Clinical and Genetic Heterogeneity, Overlap with
Other Tumor Syndromes, and Atypical Glucocorticoid
Hormone Secretion in
Adrenocorticotropin-Independent Macronodular Adrenal Hyperplasia
Compared with Other Adrenocortical Tumors
Hui-Pin Hsiao, Lawrence S. Kirschner, Isabelle Bourdeau, Margaret F. Keil, Sosipatros A. Boikos, Somya Verma, Audrey J. Robinson-White, Maria Nesterova, Andre´ Lacroix, and Constantine A. StratakisSection on Endocrinology and Genetics (H.-P.H., S.A.B., S.V., A.J.R.-W., M.N., C.A.S.), Program on Developmental Endocrinology and Genetics and Pediatric Endocrinology Interinstitute Training Program (M.F.K., S.V., C.A.S.), National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland 20892; Division of Endocrinology, Diabetes, and Metabolism (L.S.K.), Department of Internal Medicine, Ohio State University, Columbus, Ohio 43210; Endocrinology Division (I.B., A.L.), Department of Medicine, Centre Hospitalier de l’Universite´ de Montre´al, Montre´al, Que´bec, Canada H2W 1T8; and Department of Pediatrics (H.-P.H.), Kaohsiung Municipal Hsiao-Kang Hospital and Department of Pediatrics, Faculty of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 807, Taiwan
Objective: ACTH-independent macronodular adrenal hyperplasia (AIMAH) is often associated with subclinical cortisol secretion or atypical Cushing’s syndrome (CS). We characterized a large series of patients of AIMAH and compared them with patients with other adrenocortical tumors. Design and Patients: We recruited 82 subjects with: 1) AIMAH (n⫽ 16); 2) adrenocortical cortisol-producing adenoma with CS (n⫽ 15); 3) aldosterone-producing adenoma (n ⫽ 19); and 4) single adenomas with clinically nonsignificant cortisol secretion (n⫽ 32).
Methods: Urinary free cortisol (UFC) and 17-hydroxycorticosteroid (17OHS) were collected at base-line and during dexamethasone testing; aberrant receptor responses was also sought by clinical testing and confirmed molecularly. Peripheral and/or tumor DNA was sequenced for candidate genes.
Results: AIMAH patients had the highest 17OHS excretion, even when UFCs were within or close to the normal range. Aberrant receptor expression was highly prevalent. Histology showed at least two subtypes of AIMAH. For three patients with AIMAH, there was family history of CS; germline mutations were identified in three other patients in the genes for menin (one), fumarate hydratase (one), and adenomatosis polyposis coli (APC) (one); a PDE11A gene variant was found in another. One patient had a GNAS mutation in adrenal nodules only. There were no mutations in any of the tested genes in the patients of the other groups.
Conclusions: AIMAH is a clinically and genetically heterogeneous disorder that can be associated with various genetic defects and aberrant hormone receptors. It is frequently associated with atypical CS and increased 17OHS; UFCs and other measures of adrenocortical activity can be mis-leadingly normal. (J Clin Endocrinol Metab 94: 2930 –2937, 2009)
ISSN Print 0021-972X ISSN Online 1945-7197 Printed in U.S.A.
Copyright © 2009 by The Endocrine Society
doi: 10.1210/jc.2009-0516 Received March 9, 2009. Accepted May 29, 2009. First Published Online June 9, 2009
Abbreviations: ACS, Adrenocortical cortisol-producing adenoma; AIMAH, ACTH-independent macronodular adrenal hyperplasia; APA, aldosterone-producing adenoma; BAH, bilateral ad-renocortical hyperplasias; BMAH, bilateral macronodular adad-renocortical hyperplasia; CS, Cush-ing’s syndrome; GIP, gastric inhibitory polypeptide; oCRH, ovine CRH; 17OHS, 17-hydroxy-corticosteroid; SCA, single adenomas with clinically nonsignificant cortisol secretion; UFC, urinary free cortisol.
E n d o c r i n e C a r e
E
ndogenous Cushing’s syndrome (CS) is due to primary ad-renal disease in approximately 15–20% of cases; in these patients, CS is caused mainly by unilateral adenomas (1). Bi-lateral adrenal lesions occur only in 10 –15% of adrenal CS and include primary pigmented nodular adrenocortical dis-ease (PPNAD) and ACTH-independent macronodular adrenal hyperplasia (AIMAH), also known as massive macronodular adrenal disease (2, 3). PPNAD is most frequently associated with Carney complex and is caused mainly by inactivating germline mutations of PRKAR1A and allelic losses of its locus at the 17q22-24 chromosomal region (4, 5).AIMAH is a rare cause of CS, accounting for less than 1% of adrenal CS. Since the review of AIMAH by Lieberman et al. in 1994 (6), a greater number of cases have been reported (7). Several studies demonstrated that the regulation of cortisol secretion in AIMAH (as well as in some unilateral, single adenomas) is mediated by the aberrant expression and function of membrane-bound G protein-coupled receptors such as those for gastric inhibitory polypeptide (GIP), vasopressin, catecholamines, LH/human cho-rionic gonadotropin, serotonin, IL-I, and leptin (8 –14).
Establishing the diagnosis of AIMAH with CS is based on, first, the clinical phenotype of CS; this is followed by the demonstration of ACTH-independent hypercortisolism and bilateral adrenal nod-ular enlargement on radiologic imaging. More often than not, di-agnosis of AIMAH is difficult because hypercortisolism usually de-velops slowly over several years, may be cyclical and is frequently associated with subtle clinical manifestations. Radiologic imaging is helpful, but occasionally adrenal nodularity is indistinguishable from that in normal elderly persons (15). Biochemically, too, there is no specific testing for the diagnosis of AIMAH: whereas most patients with PPNAD respond to dexamethasone with a paradox-ical (unexpected) increase in glucocorticoid excretion during Lid-dle’s test, AIMAH patients respond to dexamethasone as patients with the common adrenal cortisol-producing tumors do, requiring final confirmation of the diagnosis by histological examination (16).
In this investigation, we studied genetically and clinically a large series of patients with AIMAH and compared them with patients with other adrenocortical tumors. The data prove the clinical and genetic heterogeneity of the condition in these patients but also demonstrate the large genetic component of the condition. Finally, our study proposes that urinary 17-hydroxycorticosteroid (17OHS) is the best screening text for hypercortisolism in AIMAH.
Patients and Methods
A total of 102 patients were admitted to the National Institutes of Health Warren Magnuson Clinical Center from 2000 to 2008 for the work-up and treatment of adrenocortical tumors under protocol 00CH160. Eighty-two of 102 patients (28 males and 54 females) met the following criteria: 1) evidence for the existence of an adrenocortical tumor, as indicated by imaging studies or biochemical investigation of hormonal se-cretion; 2) exclusion of Carney complex or other diseases associated with micronodular forms of adrenocortical hyperplasia. The National Institute of Child Health and Human Development Institutional Review Board ap-proved this study; informed consents were obtained from all subjects.
The 82 subjects whose data were included here were divided into four groups on the basis of their final histological examination or, for those with nonsecreting tumors, biochemical testing and radiological imaging: 1)
AIMAH (n⫽ 16); 2) adrenocortical cortisol-producing adenoma with CS (ACS) (n⫽ 15); 3) aldosterone-producing adenoma (APA) (n ⫽ 19); and 4) singleadenomaswithclinicallynonsignificantcortisolsecretion(SCA)(n⫽32). The following data were analyzed for all subjects, as we have reported elsewhere for the investigation of CS (17): diurnal variation in plasma ACTH and cortisol levels; cortisol levels before and after the overnight 8-mg dexamethasone test; and plasma ACTH and cortisol levels before and after iv administration of 1g/kg ovine CRH (oCRH; corticorelin). Urine was collected for 24 h for free cortisol excretion (UFC) and 17-hydroxycorticosteroid (17OHS), and their data are presented as the mean of two consecutive measurements.
Finally, in all patients with AIMAH, a review of their pathological reports (i.e. macroscopic appearance and weights) and histology was conducted. We recorded size and number of nodules and the presence of hyperplasia or atrophy of nonnodular adrenal cortex, as we have re-ported elsewhere (18).
Provocative tests for the detection of aberrant receptor expression
Provocative tests for the identification of aberrant receptor expres-sion was employed in the AIMAH and ACS subgroups over a 3-d period, as described previously (19, 20). The protocol is based on monitoring plasma levels of steroids at 30- to 60-min intervals for 2–3 h during tests that transiently modulate the levels of ligands for potentially aberrant receptors. Initial tests included a posture test performed in a supine po-sition for baseline, followed by a 2-h ambulatory period (to evaluate potential modulation by vasopressin, catecholamines, angiotensin II, and others); this was followed by a standard mixed meal to evaluate the response to fluctuations of gastrointestinal hormones including GIP. On the second day, the administration of 100g GnRH iv (gonadorelin, 1 g/kg, iv, testing for modulation by FSH, LH, GnRH) was followed by 200g TRH iv (modulation by TSH, prolactin, TRH) and GHRH (sermorelin, 1 g/kg, iv). Responses to 1 mg glucagon iv, 10 IU arginine vasopressin im were tested sequentially on the third day. A change of 50% or greater of plasma cortisol was defined as a positive response; a 25– 49% change, as a partial response; and a change of less than 25%, as no response; these cutoffs and their justification have been published elsewhere (19, 20).
Dexamethasone testing
A 6-d Liddle’s test was conducted for patients with AIMAH, as de-scribed elsewhere (16, 17, 21). After 2 d of baseline measurement of urinary steroid excretion, dexamethasone, 0.5 mg, was given orally every 6 h for 2 d starting at 0600 h; the dose of dexamethasone was then increased to 2 mg every 6 h for the last 2 d of the test. The 24-h UFC and urinary 17OHS were measured on each day and percentage changes from baseline were calculated. 24-h UFC were corrected for body surface area and 17OHS rates were corrected for creatinine excretion (per day per gram creatinine). In all patients, dexamethasone levels were measured on the last day of the test, and this ensured adequate absorption of the medication; in addition, ACTH levels were suppressed (⬍5 pmol/liter) in all patients on the same day of the test.
Hormone assays
Plasma cortisol and ACTH levels were measured as described elsewhere (22). UFC excretion was measured by direct RIA (23). The intraassay co-efficient of variation was 5%, and the interassay coco-efficient of variation was 10%; urinary 17OHS excretion was measured by using a modification of the colorimetric method as previously described by Murphy (24). The intraas-say and interasintraas-say coefficients of variation were 6 and 11%, respectively.
Genetic and other molecular tests
Genomic DNAs were obtained from the blood leukocytes in all subjects and the adrenocortical tissues when they underwent surgery (25, 26). Mu-tation analysis was performed for the menin (MEN1), fumarate hydratase (FH), adenomatous polyposis coli (APC), GNAS, and PDE11A (phospho-diesterase 11A) genes (26–28).
Aberrant receptor expression that was suggested by clinical testing was confirmed by molecular testing as previously published (19, 20). The snap-frozen tissue (50 –100 mg) was homogenized with 1 ml TRIZOL reagent (Invitrogen, Carlsbad, CA). The extracted RNA was treated with deoxyribonuclease digestion to eliminate contaminating genomic DNA using ribonuclease-free deoxyribonuclease set (QIAGEN, catalog no. 79254; Valencia, CA). The RNA was further cleaned up with RNeasy minikit (QIAGEN, catalog no. 74104). The integrity and concentration of the RNA were determined by agarose gel electrophoresis with ethidium bromide and ND-1000 spectrophotometer (NanoDrop, Wilmington, DE). Two micrograms of RNA was reverse transcriptased to cDNA using the oligo(dT) primers in the SuperScript III first-strand synthesis system (In-vitrogen), according the procedures by the manufacturer. PCRs were per-formed with Biolase DNA polymerase (Bioline, Randolph, MA) and prim-ers specific to the genes of individual receptors and internal control,-actin. The primers were designed for the receptors of ACTH, V1, V2, V3, GIP, and LH/human chorionic gonadotropin, either flanking the intron or on the exon/exon boundary to prevent the amplification from the genomic DNA (29–31). The amplicon products of the PCR were visualized and auto-graphed in agarose gel electrophoresis with ethidium bromide under the AlphaImiger 3400 (Imgen Technologies, Alexandria, VA).
Statistical analysis
All data were expressed as the mean⫾SD(for demographic data) or mean⫾SE(for all comparisons and the data presented in the figures). Data were analyzed by using SPSS 14.0 for Windows (SPSS Inc., Chicago, IL). The one-way ANOVA with post hoc follow-up (Tukey honestly significant difference) for multiple comparisons was used to test for any differences in demographics; for the percentage and frequency among groups in provocative tests, the2test and follow-up pairwise comparison
tests were done with cross-tabulation procedure. The Kruskal-Wallis H test was used to assess differences in Liddle test; Mann-Whitney U test was used for post hoc test among groups. For all statistical comparisons, P⬍0.05was considered significant.
Results
Demographics
The mean age of AIMAH patients was 46.8 yr (46.8⫾ 9.8 yr); this was not significantly different from the mean age of patients in
other groups (Table 1). The overall gender distribution (female to male) for this study was 54 to 28, a female predominance (65.9% females) that is similar to what has been reported for adrenal tumors in other studies. The only group of patients in which males (n⫽ 12) outnumbered females (n⫽ 7) was the APA group.
Clinical characteristics and baseline glucocorticoid hormone levels
Patients with AIMAH and ACS did not have any diurnal rhythmicity in their cortisol secretion; those with APA and SCA groups had the expected diurnal variation in their midnight to morning cortisol values. Morning cortisol values were higher for patients with AIMAH and ACS, 535.2⫾ 198.6 and 538.0 ⫾ 215.2 nmol/liter, respectively, vs. those in patients with SCA (389.0⫾ 140.7 nmol/liter, P ⬍ 0.05) (Table 1). Patients with AIMAH and ACS had a trend of higher morning cortisol levels than the APA group, but this difference did not reach statistical significance.
UFCs were significantly different in patients with AIMAH and ACS vs. those with APA and SCA (611.9 ⫾ 320.7 and 625.7⫾ 448.5 nmol/d 183.5 ⫾ 123.6 and 135.2 ⫾ 64.0 nmol/d, respectively, P⬍ 0.01). Patients in the AIMAH group had the highest urinary 17OHS excretion (43.1⫾ 20.4 mmol/d 䡠 g crn); 17OHS were 29.8⫾ 13.8 mmol/d 䡠 g crn in patients with ACS (P⫽ 0.08) and 15.2 ⫾ 10.8 and 18.2 ⫾ 12.7 mmol/d 䡠 g crn (P ⬍ 0.01) in patients with APA and SCA, respectively (Table 1). Dexamethasone and oCRH testing, and aberrant receptor expression studies
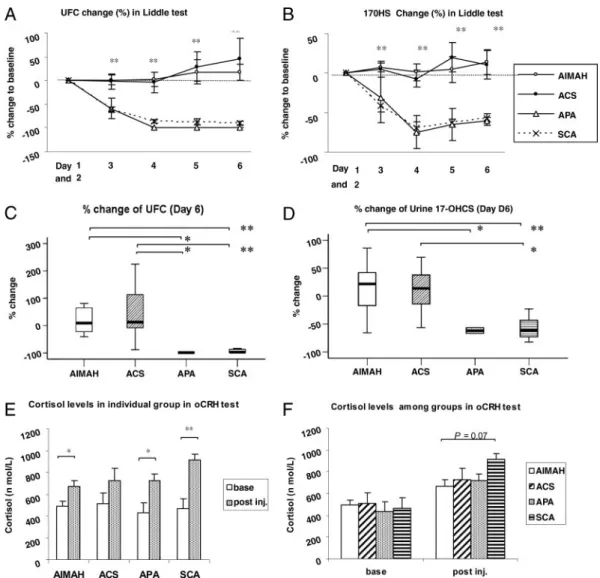
During the Liddle’s test, patients with AIMAH and ACS failed to suppress their glucocorticoid hormone serum levels and urinary excretion rates, consistent with our data in an earlier cohort of similar patients that were studied in our institution (16). In contrast, all patients with APA and SCA suppressed (Fig. 1, A and B). The percentage changes on d 6 from baseline (d 0) TABLE 1. Clinical and laboratory data for all patients with ACTH-independent macronodular adrenal hyperplasia and other adrenocortical tumors
AIMAH (nⴝ 16) ACS (nⴝ 15) APA (nⴝ 19) SCA (nⴝ 32) P value
Gender (F:M ) 11:5 11:4 7:12 25:7
Age (yr) 46.8⫾ 9.8 44.3⫾ 14.8 51.2⫾ 10.0 49.0⫾ 10.7 0.32
8 AM cortisol (nmol/liter) 535.2⫾ 198.6a 538.0⫾ 215.2a 433.2⫾ 82.8 389.0⫾ 140.7 0.03
12 midnight cortisol (nmol/liter) 435.9⫾ 242.8b,c 471.8⫾ 355.9b,c 168.3⫾ 57.9 129.7⫾ 69.0 ⬍0.01
ACTH (pmol/liter) 1.9⫾ 2.5b 0.7⫾ 0.4d 4.8⫾ 3.7 3.5⫾ 2.3 ⬍0.01
Cortisol, before DEX (nmol/liter) 527.0⫾ 157.3 405.6⫾ 126.9 485.6⫾ 209.7 449.7⫾ 182.1 0.35
Cortisol, after DEX (nmol/liter) 320.0⫾ 298.0a,b 303.5⫾ 223.5a 93.8⫾ 88.3 85.5⫾ 60.7 ⬍0.01
UFC (nmol/d䡠 g creatinine) 611.9⫾ 320.7c,d 625.7⫾ 448.5c,d 183.5⫾ 123.6 135.2⫾ 64.0 ⬍0.01
17OHS (mmol/d䡠 g creatinine) 43.1⫾ 20.4c,d 29.8⫾ 13.8b 15.2⫾ 10.8 18.2⫾ 12.7 ⬍0.01
Change at d 6 in Liddle’s test (%)
UFC 18.0⫾ 48.0b,c 44.6⫾ 109.7b,c ⫺98.7 ⫾ 0.1 ⫺90.3 ⫾ 15.6 ⬍0.01
17OHS 14.2⫾ 47.1b,c 10.6⫾ 44.9a ⫺61.2 ⫾ 7.5 ⫺57.5 ⫾ 18.8 ⬍0.01
Conversion factors to metric units are as follows: cortisol, 0.036 to micrograms per deciliter; ACTH, 5.0 to picograms per milliliter; UFC, 0.362 to micrograms per day per gram creatinine.
aP⬍ 0.05 compared with SCA. bP⬍ 0.05 compared with APA. cP⬍ 0.01 compared with SCA. dP⬍ 0.01 compared with APA.
were for patients with AIMAH, ACS, APA, and SCA; UFCs 18.0⫾ 48.0%, 44.6 ⫾ 109.7%, ⫺98.7 ⫾ 0.1%, and ⫺90.3 ⫾ 15.6% (P ⬍ 0.01), respectively; and 17OHS, 14.2 ⫾ 47.1%, 10.6⫾ 44.9%, ⫺61.2 ⫾ 7.5%, and ⫺57.5 ⫾ 48.8% (P ⬍ 0.01), respectively (Fig. 1, C and D).
Although cortisol levels responded to oCRH testing in all patients, patients with AIMAH, ACS, and APA had lower re-sponses than what was seen in patients with SCA (670.4⫾ 55.2, 725.6⫾ 110.4, and 722.8 ⫾ 60.7 nmol/liter vs. 915.9 ⫾ 52.4 nmol/liter, respectively P⫽ 0.07) (Fig. 1, E and F). It is
note-worthy that in almost none of our patients with CS there was a flat response to oCRH stimulation.
The response rates for the aberrant receptor provocative tests for patients with AIMAH and ACS are shown on Table 2; there were no statistically significant differences in response rates be-tween these two groups of patients with cortisol-producing tu-mors. In all cases, expression of the aberrant receptor was con-firmed by RT-PCR (Fig. 2) and as previously published (19, 20). There was no significant difference in the expression of the ab-errant receptors within the AIMAH group.
Clinical and molecular genetics
There were three unrelated patients among the 16 with AIMAH who had family history of adrenocortical tumors and/or ACTH-independent CS; the relatives of these patients, however, were not available for investigation. Inheritance of the condition was suggested to be in an autosomal dominant manner based on the constructed pedigrees (data not shown). There were no fa-milial cases in any of the other groups of patients (Table 3).
We identified three germline mutations in known genes in three other patients with AIMAH. Interestingly, none of these
FIG. 1. A and B, Serial percentage changes (from d 1 to d 6) compared with the baseline of UFC and 17OHS during Liddle’s test. C and D, Percentage changes
compared with baseline in UFCs and 17OHS on d 6 of the Liddle’s test. E and F, Changes in cortisol levels during the oCRH test. *, P⬍ 0.05; **, P ⬍ 0.01.
FIG. 2. The RT-PCR analysis of ACTH receptor and V1 receptor expression in
various adrenal tissue samples compared with the-actin. Lanes 1 and 2, Type 1 (BMAH) AIMAH patients; lane 3, type 2 (diffuse hyperplasia) AIMAH patient; lane 4, ACS patient; and lane 5, normal control adrenal tissue.
patients had family history of ACTH-independent CS or any other endocrine condition. One (ADT43.01) presented with AIMAH and hyperparathyroidism, and he was found to have a MEN1 mutation (Pro494Leu) (28); another (ADT06.01) pre-sented with AIMAH and the hereditary leiomyomatosis and renal cancer syndrome; she had a heterozygous fumarate hy-dratase mutation (32); a third patient (ADT52.02) had an APC gene mutation (4393_4394delAG) also in the heterozygote state, and her history included polyps and desmoids tumors. This mu-tation has been previously reported in patients with familial ad-enomatous polyposis and is predicted to produce a truncated protein (see http://www.hgmd.cf.ac.uk/ac/index.php). There was one additional patient who was found to have a somatic
GNAS mutation (Arg201His) in her ad-renocortical tumor tissue only; the patient did not have any other signs of McCune-Albright syndrome and was in that sense similar to the patients reported by Fra-goso et al. (33). There were no pathogenic mutations in the PRKAR1A gene; one of the three familial AIMAH cases was a car-rier of the R867G PDE11A gene poly-morphism (27, 34). None of the other pa-tients, in any of the groups, was found to carry a mutation in any of the tested genes in the peripheral or tumor DNA.
Tumors in other organs in our AIMAH, ACS, APA, and SCA patients were found in five, two, one, and two cases, respectively. In their families, there were three AIMAH patients with family history of tumors other than in the adre-nal gland; the SCA groups had three such patients (Table 3).
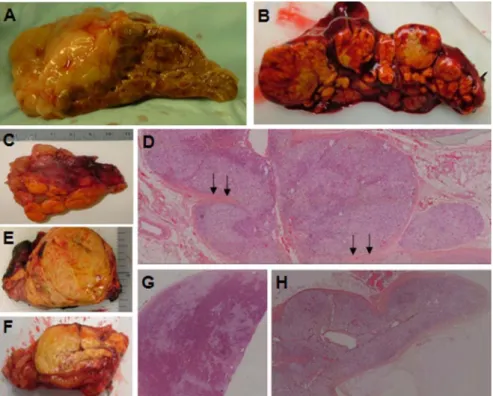
Histology of patients with AIMAH It was recognized that patients with AIMAH could generally be subgrouped in two categories (18) (Fig. 3): those with multiple nodules or discrete adenomas and intervening atrophic cortical tissue [type I AIMAH, bilateral adenomata, or bilateral macronodular adrenocortical hyperplasia (BMAH)] and those with dif-fuse hyperplasia and no residual normal or surrounding atrophic adrenal cortex (type II AIMAH). Most patients with AIMAH belonged to the second category; the three familial cases and the patients with germline MEN1 and APC and the one with the somatic GNAS mutation belonged to the first group.
Discussion
Although the majority of our patients with AIMAH presented in the fifth decade of life (mean age of 46.8 yr), most had an insid-ious and atypical form of CS for a number of years. It is char-acteristic that by the time these patients were operated at the National Institutes of Health and despite their lack of suppres-sion in response to dexamethasone as well as relative loss of their diurnal rhythm in cortisol secretion, almost all of them retained their response to oCRH. As a group, AIMAH patients had higher morning cortisol levels and lower oCRH responses than patients with APAs or SCAs, but their hypothalamic-pituitary-adrenal axis was not completely suppressed. This is a phenomenon that we have described before in patients with PPNAD (17, 35), sug-gesting that oCRH testing is not useful in the investigation of bilateral adrenocortical hyperplasias (BAH) that are generally associated with atypical, milder, chronic, or cyclical forms of CS. In the absence of the typical paradoxical rise in glucocorticoid hormone excretion rates in response to dexamethasone for
mi-FIG. 3. A, Corticotropin-induced hyperplasia. The adrenal is large and nodular, but the overall anatomy and
shape of the gland is largely preserved. B and C, Two examples, right and left adrenal gland, respectively, with multiple nodules or discrete adenomas and intervening atrophic cortical tissue (type I AIMAH, BMAH). D, Hematoxylin and eosin (H&E) staining (magnification,⫻5) of the tissue from C; multiple nodules are clearly visible, and the intervening cortex is atrophic and even sandwiched between adenomas (arrows). E and F, Two examples, right and left adrenal gland, respectively, with diffuse cortical hyperplasia and no residual normal or surrounding atrophic adrenal cortex (type II AIMAH). G, Histology of tissue from E showing a large adenoma in the context of diffuse hyperplasia (H&E staining,⫻5). H, Histology of tissue from F showing diffuse cortical hyperplasia (H&E staining,⫻5).
TABLE 2. Response to testing for aberrant receptor expression in patients with AIMAH and ACS
AIMAH (nⴝ 14) % ACS (nⴝ 12) % Posture 4/11 36.4 3/12 25.0 Meal 1/12 8.3 0/11 0.0 GnRH 1/6 16.7 0/8 0.0 TRH 1/3 33.3 1/8 12.5 GHRH 2/11 18.2 0/10 0.0 Glucagon 0/10 0.0 2/11 18.2 Vasopressin 5/11 45.5 4/10 40.0
The denominator stands for the number of patients being tested; the numerator stands for the number of patients with positive response (⬎150% increasing).
cronodular forms of BAH (16 –18, 35), how can one diagnose AIMAH in its early stages? The suggestion of BAH on radiolog-ical imaging is the first diagnostic criterion, as we proposed else-where (16, 36). The present investigation identified another use-ful diagnostic feature for patients with AIMAH: these patients had high urinary 17OHS excretion, even when their UFC levels were comparable or even lower than those of the ACS group. Urinary 17OHS has long been known to represent the fractions of the corticosteroids possessing a hydroxyl group at position 17 of the steroid structure, including cortisol, cortisone, 6-hy-droxycortisol, tetrahydrocortisol, allotetrahydrocortisol, and tetrahydrocortisone (37, 38). Thus, patients with AIMAH ap-pear to excrete in their urine various glucocorticoid metabolites that are not detected by the assays for UFCs; measurement of 17OHS can therefore be a useful and early diagnostic test for patients with suspected atypical or early CS due to AIMAH.
It should be noted that in two patients with ACS, there was a rise in glucocorticoid secretion during Liddle’s test, albeit modest compared with what has been seen in PPNAD and other mi-cronodular forms of BAH (16, 35, 39). This is consistent with our previous data (16) and may indicate that at least some patients with ACS may be affected by a mild from of micronodular BAH or that PRKAR1A mutations or related genetic defects that were not detected by routine sequencing were present at the tissue level, as we have shown before in at least some cases of AIMAH or ACS (26, 40).
Nine AIMAH patients showed aberrant cortisol responses to at least one stimulus; this response rate was consistent with what has been reported in other studies that used a similar testing protocol (10 –12, 14, 41, 42). In addition, patients with ACS also showed aberrant receptor expression; again, this has also been reported to be a relatively frequent phenomenon (14, 41– 45). To date, the unexpected regulation of cortisol secretion by receptors for various neuroendocrine substances in ACTH-independent CS remains a biological puzzle; both mouse and human studies indicate that in at least some of these patients, this phenomenon is a primary defect, causative of the CS phenotype (41).
Our clinical and molecular genetic data pointed to significant heterogeneity among our cohort of patients with AIMAH: there were three cases with some (but not extensive) family history of adrenocortical tumors. There are so far seven reports of familial AIMAH, all pointing to an autosomal dominant transmission (31, 41, 45– 47). Evidence for heterogeneity included the type of aberrant receptor expression involved in the AIMAH phenotype, the association with various tumors, and various types of germ-line mutations. First, the aberrant receptors associated with fa-milial AIMAH have most frequently be reported to be the va-sopressin (V1 and V2) and the -adrenergic receptors; the
serotonin receptor was present in one family (31, 41, 45– 47). In our study, two of the three familial cases had aberrant vasopres-sin receptors. Second, we found thyroid, parathyroid, and uter-ine leiomyomatous tumors that have all been previously reported in other patients with AIMAH (31, 41, 45– 47); we also found three cases with parotid tumors that have not been previously reported in association with AIMAH.
Finally, an astonishing three of 16 patients (19%) had germ-line genetic defects in MEN1, FH, and APC, all previously re-ported pathogenic mutations in the respective genes. A fourth patient had a GNAS somatic (tumor) mutation (R201H) like two of the patients with AIMAH reported by Fragoso et al. (33); none of these patients had any signs of McCune-Albright syndrome. Another patient (with familial AIMAH) carried the R867G PDE11A gene variant (27, 34), which, although a relatively fre-quent gene variant, is located in a highly conserved region of the PDE11A gene and affects enzymatic activity in vitro (34).
Histologically we were able to subclassify our patients with AIMAH in two groups, those with multiple nodules or discrete adenomas and intervening atrophic cortical tissue (type I AIMAH) and those with diffuse hyperplasia (type II AIMAH). Most of our patients belonged to the second category; the three familial cases and the patients with germline MEN1 and APC and somatic GNAS mutations belonged to the first group. How-ever, we found no association of this histopathological obser-vation with distinct expressed receptor pattern or any other clin-TABLE 3. Mutations and other tumors in patients with ACTH-independent macronodular adrenal hyperplasia and other
adrenocortical tumors
Familial
cases Mutation
Other tumors (number of cases)
Other tumors in family members (number)
AIMAH (n⫽ 16) 3 MEN1 (Pro494Leu)
FH (c.781del7)
APC (c. 4393_4394delAG) GNAS (Arg201His), somatic
Thyroid adenoma (1) Lymphoma (1) Uterine fibroids (5) Parotid tumor (3) Parathyroid adenoma (1) Thyroid cancer (1) in M Prostate cancer (1) in F Lung cancer (1) in M
ACS (n⫽ 15) None None Parathyroid adenoma (1)
Nodular goiter (1)
None
APA (n⫽ 19) None None Thyroid nodule (1) None
SCA (n⫽ 32) None None Thyroid nodule (1)
Parathyroid adenoma (1)
Pancreatic Ca (F, GF on maternal side) Uterine Ca (M)
Cervical Ca (S) Breast Ca (A) Pituitary tumor (F)
ical feature; it remains unclear whether this observation reflects simply different stages of changes in the adrenal cortex during the development of a chronic disease, or it is in fact a direct effect of the underlying molecular etiology.
We conclude that AIMAH is a heterogeneous disorder that is often associated with genetic defects at both the germline or the somatic level. Determination of urinary 17OHS is a useful di-agnostic test for the early detection of abnormal glucocorticoid secretion in this condition; oCRH testing, on the other hand, is not helpful. Histological subgrouping may assist in the future in further investigating the molecular causes of this fascinating condition.
Acknowledgments
We thank the patients and their families who participated in our research studies and donated their time for this investigation. We also want to thank Dr. Madson Q. Almeida for expert technical assistance with arts and graphics. We also thank Drs. Stephen Libutti and Anathea Powell from the National Cancer Institute, National Institutes of Health (NIH), for their collaboration and expert surgical assistance.
Address all correspondence and requests for reprints to: Constantine A. Stratakis, M.D., D(Med)Sc., Section on Endocrinology and Genetics, Program on Developmental Endocrinology and Genetics, National In-stitute of Child Health and Human Development, National InIn-stitutes of Health, 10 Center Drive, Building 10, Clinical Research Center, Room 1-3330, MSC1103, Bethesda, Maryland 20892. E-mail: stratakc@ mail.nih.gov.
This work was supported in part by the National Institute of Child Health and Human Development, intramural NIH project Z01-HD-000642-04 (to Dr. C.A.S.) and in part by the NIH Clinical Center. H.-P.H. was supported for a fellowship at the laboratory of C.A.S. by the Department of Pediatrics, Kaohsiung Municipal Hsiao-Kang Hospital, and College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan.
Disclosure Summary: The authors have nothing to disclose.
References
1. Nieman LK 2001 Cushing’s syndrome. In: Endocrinology. Philadelphia: W.B. Saunders; 1691–1720
2. Stratakis CA, Kirschner LS 1998 Clinical and genetic analysis of primary bi-lateral adrenal diseases (micro- and macronodular disease) leading to Cushing syndrome. Horm Metab Res 30:456 – 463
3. Bourdeau I, Stratakis CA 2002 Cyclic AMP-dependent signaling aberrations in macronodular adrenal disease. Ann NY Acad Sci 968:240 –255 4. Stratakis CA, Kirschner LS, Carney JA 2001 Clinical and molecular features
of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 86:4041– 4046
5. Groussin L, Kirschner LS, Vincent-Dejean C, Perlemoine K, Jullian E, Delemer
B, Zacharieva S, Pignatelli D, Carney JA, Luton JP, Bertagna X, Stratakis CA, Bertherat J 2002 Molecular analysis of the cyclic AMP-dependent protein
kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Car-ney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA sig-naling is associated with adrenal tumorigenesis in PPNAD. Am J Hum Genet 71:1433–1442
6. Lieberman SA, Eccleshall TR, Feldman D 1994 ACTH-independent massive bilateral adrenal disease (AIMBAD): a subtype of Cushing’s syndrome with major diagnostic and therapeutic implications. Eur J Endocrinol 131:67–73 7. Christopoulos S, Bourdeau I, Lacroix A 2005 Clinical and subclinical ACTH-independent macronodular adrenal hyperplasia and aberrant hormone recep-tors. Horm Res 64:119 –131
8. Horiba N, Suda T, Aiba M, Naruse M, Nomura K, Imamura M, Demura H
1995 Lysine vasopressin stimulation of cortisol secretion in patients with ad-renocorticotropin-independent macronodular adrenal hyperplasia. J Clin En-docrinol Metab 80:2336 –2341
9. Lacroix A, Bolte E, Tremblay J, Dupre J, Poitras P, Fournier H, Garon J, Garrel
D, Bayard F, Taillefer R 1992 Gastric inhibitory polypeptide-dependent
cor-tisol hypersecretion—a new cause of Cushing’s syndrome. N Engl J Med 327: 974 –980
10. Lacroix A, Hamet P, Boutin JM 1999 Leuprolide acetate therapy in luteinizing hormone-dependent Cushing’s syndrome. N Engl J Med 341:1577–1581 11. Lacroix A, Tremblay J, Rousseau G, Bouvier M, Hamet P 1997 Propranolol
therapy for ectopic-adrenergic receptors in adrenal Cushing’s syndrome. N Engl J Med 337:1429 –1434
12. Pralong FP, Gomez F, Guillou L, Mosimann F, Franscella S, Gaillard RC 1999 Food-dependent Cushing’s syndrome: possible involvement of leptin in cor-tisol hypersecretion. J Clin Endocrinol Metab 84:3817–3822
13. Reznik Y, Allali-Zerah V, Chayvialle JA, Leroyer R, Leymarie P, Travert G,
Lebrethon MC, Budi I, Balliere AM, Mahoudeau J 1992 Food-dependent
Cushing’s syndrome mediated by aberrant adrenal sensitivity to gastric inhib-itory polypeptide. N Engl J Med 327:981–986
14. Willenberg HS, Stratakis CA, Marx C, Ehrhart-Bornstein M, Chrousos GP,
Bornstein SR 1998 Aberrant interleukin-1 receptors in a cortisol-secreting
adrenal adenoma causing Cushing’s syndrome. N Engl J Med 339:27–31 15. Doppman JL 1997 Problems in endocrinologic imaging. Endocrinol Metab
Clin North Am 26:973–991
16. Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Nieman LK,
Chrousos GP, Papanicolaou DA 1999 Paradoxical response to dexamethasone
in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med 131:585–591
17. Batista DL, Riar J, Keil M, Stratakis CA 2007 Diagnostic tests for children who are referred for the investigation of Cushing syndrome. Pediatrics 120:e575– e586
18. Stratakis CA 2009 New genes and/or molecular pathways associated with adrenal hyperplasias and related adrenocortical tumors. Mol Cell Endocrinol 300:152–157
19. Bourdeau I, D’Amour P, Hamet P, Boutin JM, Lacroix A 2001 Aberrant membrane hormone receptors in incidentally discovered bilateral macronodu-lar adrenal hyperplasia with subclinical Cushing’s syndrome. J Clin Endocrinol Metab 86:5534 –5540
20. Lacroix A, Mircescu H, Jamet P 1999 Clinical evaluation of the presence of abnormal hormone receptors in adrenal Cushing’s syndrome. Endocrinologist 9:9 –15
21. Liddle GW 1960 Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 20:1539 –1560
22. Papanicolaou DA, Yanovski JA, Cutler Jr GB, Chrousos GP, Nieman LK 1998 A single midnight serum cortisol measurement distinguishes Cushing’s syn-drome from pseudo-Cushing states. J Clin Endocrinol Metab 83:1163–1167 23. Gomez MT, Malozowski S, Winterer J, Vamvakopoulos NC, Chrousos GP 1991 Urinary free cortisol values in normal children and adolescents. J Pediatr 118:256 –258
24. Murphy BE 1968 Clinical evaluation of urinary cortisol determinations by competitive protein-binding radioassay. J Clin Endocrinol Metab 28:343–348 25. Gunther DF, Bourdeau I, Matyakhina L, Cassarino D, Kleiner DE, Griffin K,
Courkoutsakis N, Abu-Asab M, Tsokos M, Keil M, Carney JA, Stratakis CA
2004 Cyclical Cushing syndrome presenting in infancy: an early form of pri-mary pigmented nodular adrenocortical disease, or a new entity? J Clin En-docrinol Metab 89:3173–3182
26. Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S,
Papageorgiou T, Bourdeau I, Kirschner LS, Vincent-Dejean C, Perlemoine K, Gicquel C, Bertagna X, Stratakis CA 2003 Molecular and functional analysis
of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Can-cer Res 63:5308 –5319
27. Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ,
Stein E, Levine E, Delimpasi G, Hsiao HP, Keil M, Heyerdahl S, Matyakhina L, Libe R, Fratticci A, Kirschner LS, Cramer K, Gaillard RC, Bertagna X, Carney JA, Bertherat J, Bossis I, Stratakis CA 2006 A genome-wide scan
identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet 38:794 – 800 28. Lemos MC, Thakker RV 2008 Multiple endocrine neoplasia type 1 (MEN1):
analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 29:22–32
29. Miyamura N, Taguchi T, Murata Y, Taketa K, Iwashita S, Matsumoto K,
Nishikawa T, Toyonaga T, Sakakida M, Araki E 2002 Inherited
cortisol secretion by vasopressin and catecholamines: detection of the aberrant hormone receptors on adrenal gland. Endocrine 19:319 –326
30. Perraudin V, Delarue C, Lefebvre H, Do Rego JL, Vaudry H, Kuhn JM 2006 Evidence for a role of vasopressin in the control of aldosterone secretion in primary aldosteronism: in vitro and in vivo studies. J Clin Endocrinol Metab 91:1566 –1572
31. Vezzosi D, Cartier D, Regnier C, Otal P, Bennet A, Parmentier F, Plantavid M,
Lacroix A, Lefebvre H, Caron P 2007 Familial
adrenocorticotropin-indepen-dent macronodular adrenal hyperplasia with aberrant serotonin and vaso-pressin adrenal receptors. Eur J Endocrinol 156:21–31
32. Matyakhina L, Freedman RJ, Bourdeau I, Wei MH, Stergiopoulos SG,
Chidakel A, Walther M, Abu-Asab M, Tsokos M, Keil M, Toro J, Linehan WM, Stratakis CA 2005 Hereditary leiomyomatosis associated with bilateral,
massive, macronodular adrenocortical disease and atypical cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab 90: 3773–3779
33. Fragoso MC, Domenice S, Latronico AC, Martin RM, Pereira MA, Zerbini MC,
Lucon AM, Mendonca BB 2003 Cushing’s syndrome secondary to
adrenocorti-cotropin-independent macronodular adrenocortical hyperplasia due to activating mutations of GNAS1 gene. J Clin Endocrinol Metab 88:2147–2151 34. Horvath A, Giatzakis C, Robinson-White A, Boikos S, Levine E, Griffin K,
Stein E, Kamvissi V, Soni P, Bossis I, de Herder W, Carney JA, Bertherat J, Gregersen PK, Remmers EF, Stratakis CA 2006 Adrenal hyperplasia and
ad-enomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Res 66:11571–11575
35. Powell AC, Stratakis CA, Patronas NJ, Steinberg SM, Batista D, Alexander
HR, Pingpank JF, Keil M, Bartlett DL, Libutti SK 2008 Operative management
of Cushing syndrome secondary to micronodular adrenal hyperplasia. Surgery 143:750 –758
36. Doppman JL, Chrousos GP, Papanicolaou DA, Stratakis CA, Alexander HR,
Nieman LK 2000 Adrenocorticotropin-independent macronodular adrenal
hyperplasia: an uncommon cause of primary adrenal hypercortisolism. Radi-ology 216:797– 802
37. Hoshiro M, Ohno Y, Masaki H, Iwase H, Aoki N 2006 Comprehensive study of urinary cortisol metabolites in hyperthyroid and hypothyroid patients. Clin Endocrinol (Oxf) 64:37– 45
38. Briggs MH, Christie GA, eds 1972 Advances in steroid biochemistry and pharmacology. In. London and New York: Academic Press; 75–77 39. Bourdeau I, Lacroix A, Schurch W, Caron P, Antakly T, Stratakis CA 2003
Primary pigmented nodular adrenocortical disease: paradoxical responses of cortisol secretion to dexamethasone occur in vitro and are associated with increased expression of the glucocorticoid receptor. J Clin Endocrinol Metab 88:3931–3937
40. Bourdeau I, Matyakhina L, Stergiopoulos SG, Sandrini F, Boikos S, Stratakis
CA 2006 17q22–24 chromosomal losses and alterations of protein kinase a
subunit expression and activity in adrenocorticotropin-independent ma-cronodular adrenal hyperplasia. J Clin Endocrinol Metab 91:3626 –3632 41. Lacroix A, Baldacchino V, Bourdeau I, Hamet P, Tremblay J 2004 Cushing’s
syndrome variants secondary to aberrant hormone receptors. Trends Endo-crinol Metab 15:375–382
42. Mircescu H, Jilwan J, NⴕDiaye N, Bourdeau I, Tremblay J, Hamet P, Lacroix
A 2000 Are ectopic or abnormal membrane hormone receptors frequently
present in adrenal Cushing’s syndrome? J Clin Endocrinol Metab 85:3531– 3536
43. Arnaldi G, Gasc JM, de Keyzer Y, Raffin-Sanson ML, Perraudin V, Kuhn JM,
Raux-Demay MC, Luton JP, Clauser E, Bertagna X 1998 Variable expression
of the V1 vasopressin receptor modulates the phenotypic response of steroid-secreting adrenocortical tumors. J Clin Endocrinol Metab 83:2029 –2035 44. Bertherat J, Contesse V, Louiset E, Barrande G, Duparc C, Groussin L, Emy
P, Bertagna X, Kuhn JM, Vaudry H, Lefebvre H 2005 In vivo and in vitro
screening for illegitimate receptors in adrenocorticotropin-independent ma-cronodular adrenal hyperplasia causing Cushing’s syndrome: identification of two cases of gonadotropin/gastric inhibitory polypeptide-dependent hyper-cortisolism. J Clin Endocrinol Metab 90:1302–1310
45. Tatsuno I, Uchida D, Tanaka T, Koide H, Shigeta A, Ichikawa T, Sasano H,
Saito Y 2004 Vasopressin responsiveness of subclinical Cushing’s syndrome
due to ACTH-independent macronodular adrenocortical hyperplasia. Clin Endocrinol (Oxf) 60:192–200
46. Lee S, Hwang R, Lee J, Rhee Y, Kim DJ, Chung UI, Lim SK 2005 Ectopic expression of vasopressin V1b and V2 receptors in the adrenal glands of fa-milial ACTH-independent macronodular adrenal hyperplasia. Clin Endocri-nol (Oxf) 63:625– 630
47. Bourdeau I 2004 Clinical and molecular genetic studies of bilateral adrenal hyperplasias. Endocr Res 30:575–583