become turned on during the early stages of protein folding.By combining site-directed mutagenesis, peptide dissection studies, structural analyses, and equilibrium and kinetic folding studies, we hope to gain greater understanding about the early kinetic events during protein folding as well as the interrelationship between pro-tein sequence and propro-tein structure and folding.
Materials and methods
Mutagenesis, expression, and purification of mutant proteins
The plasmid pNMHUB containing the human ubiquitin gene was kindly provided by Dr.Ernest Laue, and it was obtained originally from his colleagues at Du Pont-Merck (Wilmington, DE, USA). The gene with its cII ribosomal binding site was cloned into pAL-TER-1, and site-directed mutagenesis was carried out.Mutated vectors pALTER-e18d, pALTER-dg10e18d, pALTER-f45w, and pALTER-dg10f45w, corresponding to ubiquitin with a Glu-18 to Asp substitution only, with the E18D substitution plus a deletion at Gly-10, with a Phe-45 to Trp substitution, and with the F45W mutation plus the deletion at Gly-10, respectively, were trans-formed to E. coli JM109 (DE3) or BL21 (DE3) for protein ex-pression.The expression was performed by simply incubating the transformed clone in 2 × TY medium at 37°C overnight.
Cells were harvested and lysed by the freeze-thaw method.The harvested cells were frozen in liquid nitrogen, and kept at −80°C for at least 24 h.Twenty mL of freeze-thaw buffer (50 mM Tris-HCl, 2 mM EDTA, pH 7.8) was then used to suspend 1 g of the frozen cells.The suspension was stirred gently at room tempera-ture for 2 h, and then centrifuged at 16,000g for 30 min.To the supernatant, 20% polyethylenimine was added dropwise to a final concentration of 0.2% and stirred on ice for 15 min to precipitate
the DNA.The mixture was then spun at 30,000g for 20 minutes. Ammonium sulfate was added to the supernatant until it was 45% saturated, and the mixture was then centrifuged at 11,000g for 15 minutes.The supernatant was then passed through a 0.45 m syringe filter to remove any cell debris or precipitated protein particles.Next, 150 mL of the protein solution was pumped at a flow rate of 2.5 mL/min into a prepacked Hi-Load phenyl-sepha-rose column (16/10) that was preequilibrated with 45 % ammo-nium sulfate saturated freeze-thaw buffer, washed with at least 2 column-bed volumes of the same buffer, and then eluted by de-creasing the ammonium sulfate concentration.The mutant proteins were eluted by 38–26% saturated ammonium sulfate.The correct fractions were analyzed by SDS/PAGE, pooled together, and con-centrated by ultrafiltration.During the final stage, at most 3 mL of concentrate was applied to a prepacked Superdex 75 (16/60) gel filtration column preequilibrated with 0.15 M phosphate buffer (pH 7.0).The flow rate was 1 mL/min.Ubiquitin eluted in the fractions from 80 to 88 mL.The fractions were pooled, dialyzed against water overnight, and lyophilized.The purity and the yield of the final product proteins were checked using SDS/PAGE, MALDI-mass spectrometry, and amino acid analysis.
Peptide synthesis
The peptides desG10(1–16), U(1–17), and U(1–34) were synthe-sized by the fmoc-polyamide method.The peptide sequences are: desG10(1–16), MQIFVKTLTKTITLEV-NH2; U(1–17), MQIFVK
TLTGKTITLEV-NH2; U(1–34), MQIFVKTLTGKTITLEVEPSD
TIENVKAKIQDKE-NH2.The C-terminal ends of the peptides
were amidated to prevent the formation of nonnative interactions, including the potential salt bridge between the two ends of the sequence.The synthesized products were purified by reverse-phase HPLC as described for related peptides (Cox et al.1993). The final products were analyzed using amino acid analysis and positive ion-electrospray mass spectroscopy.The fractions con-taining the desired products were lyophilized and stored at −20°C.
Equilibrium foldingstudies followed by spectrofluorometry
Fluorescence measurements were performed in a 1 cm-pathlength quartz cuvette at room temperature using a JASCO FP-777 spec-trofluorometer.A 20 mg/mL protein solution was prepared in wa-ter, diluted 100 times into native buffer (20 mM glycine/HCl, pH 3.0) and unfolding buffer (20 mM glycine/HCl with 7 M GdnHCl, pH 3.0), which were prefiltered through a 0.22m membrane, to a final concentration of 0.2 mg/mL. Appropriate ratios of the na-tive and unfolding protein solution were mixed to prepare samples at different concentrations of GdnHCl.The final GdnHCl concen-tration of each sample was determined by refractometry.The samples were individually equilibrated at room temperature for at least 1 h before measurements.They were then excited at 277 nm (3 nm bandwidth), and the Tyr fluorescence emission was recorded at 303 nm (10 nm bandwidth).
The free energy change (⌬GH2O) and the m value were obtained by a nonlinear least-squares fit of the data to the equation:
Y=
exp共−共⌬GH2O+m关GdnHCl兴兲ⲐRT兲
共Yf+mf关GdnHCl兴兲−共Yu+mu关GdnHCl兴兲
where R is the gas constant (1.987 cal/mol−1
K−1
) and T is the absolute temperature.Y is the fluorescence intensity for a given [GdnHCl]; Yfand Yuare the fluorescence intensities of the folded
and unfolded state extrapolated to 0 M GdnHCl, respectively; and m, mfand mudenote the [GdnHCl] dependence of the transition,
pretransition and posttransition regions, respectively.Cm is the
[GdnHCl] at the midpoint of the transition.
2D-NMR spectroscopy
All NMR spectra were recorded on a Bruker AM 500 NMR spec-trometer.Samples were dissolved in 0.5 mL of H2O/D2O (9/1)
unless otherwise noted.The concentrations of the protein samples were in the range of 2.4 to 4 mM. The concentration of the peptide desG10(1–16) was 0.1 mM. A 1/50 volume of TSP solution (0.75%) was added as an internal reference. A 1/100 volume of 30 mM NaN3was also added as a preservative in the peptide samples.
The pH values of the protein samples and the peptide sample were adjusted with deuterium chloride to 7.2. desG10(1–16) is more soluble at low pH; hence the pH value was adjusted to 1.9. Quoted pH values were not corrected for the D/H isotope effect.Finally, each sample was spun down for 3 min at full speed in a benchtop centrifuge to remove insoluble material.
2D-TOCSY and NOESY were recorded using standard phase-cycling sequences at 295 K (protein) and 280 K (peptide).Usually, spectra were acquired with 2K data points in the direct dimension and 512 increments in the indirect dimension.Typically, 64 scans were collected per increment.For protein samples, 60 msec mixing time in TOCSY and 100, 125, or 150 msec mixing time in NOESY were used.For peptide samples, 80 msec mixing time in TOCSY and 300 msec mixing time in NOESY were used.For diagonal subtraction, a mixing time of 30 msec was used for the control spectrum.Data were processed on Silicon Graphics Indigo com-puters usingAzarasoftware (written by Wayne Boucher, Depart-ment of Biochemistry, University of Cambridge).Data were zero-filled once and twice in the direct and indirect dimensions, respec-tively, to obtain a final matrix size of 2K × 1K words.The shifted square sine bell window functions in both dimensions were applied for all spectra.The Ansig program (version 3.3) was used to assign the spectra (Kraulis 1989).
NMR titration
For the 1D NMR studies in the GdnDCl denaturation experiments, 2–3 mM solutions of protein and peptide were used.GdnDCl was obtained by repeatedly lyophilizing GdnHCl in D2O.For exchange
of the amide protons in the protein solutions, an unbuffered D2O
solution of the protein was titrated to pH 7.0 and incubated at room temperature overnight, then adjusted to pH 3.0 using DCl before measurement.TSP was added as an internal reference standard. After each measurement, about 10L of sample was taken out to measure the concentration of GdnDCl by refractometry, and GdnDCl was added to increase the concentration; the pH was also readjusted each time.NMR spectra were recorded at 290 K.The NMR probe was tuned and rematched every two or three samples. The length of the 90° pulse was found to increase with increasing ionic strength.In each spectrum, 8K data points were acquired, but the number of scans was increased from 32 to 192 with decreasing protein concentration (due to adjustments in the GdnDCl concen-tration and pH) and the impaired tuning at high salt concenconcen-trations. 1D spectra were recorded, using a NOESY-type pulse sequence to obtained a good baseline.
1-D magnetization transfer spectroscopy
About 2 mM of wild-type ubiquitin in the presence of 3.1 M GdnDCl was prepared in D2O.TSP was included as an internal
reference.The pH value was adjusted to 3.0 with 1 M DCl.1D spectra were recorded at 320 K, and 8K data points were collected. The sample was preirradiated with long, low-power pulses at the frequencies of the native Thr-12 and Thr-14␥-methyl proton reso-nances.Another control spectrum was recorded with presaturation in a blank region of the spectrum.The spectra were recorded in an interleaved fashion in blocks of 8 scans.Four hundred scans were recorded to improve the signal-to-noise ratio.The difference spec-trum was obtained by subtracting the target proton-presaturated spectrum from the control spectrum.
Stopped-flow fluorescence measurements
Fluorescence-detected refolding and unfolding experiments were performed on an SX.18MV stopped-flow instrument (Applied Photophysics, UK).A 150W xenon lamp was used.Phe-45 was replaced with tryptophan and used as a probe in the fluorescence experiments.Proteins were excited at 295 nm (4.65 nm band-width), and emission was recorded at 349 nm (9.3 nm bandwidth, 2 mm pathlength).The stopped-flow apparatus and observation cell were thermostated at 25°C by circulating water from a tem-perature-controlled water bath.The dead time of the stopped flow instrument for water is about 1.1 msec for 1:1 dilution and 1.5 msec for 1:10 dilution.For refolding experiments, a solution of 2 mg/mL unfolded protein in 6 M GdnHCl (0.1 M citrate buffer, pH 3) was diluted 11-fold with refolding buffer containing different amounts of GdnHCl.For unfolding experiments, a solution of native protein in 2.4 M GdnHCl was unfolded by a one-to-one dilution with concentrated GdnHCl buffer solution.Kinetic traces were recorded over intervals of 0.2 sec and 10 sec for refolding and unfolding studies, respectively.Five kinetic traces were averaged at each GdnHCl concentration and fitted using single exponential software provided by the software package of this instrument.The first 3 msec of the refolding traces and first 10 msec of the un-folding traces were discarded in the fitting.
Acknowledgments
We thank Dr.Leonard C.Packman for assistance in the peptide synthesis and purification, and Drs.Andrew R.C.Raine, Bill Broadhurst, and Daniel Nietlispach for their advice on the use of the NMR and computer programs. P.Y.C. acknowledges scholar-ship support from the Government of the Republic of China that allowed her to pursue her graduate studies at the University of Cambridge.Part of this research was also supported by the Insti-tute of Chemistry, Academia Sinica and a grant from the National Science Council, Republic of China (Grant No.NSC 89–2113-M-001–092).
The publication costs of this article were defrayed in part by payment of page charges.This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
Arcus, V.L., Vuilleumier, S., Freund, S.M.V., Bycroft, M., and Fersht, A.R. 1995.A comparison of the pH, urea, and temperature-denatured states of barnase by heteronuclear NMR: Implications for the intiation of protein folding. J. Mol. Biol. 254: 305–321.
Axe, D.D., Foster, N.W., and Fersht, A.R. 1999. An irregular-bulge common to a group of bacterial RNase is an important determinant of stability and function in barnase. J. Mol. Biol. 286: 1471–1485.
Blanco, F.J., Jimenez, M.A., Rico, M., Santoro, J., Herranz, J., and Nieto, J.L. 1991.Tendamistat (12-26) fragment — NMR characterization of isolated
-turn folding intermediates. Eur. J. Biochem. 200: 345–351.
Blanco, F.J., Jimenez, M.A., Herranz, J., Rico, M., Santoro, J., and Nieto, J.L. 1993.NMR evidence of a short linear peptide that folds into a-hairpin in aqueous-solution. J. Am. Chem. Soc. 115: 5887–5888.
Blanco, F.J.and Serrano, L.1995.Folding of protein-G B1 domain studied by the conformational characterization of fragments comprising its secondary structure elements. Eur. J. Biochem. 230: 634–649.
Bundi A.and Wüthrich K.1979.1H NMR parameters of the common amino acid residues measured in aqueous solution of the linear tetrapeptides H-Gly-Gly-X-L-Ala-OH. Biopolymers 18: 285–297.
Chan, A.W.E., Hutchinson, E.G., Harris, D., and Thornton, J.M. 1993. Identi-fication, classiIdenti-fication, and analysis of-bulges in proteins. Protein Sci. 2: 1574–1590.
Chandrasekhar, K., Profy, A.T., and Dyson, H.J. 1991. Solution conformational preferences of immunogenic peptides derived from the principal neutraliz-ing determinant of the HIV-1 envelope glycoprotein gp120. Biochemistry
30: 9187–9194.
Cox, J.P.L., Evans, P.A., Packman, L.C., Williams, D.H., and Woolfson, D.N. 1993.Dissecting the structure of a partially folded protein - circular dichro-ism and nuclear magnetic resonance studies of peptides from ubiquitin. J. Mol. Biol. 234: 483–492.
de Alba, E., Jiménez, M.A., Rico, M., and Nieto, J.L. 1996. Conformational investigation of designed short linear peptides able to fold into-hairpin structures in aqueous solution. Folding& Design 1: 133–144.
de Alba, E., Jiménez, M.A., and Rico, M. 1997. Turn residue sequence deter-mines-hairpin conformation in designed peptides. J. Am. Chem. Soc. 119: 175–183.
de Alba, E., Rico, M., and Jiménez, M.A. 1999. The turn sequence directs
-strand alignment in designed -hairpins. Protein Sci. 8: 2234–2244.
Dyson, H.J., Cross, K.J., Houghten, R.A., Wilson, I.A., Wright, P.E., and Le-rner, R.A.1985.The immunodominant site of a synthetic immunogen has a conformational preference in water for a type-II reverse turn. Nature 318: 480–483.
Evans, P.A., Topping, K.D., Woolfson, D.N., and Dobson, C.M. 1991. Hydro-phobic clustering in nonnative states of a protein — Interpretation of chemi-cal-shifts in NMR spectra of denatured states of lysozyme. Proteins 9: 248–266.
Freund, S.M.V., Wong, K.-B., and Fersht, A.R. 1996. Initiation sites of protein folding by NMR analysis. Proc. Natl. Acad. Sci. 93: 10600–10603. Griffiths-Jones, S.R., Maynard, A.J., and Searle, M.S. 1999. Dissecting the
stability of a-hairpin peptide that folds in water: NMR and molecular dynamics analysis of the-turn and -strand contributions to folding. J. Mol. Biol. 292: 1051–1069.
Hynes, T.R., Kautz, R.A., Goodman, M.A., Gill, J.F., and Fox, R.O. 1989. Transfer of a-turn structure to a new protein context. Nature 339: 73–76. Ibarra-Molero, B., Loladze, V.V., Makhatadze, G.I., and Sanchez-Ruiz, J.M. 1999.Thermal versus guanidine-induced unfolding of ubiquitin.An analy-sis in terms of the contributions from charge–charge interactions to protein stability. Biochemistry 38: 8138–8149.
Jenson, J, Goldstein, G., and Breslow, E.1980.Physical-chemical properties of ubiquitin. Biochim. Biophys. Acta. 624: 378–385.
Khorasanizadeh, S., Peters, I.D., Butt, T.R., and Roder, H. 1993. Folding and stability of a tryptophan-containing mutant of ubiquitin. Biochemistry 32: 7054–7063.
Kim, K., Ramanathan, R., and Frieden, C. 1997. Intestinal fatty acid binding protein: A specific residue in one turn appears to stabilize the native struc-ture and be responsible for slow refolding. Protein Sci. 6: 364–372. Kraulis, P.J.1989.Ansig — A program for the assignment of protein H-1
2D-NMR spectra by interactive computer-graphics. J. Magn. Reson. 84: 627–633.
Kraulis, P.J.1991.MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures, J. Appl. Cryst. 24: 946–950. Lenkinski, R.E., Chen, D.M., Glickson, J.D., and Goldstein, G. 1977. Nuclear
magnetic resonance studies of the denaturation of ubiquitin. Biochim. Bio-phys. Acta 494: 126–130.
Levinthal, C.1968.Are these pathways for protein folding? J. Chem. Phys. 85: 44–45.
Milner-white, E.J.and Poet, R.1987.Loops, bulges, turns and hairpins in proteins. Trends Biochem. Sci. 12: 189–192.
Predki, P.F., Agrawal, V., Brunger, A.T., and Regan, L. 1996. Amino-acid substitutions in a surface turn modulate protein stability. Nat. Struct. Biol. 3: 54–58.
Ramirez-Alvarado, M., Serrano, L., and Blanco, F.J. 1997. Conformational analysis of peptides corresponding to all the secondary structure elements of protein L B1 domain: Secondary structure propensities are not conserved in proteins with the same fold. Protein Sci. 6: 162–174.
Searle, M.S., Williams, D.H., and Packman, L.C. 1995. A short linear peptide derived from the N-terminal sequence of ubiquitin folds into a water-stable nonnative-hairpin. Nat. Struct. Biol. 2: 999–1006.
Searle, M.S., Zerella, R., Dudley, D.H., and Packman, L.C. 1996. Native-like
-hairpin structure in an isolated fragment from ferredoxin — NMR and CD
studies of solvent effects on the N-terminal- 20 residues. Protein Eng. 9: 559–565.
Shin, H.C., Merutka, G., Waltho, J.P., Wright, P.E., and Dyson, H.J. 1993. Peptide models of protein-folding initiation sites.2.The G-H turn region of myoglobin acts as a helix stop signal. Biochemistry 32: 6348–6355. Sibanda, B.L.and Thornton, J.M.1991.Conformation of-hairpins in protein
structures — Classification and diversity in homologous structures. Methods Enzymol. 202: 59–82.
Vijay-kumar, S., Bugg, C.E., and Cook, W.J. 1987. Structure of ubiquitin re-fined at 1.8 Å resolution. J. Mol. Biol. 194: 531–544.
Wishart, D.S., Sykes, B.D., and Richards, F.M. 1991. Relationship between nuclear-magnetic-resonance chemical-shift and protein secondary structure. J. Mol. Biol. 222: 311–333.
Zerella, R., Chen, P.Y., Evans, P.A., Raine, A., and Williams, D.H. 2000. Structural characterization of mutant peptides derived from ubiquitin and implications for protein folding. Protein Sci. 8: 1320–1331.
Zerella, R., Evans, P.A., Ionides, J.M.C., Williams, D.H., Trotter, B.W., and Mackay, J.P.1999.Autonomous folding of a peptide corresponding to the N-terminal-hairpin from ubiquitin. Protein Sci. 8: 1320–1331. Zimmerman, S.S.and Scheraga, H.A.1977.Local interactions in bends of