Zhi-Qiang You, Chao-Ping Hsu, and Graham R. Fleming
Citation: The Journal of Chemical Physics 124, 044506 (2006); doi: 10.1063/1.2155433 View online: http://dx.doi.org/10.1063/1.2155433
View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/124/4?ver=pdfcov Published by the AIP Publishing
Articles you may be interested in
Electronic couplings for molecular charge transfer: Benchmarking CDFT, FODFT, and FODFTB against high-level ab initio calculations
J. Chem. Phys. 140, 104105 (2014); 10.1063/1.4867077
Definition and determination of the triplet-triplet energy transfer reaction coordinate J. Chem. Phys. 140, 034102 (2014); 10.1063/1.4861560
The fragment spin difference scheme for triplet-triplet energy transfer coupling J. Chem. Phys. 133, 074105 (2010); 10.1063/1.3467882
Structure and excited-state dynamics of anthracene: Ultrahigh-resolution spectroscopy and theoretical calculation
J. Chem. Phys. 130, 134315 (2009); 10.1063/1.3104811
Ab initio calculations of triplet excited states and potential-energy surfaces of vinyl chloride: Insights into spectroscopy and photodissociation dynamics
J. Chem. Phys. 122, 194321 (2005); 10.1063/1.1898208
Triplet-triplet energy-transfer coupling: Theory and calculation
Zhi-Qiang YouInstitute of Chemistry, Academia Sinica, 128 Academia Road Section 2, Nankang, Taipei 115, Taiwan Chao-Ping Hsua兲
Institute of Chemistry, Academia Sinica, 128 Academia Road Section 2, Nankang, Taipei 115, Taiwan and Institute of Molecular Sciences, National Chiao-Tung University, 1001 Ta Hsue Road,
Hsinchu 300, Taiwan Graham R. Fleming
Department of Chemistry, University of California, Berkeley, California 94720
and Lawrence Berkeley National Laboratory, Physical Biosciences Division, Berkeley, California 94720 共Received 3 October 2005; accepted 22 November 2005; published online 25 January 2006兲 Triplet-triplet 共TT兲 energy transfer requires two molecular fragments to exchange electrons that carry different spin and energy. In this paper, we analyze and report values of the electronic coupling strengths for TT energy transfer. Two different methods were proposed and tested: 共1兲 Directly calculating the off-diagonal Hamiltonian matrix element. This direct coupling scheme was generalized from the one used for electron transfer coupling, where two spin-localized unrestricted Hartree-Fock wave functions are used as the zero-order reactant and product states, and the off-diagonal Hamiltonian matrix elements are calculated directly. 共2兲 From energy gaps derived from configuration-interaction-singles共CIS兲 scheme. Both methods yielded very similar results for the systems tested. For TT coupling between a pair of face-to-face ethylene molecules, the exponential attenuation factor is 2.59 Å−1共CIS/6-311+G**兲, which is about twice as large as typical values for electron transfer. With a series of fully stacked polyene pairs, we found that the TT coupling magnitudes and attenuation rates are very similar irrespective of their molecular size. If the polyenes were partially stacked, TT couplings were much reduced, and they decay more rapidly with distance than those of full-stacked systems. Our results showed that the TT coupling arises mainly from the region of close contact between the donor and acceptor frontier orbitals, and the exponential decay of the coupling with separation depends on the details of the molecular contacts. With our calculated results, nanosecond or picosecond time scales for TT energy-transfer rates are possible. © 2006 American Institute of Physics.关DOI:10.1063/1.2155433兴
I. INTRODUCTION
Triplet-triplet 共TT兲 energy transfer is a process of ex-changing both spin and energy between a pair of molecules or molecular fragments. It plays an important role in many photophysical processes in chemistry1–4 and biology.5–7 When a closed-shell molecule is photoexcited to its singlet excited state, it may undergo intersystem crossing 共ISC兲 to reach a triplet state. TT energy transfer may subsequently occur, and this provides a chance to design materials with interesting properties for potentially useful applications共see, for example, Refs. 2–4 and 8兲. In photosynthetic organisms, photoexcitation of chrolophylls 共Chls兲 or bacteriochloro-phylls共Bchls兲 under sunlight inevitably leads to the forma-tion of triplet Chls or Bchls. For the sake of definiteness we focus only on Bchl: Bchl→ h 1 Bchlⴱ→ ISC 3Bchlⴱ. 共1兲
Through spin exchange共SE兲, reactive singlet oxygen is gen-erated:
3
Bchlⴱ+3O2→ SE
Bchl +1O2ⴱ→ oxidative damages. 共2兲 Carotenoids 共Cars兲 in photosynthetic proteins can directly quench triplet Bchls through a TT spin-exchange process, and thereby avoid the formation of reactive singlet oxygen:9
3
Bchlⴱ+1Car→ SE 1
Bchl +3Car, 共3兲
or can quench singlet oxygen directly:10 1O 2 ⴱ+1Car→ SE 3O 2+3Car. 共4兲
The TT energy-transfer process can be viewed as two simultaneous electron transfers with different spin 共␣→␣,→兲共Fig. 1兲. It is similar to the Dexter exchange coupling in the singlet-singlet energy transfer,11which arises from exchanging electrons of the same spin but different energies. Therefore, in addition to the intrinsic importance of understanding the TT energy transfer, an understanding of the coupling mechanism should provide insight into Dexter exchange coupling.
In the singlet-singlet energy transfer, where the spin state of each fragment is conserved, it can be shown that the elec-tronic coupling arises from:11–15 共1兲 Coulombic coupling,
a兲Author to whom correspondence should be addressed. Electronic
mail:[email protected]
THE JOURNAL OF CHEMICAL PHYSICS 124, 044506共2006兲
0021-9606/2006/124共4兲/044506/10/$23.00 124, 044506-1 © 2006 American Institute of Physics This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
which is the Coulomb interaction between electronic transi-tions. Under the dipole approximation, this interaction re-duces to the well-known Förster dipole-dipole coupling;共2兲 Dexter exchange coupling, arising from the exchange inte-grals that account for the indistinguishability of the electrons in many-electron wave functions.11The Dexter coupling ex-ists only at short donor-acceptor distance, since the exchange integrals are expected to decrease steeply with separation. At short distances, or for dipole-forbidden transitions, the dipole approximation breaks down, and the full Coulombic cou-pling may not follow a typical R−6 dipole-dipole form. Higher-order multipole interactions may lead to R−8 or steeper distance dependence. Moreover, if the close-contact geometry does not allow spherical harmonic expansion, there is no simple polynomial distance dependence.16,17Therefore, observation of a distance dependence that is steeper than R−6 is not sufficient to conclude that the Dexter exchange cou-pling is the relevant mechanism 共see, for example, Refs. 18 and 19兲. To help resolve the difficulty of deducing the origin of electronic coupling from the experimental data, it is im-portant to find reliable theoretical estimates of the magnitude of the Dexter coupling strength. Estimating magnitude of the TT coupling, a similar quantity, could shed light on this issue.
Experimental studies have shown that the TT exchange process behaves like two simultaneous electron transfers 共ETs兲.20,21
Recent findings using phenylene oligomers as spacers further support this picture. TT energy-transfer rates between Ru and Os dinuclear metal complexes were reported to decrease with separation exponentially by 0.32, 0.44, and 0.50 Å−1,2,3while optically induced intervalence ET between Ru complexes had exponents of 0.084 or 0.118 Å−1共Ref. 22兲 for the electronic couplings. If the latter values are multiplied by a factor of 2 to convert to ET rates, the exponents for the ET are roughly half of those for the TT energy transfer.
Despite the fundamental importance of TT energy trans-fer in photophysics, theoretical characterization of TT cou-pling has been rather rare. In the 1960s, Jortner and co-workers studied the effect of exchange coupling on charge mobility in organic molecular crystals.23–25Using hydrogen-like orbitals, Levy and Speiser calculated the exchange inte-grals in covalently linked␣-diketones and a number of aro-matic fragments.26 In Ref. 27, the TT exchange coupling between the two radical methylene groups of 1,4-dimethylenecyclohexane was estimated to demonstrate the relationship of TT energy transfer with electron and hole transfers. The Dexter exchange coupling was estimated to be
of the order of 10−4 eV between a Bchl and a neurosporene in a number of arrangements.28 Calculations using the Praiser-Parr-Pople 共PPP兲 Hamiltonian yielded values on the order of 10−5 eV for the TT coupling between Bchls and lycopenes in the bacterial LH2 light-harvesting complex.29 Such coupling strengths lead to transfer rates that are much smaller than those observed experimentally, which are on the nanosecond time scale.8–10 A serious underestimate of the Coulomb coupling from the forbidden S1state of carotenoid is found in Refs. 28 and 29, possibly due to deficiencies in the semiempirically parametrized Hamiltonians.17Therefore, it is important to establish an ab initio based methodology to determine whether the mechanism or calculation method is responsible for the underestimates of exchange couplings.
Recent calculations employing the quantum Monte Carlo 共QMC兲 approach have obtained a singlet-triplet energy gap within experimental errors共0.01 eV兲 for a porphyrin.30Such energy gaps result from the exchange integrals, and therefore provide a route to both TT and Dexter coupling magnitudes. No QMC estimate for TT coupling is yet available, and it is desirable to derive TT coupling values from wave-function-based models for future comparison.
ET coupling has been calculated via quantum chemistry methods, either by using a resonant condition at the transi-tion state, and taking half of the energy gap between the lowest two adiabatic states as the coupling strength,31 or by directly calculating the coupling matrix element term by term using unrestricted Hartree-Fock共UHF兲 charge-localized solutions.32–34In this work we explore whether the methods used for ET coupling can be generalized to calculate TT coupling. In particular, we discuss the theoretical grounds for using the direct coupling 共DC兲 scheme and the energy-gap-based method to obtain TT energy-transfer coupling. Results for both symmetric and asymmetric small test systems will also be discussed.
II. THEORY AND METHODS A. Direct coupling
The DC scheme is described in Refs. 32–35 for ET sys-tems. It has also been used to estimate TT coupling for cyclohexane-spaced methylene radicals.32In this section we discuss the theoretical background of this coupling scheme.
To calculate the off-diagonal Hamiltonian matrix ele-ment, we need two zero-order wave functions that can prop-erly represent the reactant and product states 共“diabatic states” in the ET literature兲.36
In the ET systems, it has been shown that UHF solutions can often provide good approxi-mation for such charge-localized states. We propose that a similar scheme can be used for TT coupling. Namely,⌿r, the reactant state, is modeled by an UHF solution such that the donor fragment is in its triplet state, while the acceptor is in singlet state, and vice versa for⌿p, the product state.
To evaluate the transfer coupling between spin-localized states, we have Trp= Hrp− Srp共Hrr+ Hpp兲/2 1 − Srp 2 , 共5兲 where FIG. 1. A schematic picture of TT energy transfer as two simultaneous
electron transfers between the donor共D兲 and acceptor 共A兲.
Hrp=具⌿r兩Hˆ兩⌿p典 =
冕
dx⌿r *共x兲Hˆ⌿ p共x兲 共6兲 and Srp=具⌿r兩⌿p典 =冕
dx⌿r *共x兲⌿ p共x兲, 共7兲where xiis the spin and spatial coordinates of electron i , H is the Hamiltonian for the system, and Trpis the transfer inte-gral, or transfer-matrix element defined in scattering prob-lems, which is the effective full coupling.⌿rand⌿pare the spin-localized wave functions before and after energy trans-fer, respectively.
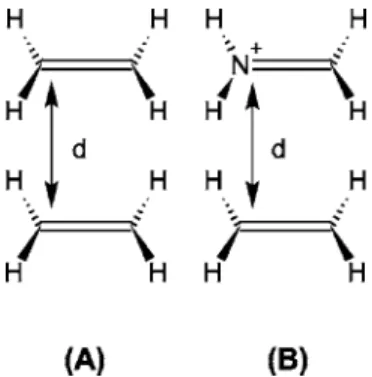
As an illustrative example, the TT coupling of a face-to-face arrangement of two ethylene molecules 关Fig. 2共A兲兴 is discussed. To simplify the problem, we assume that⌿r and ⌿p are composed of the same set of core orbitals, and the differences are only in the four highest occupied spin-orbitals. Assuming that the reactant state is composed of one molecule 共denoted as D兲 in its -* triplet state while the other共denoted as A兲 is in its singlet ground state, 共and vice versa for the the product state兲, we have
⌿r=兩⌿coreDD *
A¯A兩, 共8兲
⌿p=兩⌿coreD¯DAA*兩, 共9兲 where兩¯兩 represents a Slater determinant, a short bar above an orbital共¯兲 denotes aspin-orbital, while an orbital with-out the short bar 共兲 is an ␣ spin-orbital. The leading con-tribution in Trpis37 Trp⬇ Hrp=关D*¯A 储¯DA*兴 =关D *¯ A兩¯DA *兴 − 关 D * A *兩¯ D¯A兴 = −关D * A *兩¯ D¯A兴, 共10兲
where the two-electron integrals are defined as
关ij储kl兴 ⬅ 关ij兩kl兴 − 关il兩kj兴 共11兲
and 关ij兩kl兴 = 关ij兩kl兴 =
冕
dx1dx2*i共x1兲j共x1兲 1 r12 k*共x2兲l共x2兲. 共12兲 As shown in Eq.共10兲, the leading term in Trpis an exchangeintegral, which gives rise to the “exchange” nature of the TT coupling, and is essentially the same as the Dexter coupling integral.11
In DC calculations, the expression in Eq.共5兲 is used. The effects of Srp, as well as details in the different core orbitals, are fully accounted for. The spin-localized UHF solutions⌿r and⌿pwere typically calculated using transition-state geom-etry 关R=0.5 in Eq. 共25兲 below for symmetric system, or at the minimun-energy gap for asymmetric systems兴 from ini-tial solutions that are spin localized, which were obtained from a geometry composed of an optimized triplet and a singlet molecules. For jobs with asymmetric molecules, a quintet state was sometimes used as a starting point to find the spin-localized triplet state.
B. Energy-gap-based method: Configuration-interaction singles
In a two-state model, if the noninteracting states are de-generate, half of the eigenenergy difference is exactly the coupling between reactant and product states. We tested to see if a simple ab initio configuration-interaction-singles 共CIS兲 scheme38
can give reasonable results for exchange couplings.
We performed a CIS calculation with a singlet reference while solving for the lowest two triplet states. For two eth-ylenes separated at 4.5 Å, the first two triplet states obtained via CIS/ 6-31G* from the singlet ground state reference, in their Ms= 1 configurations, are
⌿1 CIS= − 0.6752兩⌿ core12¯24兩 + 0.7217兩⌿core1¯123兩 + ¯ , 共13兲 ⌿2CIS= − 0.6558兩⌿core12¯23兩 + 0.7019兩⌿core1¯124兩 + ¯ , 共14兲 where⌿coredenotes a collection of core molecular orbitals. To gain insights from the two solutions, we assumed that the ideal solutions are composed of two equally populated single-excitation configurations, with other minor contribu-tions ignored: ⌿1 CIS⬇ − 共2兲−1/2兩⌿ core12¯24兩 +共2兲−1/2兩⌿core1¯123兩, 共15兲 ⌿2 CIS⬇ − 共2兲−1/2兩⌿ core12¯23兩 +共2兲−1/2兩⌿core1¯124兩. 共16兲 In the following, we further analyze the lowest two CIS trip-let wave functions to see if they are linear combinations of spin-localized transitions.
In a symmetric arrangement, the molecular orbitals are delocalized. The lowest CIS excited states are excitations from the delocalized orbitals 共denoted as below兲, mainly composed of the localized orbitals and*orbitals: FIG. 2. Face-to-face arrangements of共A兲 two ethylenes, and 共B兲 an ethylene
and a methaniminium cation. d denotes the intermolecular distance.
044506-3 Triplet-triplet energy-transfer coupling J. Chem. Phys. 124, 044506共2006兲
⬇
冑
12 ± 2s共D±A兲. 共17兲
Here, s =兰dxD共x兲A共x兲 is the overlap integral of the two localized orbitals. In order to keep only the major character-istics, the overlap integral s is ignored as a further simplifi-cation. The approximate compositions of the four delocalized orbitals involved in the two lowest CIS excited states are
1⬇ 2−1/2共A−D兲, ¯1⬇ 2−1/2共¯A−¯D兲, 2⬇ 2−1/2共D+A兲, ¯2⬇ 2−1/2共¯D+¯A兲, 共18兲 3⬇ 2−1/2共D * −A *兲, ¯ 3⬇ 2−1/2共¯D * −¯A*兲, 4⬇ 2−1/2共D * +A *兲, ¯ 4⬇ 2−1/2共¯D * +¯A*兲,
where D denotes one of the fragment named donor, and A is the other共acceptor兲. A/D andA/D
*
are localized molecular orbitals involved in the TT energy-transfer process.
We next show that spin-localized states can be obtained from a linear combination of⌿1CISand⌿2CIS:
1
冑
2共⌿1 CIS +⌿2CIS兲 =14共− 兩⌿core共A−D兲共D+A兲共¯D+¯A兲D*兩 +兩⌿core共A−D兲共¯A−¯D兲共D+A兲D *兩兲 =12兩⌿core共A−D兲¯A共D+A兲D *兩 =兩⌿coreDD * A¯A兩 ⬇ ⌿r loc , 共19兲which is a configuration with its spin localized in the donor fragments. We therefore assigned it as the reactant state. In the derivation of Eq. 共19兲, standard operations for determi-nants were used 共for example, see Ref. 39兲. Similarly, the other state from a linear combination of⌿1CISand⌿2CISis
1
冑
2共⌿1 CIS−⌿ 2 CIS兲 =14共兩⌿core共A−D兲共D+A兲共¯D+¯A兲A*兩 +兩⌿core共A−D兲共¯A−¯D兲共D+A兲A *兩兲 =兩⌿coreD¯DAA*兩 ⬇ ⌿ploc. 共20兲 Therefore, the two CIS triplet states are mainly com-posed of two spin-localized states. To see if the energy gap derived from the two states gives rise to TT coupling, we have37E1CIS=具⌿1CIS兩Hˆ兩⌿1CIS典
=12共EHF+⑀¯2−⑀3−关33 储¯2¯2兴 + EHF+⑀1¯−⑀
4−关44 储¯1¯1兴
− 2关43 储¯2¯1兴兲, 共21兲
E2CIS=具⌿2CIS兩Hˆ兩⌿2CIS典
=12共EHF+⑀¯1−⑀3−关33 储¯1¯1兴 + EHF+⑀2¯−⑀4−关44 储¯2¯2兴
− 2关43 储¯1¯2兴兲, 共22兲 where⑀iis the molecular-orbital energy fori共i.e., an eigen-value of the Fock matrix兲, and the two-electron integral 关¯储¯兴 is as defined in Eq. 共11兲. The energy splitting of two states in the molecular-orbital representation is therefore
E2CIS− E1CIS=21共关33兩¯2¯2兴 − 关33兩¯1¯1兴
+关44兩¯1¯1兴 − 关44兩¯2¯2兴兲. 共23兲 With Eq.共18兲, we convert the delocalized orbitals in Eq. 共23兲 to the localized donor and acceptor molecular orbitals:
E2CIS− E1CIS= − 2关D*A*兩¯D¯A兴, 共24兲 which is twice the coupling in Eq. 共10兲, and again it is ap-proximately an exchange integral, arising from the indistin-guishability of electrons.
C. Computational details
A developmental version of theQ-CHEMquantum chem-istry program package was used for all calculations presented in this work.40 The direct coupling scheme was integrated with formulas previosly reported.35The optimized geometry for singlet and triplet single molecules was calculated using density-functional theory共DFT兲/B3LYP with DZP basis sets. For simple test systems composed of small molecules, an approximate reaction coordinate R is often used in the literature:34 Qi共R兲 = 共1 − R兲Qi r + RQi p , 0艋 R 艋 1, 共25兲
where Qirepresents the ith nuclear coordinate, superscripts r and p refer to the reactant and product nuclear coordinates that are composed of optimized singlet and triplet molecules, respectively, and R is the reaction coordinate 共R=0 for the reactant and R = 1 for the product兲. In the present work, we simply used the Cartesian coordinates 共xi, yi, zi兲, with the symmetric center as the origin, for Qi. For a symmetric sys-tem such as the one shown in Fig. 2共A兲, R=0.5 was used for an approximate transition state.
III. RESULTS AND DISCUSSION A. Direct coupling
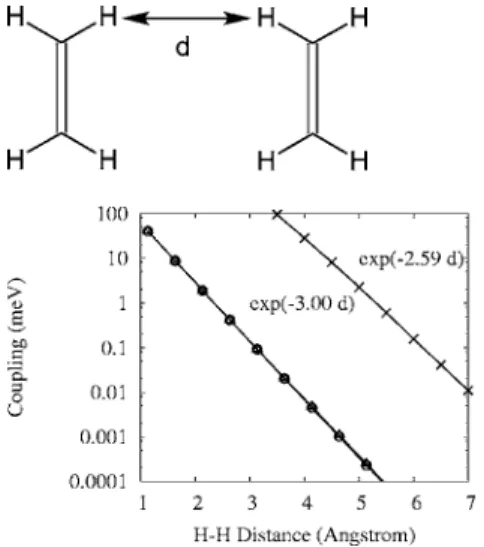
The TT electronic coupling between two face-to-face ethylenes共as depicted in Fig. 2兲 was calculated using Eq. 共5兲. Results with different basis sets and as a function of inter-molecular distance are shown in Fig. 3. Results from small basis sets show a deviation from a straight line in the semilog plot; this reflects the limitations of Gaussian basis functions. The couplings calculated from large, diffusive basis sets ex-hibit an exponential distance dependence, a characteristic property of TT electronic coupling arising from the exchange integral.11,20
According to Eq.共10兲, the major term in the TT coupling depends on the overlaps of the two highest occupied molecu-lar orbitals共HOMOs兲 and two lowest unoccupied molecular orbitals共LUMOs兲 of the two interacting fragments; while for ET, the coupling is roughly the Fock matrix element between two HOMOs关for hole transfer 共HT兲兴 or LUMOs 共for ET兲.35 Therefore, the exponential distance dependence is an impor-tant characteristic of the TT coupling. For our results, the slope of the distance dependence of the coupling was fitted to be 2.82 Å−1共DZP basis兲 or 2.59 Å−1共6-311+G*basis兲 for couplings with d = 3.5–6.5 Å. ET of the same two-ethylene system gives an exponent of 1.20 Å−1 共DZP兲 for ET and 1.44 Å−1共DZP兲 or 1.23 Å−1共6-311+G*兲 for HT.41The expo-nent of TT coupling is slightly more than the sum of the exponents derived from the ET and HT couplings. This result is similar to those reported previously. With saturated hydro-carbon spaced electron donors and acceptors, a slope of 2.6 per bond was reported for the TT energy-transfer rates,20 while for ET it was 1.15 perbond. TT energy-transfer rates in phenylene oligomer-spaced Ru and Os dinuclear metal complexes were reported to attenuate exponentially by 0.32, 0.44, and 0.50 Å−1.2,3
Optically induced intervalence ET couplings between phenylene oligomer-spaced Ru com-plexes decay exponentially with coefficients of 0.084 or 0.118 Å−1.22
Multiplying the latter values by a factor of 2 to convert to the ET rates, we can see that the decay coefficients for the ET rates are roughly half of those for the TT energy transfer.
B. Couplings derived from CIS
In Fig. 4, half of the CIS energy gaps between the two lowest triplet states of two ethylenes are presented. In all
cases, the two lowest triplet states are mainly composed of configurations where electrons are excited from the two highest occupied MOs to the two lowest unoccupied MOs. These resulst are very close to those from the DC scheme 共deviations are smaller than 5%: see Table I兲. This result confirms the two-state approximation employed in Sec. II and shows that both DC and CIS can properly describe TT coupling.
In Fig. 4, we have included the results using 6-311 + G* and aug-cc-pVTZ, two different basis sets that include diffusive functions. These calculations were performed as a test to see whether the long-range exponential decay is an artificial result that depends on the basis function used. The diffusive functions for carbon atoms in the 6-311+ G*set are
s and p type functions, both with a Gaussian exponent of
0.0438,42 while those in the aug-cc-pVTZ set are an s func-tion with an exponent of 0.044 02, and a set of p funcfunc-tions with an exponent of 0.035 69.43 As shown in Fig. 4, the different Gaussian exponents did not change the magnitude nor the slope of the exponential decay of TT coupling. Therefore, we conclude that the couplings obtained are not artifacts of the basis functions.
C. Moving along the reaction coordinate
The energy-gap-based method relies on a resonance be-tween the zero-order states to obtain the coupling. For asym-metric systems, this condition is not automatically fulfilled. In calculating ET couplings, an external electric field is often used to achieve the resonance condition. When scanning over the field strength, the minimal energy gap gives twice TABLE I. TT energy transfer coupling共in meV兲 for a pair of ethylenes 关Fig. 2共A兲兴 calculated by direct coupling
共DC兲 or configuration-interaction singles 共CIS兲.
Basis set 6 − 31G* DZP 6 − 311+ G*
Distance共Å兲 DC CIS DC CIS DC CIS
3.5 85.9 82.7 96.5 92.3 99.1 94.2
4.0 20.6 20.1 26.9 26.0 28.5 27.6
4.5 4.03 3.95 7.44 7.23 8.15 7.97
5.0 0.590 0.581 2.01 1.97 2.28 2.24
FIG. 3. Distance and basis set dependence of TT coupling, calculated from the DC scheme for the two-ethylene system. Bases sets used are as follows: 3-21G 共filled squares兲, 6-31G* 共open squares兲, DZP 共filled circles兲, and
6-311+ G*共open circles兲.
FIG. 4. Distance and basis set dependence of TT coupling. Shown are half of the CIS energy gaps between the two lowest triplet states for the two-ethylene system. Basis sets used are as follows: 3-21G 共filled squares兲, 6-31G*共open squares兲, DZP 共filled circles兲, 6-311+G*共open circles兲, and
aug-cc-pVTZ共open triangles兲.
044506-5 Triplet-triplet energy-transfer coupling J. Chem. Phys. 124, 044506共2006兲
the coupling value. For TT coupling, we propose varying the geometry along the reaction coordinate to control the spin localization. In Fig. 5 we report the potential-energy curves of the two lowest triplet states of an asymmetric system which is composed of a pair of isoelectronic fragments, an ethylene and a methaniminium cation, as depicted in Fig. 2共B兲.44
To find the minimum-energy gap in the ethylene-methaniminium ion, we have calculated the energies of the two lowest triplet states along the reaction coordinate. Both the CIS and DC results are included in Fig. 6. It is seen that, at separations of 3.5–5.0 Å, the CIS data closely follow the DC couplings, but are reduced by 15%–30%. Compared to the results in Table I, the discrepancy between the CIS and DC data was larger but still within the same order of magni-tude. In this case, CIS and DC yielded different potential-energy curves for the two states, leading to different posi-tions 共R values兲 for the transition state. Such an inconsistency may increase the discrepancy in coupling, as observed in Fig. 6.
D. Effects of size and intermolecular contacts
The TT coupling strengths decay steeply with increasing separation. Therefore, we expect them to depend strongly on the intermolecular contacts, since only the interactions of the nearest portions of orbitals contribute. We tested to see how the TT coupling magnitudes vary with molecular size and contact area. TT coupling between two all-trans polyenes was calculated for three different conformations, as depicted in Fig. 7. In one conformation 共A兲, maximum contact be-tween the planar molecules was created by having them fully stacked, while in the other two conformations关共B兲 and 共C兲兴, only the terminal C⫽C double bonds of the molecule were placed on top of each other.
In Fig. 8共A兲, we show the TT coupling of two polyenes derived from the CIS energy gaps with a large basis set 共6-311+G*兲 over a range of separations. For the fully stacked configuration, the coupling strengths from different molecules are very similar, irrespective of their sizes. The results for partial contact are shifted towards smaller values. The ratios of the coupling strengths from the two configura-tions vary. For butadiene, for example, the TT coupling from the fully stacked conformation is about 2.8–3.0 times that of the partially stacked one. The extent of this reduction varies as the configuration changes. In Fig. 8共B兲, we see that, while keeping one pair of CvC bonds stacked, but changing the overall configuration关as depicted in Fig. 7共C兲兴, the coupling magnitudes are further reduced, and the exponential distance dependence becomes steeper.
The weak dependence on molecular size in the full-stacked configurations can be shown to result from molecular-orbital normalization. In a simplified representa-tion, the HOMO and LUMO of polyene oligomers are ap-proximated as normalized linear combinations of the and
*of every ethylenelike unit, which are
*⬇ 1
冑
2n兺
i n 共− 1兲i−1 i *, 共26兲FIG. 5. Potential-energy curves of the two lowest triplet states for the ethylene-methaniminium cation system关as in Fig. 2共B兲兴. The reaction coor-dinate is as described in Eq.共25兲. Calculations were performed at CIS/6 -31G*level, with an intermolecular distance fixed at 4.5 Å. The inset is a
magnified plot of the region near the curve crossing, with grids representing 0.01共abscissa兲 and 0.002 hartrees 共ordinate兲.
FIG. 6. TT energy-transfer coupling for the asymmetric ethylene-methaniminium ion system, face to face stacked, as shown in Fig. 2共B兲. The coupling calculated by HF-CIS共open symbols with dashed lines兲 and DC 共closed symbols and solid lines兲 with 6-31G*共squares兲, DZP 共circles兲, and
6-311+ G*共triangles兲 basis sets. DC results were obtained at the
transition-state geometry, i.e., where the minimum CIS energy gap was found.
FIG. 7. Stacked pairs of hexatrienes in three different arrangements.共A兲 A maximum-contact is allowed. Panels共B兲 and 共C兲 show two configura-tions where the close-contact area exists only in the terminal CvC bonds. Pairs of all-trans butadienes and octatetraenes in similar arrange-ments were also studied.
¯ ⬇ 1
冑
2n兺
i n 共− 1兲i−1¯ i, 共27兲where n is the number of double-bond units in the molecule.
i and i* are the and* orbitals of the ith ethylenelike unit. Thus, the overlap integrals between two HOMOs or LUMOs are 具¯D兩¯A典 = 1 2n
冉
兺
i n 具¯D,i兩¯A,i典 +兺
i⫽j n 共− 1兲i+j具¯ D,i兩¯A,j典冊
, 共28兲 具D *兩 A *典 = 1 2n冉
兺
i n 具D,i * 兩 A,i * 典 +兺
i⫽j n 共− 1兲i+j具 D,i * 兩 A,j * 典冊
, 共29兲 where具i兩j典 denotes the inner product, or the overlap inte-gration over spin and spacial coordinates fori共x兲 andj共x兲. The factor 1 / 2n outside the parentheses is the normalization factor. The first summation in the parentheses is the overlap arising from the directly stacked - overlaps, while the other term is the sum of all nondirectly stacked-overlap integrals.Using a small STO-3G basis, we estimated the contribu-tions of the leading terms. At a distance of 3.5 Å, the overlap
integral for directly stackedbonding orbitals is 0.385, and for * antibonding orbitals, it is 0.148. The overlap integral values for a 共*兲 orbital to the next neighboring 共*兲 orbital drop by a large factor, to 0.007 56 共兲 and 0.003 63 共* orbitals兲. Therefore, the contributions of nondirectly stacked orbitals are almost negligible in the overlap inte-grals. The overlap integrals as shown in Eqs. 共28兲 and 共29兲 essentially become independent of n, the size of the mol-ecule. The TT coupling, which is essentially a Coulomb interaction between two overlap densities 关Eq. 共10兲兴, is therefore weakly dependent on molecular size, as seen in Fig. 8共A兲.
In a partially contacted configuration as in Fig. 7共B兲, when the contact area remains the same 共a pair of carbon atoms stacked兲, the coupling strengths become smaller with larger molecules 关Fig. 8共A兲兴. This result indicates that the
relative fraction of the contacting region compared to the
delocalized molecular orbital is a determining factor for TT coupling.
In Fig. 8共B兲, it is shown that the distance attenuation factor becomes larger when there is only a partial contact between the molecules. This result, in part, reflects the com-plexity of the intermolecular coupling. With weakly interact-ing molecules, there exists an inductive effect, where the asymptotic decay of molecular orbitals is affected by the geometrical arrangements. The asymptotic potential is re-duced by the presence of another molecule, leading to a slower decay in the orbital than in a vacuum. Therefore, when the two molecules are only partially stacked, with the nonstacked parts of the molecule isolated in space, the cou-pling extension over the space is reduced. From Figs. 8共A兲 and 8共B兲 we conclude that both the TT coupling strength and exponential decay slope depend strongly on the intermolecu-lar configurations.
Another determining factor is the orientation of molecu-lar contacts. Since the characteristic length of H-like orbitals does not vary with the orientation, one may expect that the distance dependence is weakly influenced by different orien-tations. However, for anisotropic molecules, this property will no longer hold, as is seen in the results of our tests on a pair of side-by-side ethylenes 共Fig. 9兲. Again, with two dif-ferent diffusive basis sets共6-311+G**and DZ+**兲, we find that the results are not dependent on the different diffusive Gaussian exponents.
Optimal overlap gives a coupling strength of⬃100 meV at a distance of 3.5 Å, which can serve as an upper bound in estimating TT coupling for -conjugated molecules. Using the golden rule rate expression, assuming that the overlap of density of states of the reactants and products is of the order of 0.1 eV−1, and using a coupling strength of 100 meV, gives a TT exchange lifetime of 0.1 ps. A nanosecond TT energy-transfer lifetime corresponds to a coupling of about 1 meV, which is roughly the coupling arising from 1 / 4 of intermo-lecular contact as in the nonstacked configuration of octatet-raenes at a distance of 4.5 Å.
E. Nature of TT couplings
We have reported TT energy-transfer coupling strengths with two different Hartree-Fock-based approaches. TT cou-FIG. 8. Dependence of molecular size and intermolecular contacts for TT
couplings.共A兲 Couplings are for a pair of butadienes 共triangles兲, hexatrienes 共inverse triangles兲, and octatetraenes 共squares兲 at a number of intermolecular distances. The open symbols are for the results from a fully stacked con-figuration关Fig. 7共A兲兴 and the closed symbols represent the results from the flipped configuration关Fig. 7共B兲兴. Data for a pair of ethylenes 共open circles兲 were also included for comparison.共B兲 For a pair of hexatrienes, we show the coupling of fully stacked共open squares兲, partially stacked as in Fig. 7共B兲 共filled squares兲, and as in Fig. 7共C兲 共open diamonds兲 configurations. All the results are from CIS/ 6-311+ G*calculations.
044506-7 Triplet-triplet energy-transfer coupling J. Chem. Phys. 124, 044506共2006兲
pling mainly arises from an exchange integral, with a steep distance dependence. We showed that the DC scheme, which has been mainly used for the ET coupling, can also be used to calculate the TT coupling if spin-localized UHF solutions are used. The calculated values from two distinct methods, the DC method and the CIS energy-gap method, agree well for both symmetric and asymmetric test systems, indicating the intrinsic consistency of the two methods.
Compared with previous calculations,28,29our ab initio TT couplings are larger by at least two orders of magnitude. The previous work was based on the PPP Hamiltonian, which ignores terms arising from the exchange integral in Eq.共10兲. Therefore, it is impossible to calculate the exchange interaction directly from the Hamiltonian. Instead, in Refs. 28 and 29, the authors directly integrated the necessary terms, assuming certain linear combination of Gaussian or Slater-type orbitals. In Ref. 29, the influence of superex-change through other atoms was also included. It is not clear to us what the exact source of the large discrepancy between our ab initio results and the previous semiempirical calcula-tions is, and below we discuss more possibilities.
For ET couplings, calculations using semiempirical Hamiltonians typically yield values similar to those of
ab initio models.45,46 Recently, Chen and Hsu found that
ab initio ET couplings are larger by a factor of 3 and decay
more slowly than those from a semiempirical Hamiltonian.47 Using H-like orbitals, Levy and Speiser’s data26 found an exponent for the exchange integral to be about 4.5 Å−1, as inferred from Fig. 6 of Ref. 26, a much larger value than we found. In Ref. 29, the smallest exponent used for the 2p atomic orbitals was 1.054 bohrs−1, leading to a steep expo-nential distance attenuation共about 4 Å−1 for exchange inte-grals兲. The differences in attenuation rates may in part ac-count for the large discrepancy between our results and previous calculations. The weaker distance dependence we observe may also arise from the change of the asymptotic potential due to the presence of another molecule. All atoms or molecules have positive electron affinities, and therefore
the asymptotic potential is lower than in a vacuum. As a result, an electron is likely to extend to a larger distance when it is mediated by an atom or a molecule. With semi-empirical Hamiltonians, the interactions among orbitals are limited by the parametrization, which may fall off steeply with distance since the presence of a neighboring molecule is not considered. Such an effect may be more significant for TT coupling than for ET, since the exchange integral is com-posed of four molecular orbitals, while only two molecular orbitals are involved in the major term of ET coupling,.
The results from varying molecular sizes and contact area indicate that the TT coupling is mainly determined by the relative contact area, with respect to the size of molecular orbitals. When the polyene molecules were 100% stacked, the coupling strengths and attenuation rates were very simi-lar irrespective of their sizes. On the other hand, when there was only a fraction of the molecules in close contact, the coupling strengths dropped by large factor. The two different partially stacked configurations exhibited different exponents for the coupling, and both were larger than that of the fully stacked molecules. A pair of H–H 共side-by-side兲 contacting ethylenes also gave rise to a steeper distance dependence. This indicates that the the rate of exponential attenuation depends on how the molecules are stacked and on the orien-tation of the interactingorbitals.
With hydrogen-like orbitals, the distance dependence of exchange rate can be estimated to follow exp共−2R/L兲, where
L is the average orbital radius involved in the electron
exchange.26 In much of the experimental literature, L has been replaced by the van der Waals or Bohr radii for estimat-ing the Dexter couplestimat-ing共e.g., Refs. 18 and 19兲. For example, for porphyrins, L = 4.8 Å was used as a guide to interpret the experimental results,18 which led to an attenuation rate of 0.416 Å−1, much smaller than our calculated value, 2.6 Å−1. The most serious problem in using this rough estimate is that it is very difficult to properly estimate L. The assumption of hydrogenlike orbitals is only valid for transitions to Rydberg states. For valence triplet states, L is probably close to the characteristic radius of a carbon p orbital, since theand* orbitals involved are mainly composed of these atomic orbit-als, instead of the size of the full molecule. This is also illustrated in Fig. 8共A兲, where the exponential decay slopes are very similar, irrespective of their sizes.
For the singlet-singlet energy transfer, we would like to stress that a deviation from R−6dependence at short distance does not rule out the Coulomb contribution to the coupling. We showed that a reasonable upper bound for exchange cou-pling is about 100 meV, obtained at 3.5 Å, with a full - contact. The Coulomb couplings can be at least this large at short distances. When separated by short distances, the singlet-singlet energy-transfer couplings may be a mixture of Coulomb and exchange elements, both with distance depen-dence that is steeper than the typical dipole-dipole R−6.
F. TT energy-transfer rates
TT energy transfer between Chls and Cars is of great importance in the photoprotection of plants and photosyn-thetic bacteria. With previously calculated values it was dif-FIG. 9. Effects of orientations in TT couplings. Couplings are for a pair of
side-by-side ethylenes intermolecular distances obtained by CIS/ DZ+**
共circles兲 and 6-311+G** 共triangles兲. For comparison, the CIS/6-311+G*
results for face-to-face ethylenes are included共crosses兲.
ficult to understand the difference in magnitudes of the ob-served rates and the theoretically calculated ones. In this section we briefly explore whether our calculated coupling is adequate to explain the experimental data.
The recently published crystal structure of the major light-harvesting complex II 共LHCII兲 of green plants shows many close contacts between Chls and Cars.48–50The LHCII crystal was further shown to be in a dissipative state where chlorophyll fluorescence is quenched.50 In the crystal struc-ture, the two luteins are at a distance of 3.6 Å from two Chl
a molecules.49The-contact is about 1 / 2 − 1 / 3 of the full
delocalization area in the two pairs of molecules. From our results described above, we can roughly estimate the TT cou-pling between the lutein and Chl a to be of the order of 10 meV. The actual TT transfer rate depends on the degree of overlap in the density of states, but the coupling in this range suggests the TT energy transfer could be in the picosecond range.
The triplet state kinetics of Car and Bchl a phosphores-cence in the LH2 antenna complex from purple bacteria was measured recently.8 The decay times of the triplet states of Car and Bchl are 2.0 and 1.8 ns, respectively. A triplet-triplet annihilation reaction was proposed. In this mechanism, both triplet states Bchl and Car simultaneously annihilate, and Qy singlet Bchl is generated. TT annihilation is similar to TT energy-transfer, which also involves the exchange of two electrons of different spin and energy. An electronic coupling of 10 meV could easily lead to the observed nanosecond lifetimes of the triplet state species.
IV. CONCLUSIONS
We have developed two different approaches to calculate TT energy-transfer couplings between a pair of molecules. For both asymmetric and symmetric test systems, the direct coupling method and the energy-gap-based CIS scheme yielded very similar results. Tests using basis sets with dif-ferent diffusive functions yielded essentially the same re-sults, indicating that the values we obtained are consistent within the Hartree-Fock theoretical framework. With a series of fully stacked polyene oligomer pairs, we found that the TT coupling strengths and attenuation rates are very similar, irrespective of the size of the -conjugated molecules. For partially stacked configurations, the coupling magnitudes are reduced as the relative sizes of the contact regions are re-duced. For closely spaced molecules, the calculated TT cou-pling values imply picosecond or nanosecond TT exchange time scales.
ACKNOWLEDGMENTS
Two of the authors共C.P.H. and Z.Q.Y.兲 acknowledge the National Science Council of Taiwan 共Grant No. NSC93-2113-M-001-012兲. One of the authors 共Z.Q.Y.兲 wishes to ac-knowledge financial support from Academia Sinica. The work at Berkeley was supported by the Director, Office of Science, Office of Basic Energy Sciences, Chemical Sci-ences Division, of the U.S. Department of Energy under Contract No. DE-AC03-76SF00098.
1R. Ziessel, M. Hissler, A. El-Ghayoury, and A. Harriman, Coord. Chem.
Rev. 178, 1251共1998兲.
2B. Schlicke, P. Nelser, L. De Cola, E. Sabbioni, and V. Balzani, J. Am.
Chem. Soc. 121, 4207共1999兲.
3A. D’Aéo, S. Welter, E. Cecchetto, and L. De Cola, Pure Appl. Chem. 77, 1035共2005兲.
4R. R. Islangulov, D. V. Kozlog, and F. N. Castellano, Chem. Commun.
共Cambridge兲 30, 3776 共2005兲.
5The Photochemistry of Carotenoids, edited by H. A. Frank, A. J. Young,
G. Nritton, and R. J. Cogdell共Kluwer, The Netherlands, 1999兲.
6N. I. Krinsky, Philos. Trans. R. Soc. London, Ser. B 284, 581共1978兲. 7R. J. Cogdell and H. A. Frank, Biochim. Biophys. Acta 895, 63共1987兲. 8F. S. Rondonuwu, T. Taguchi, R. Fujii, K. Yokoyama, Y. Koyama, and Y.
Watanabe, Chem. Phys. Lett. 384, 364共2004兲.
9R. J. Cogdell, T. D. Howard, R. Bittl, E. Schlodder, I. Geisenheimer, and
W. Lubitz, Philos. Trans. R. Soc. London, Ser. B 355, 1345共2000兲.
10C. S. Foote and R. W. Denny, J. Am. Chem. Soc. 90, 6233共1968兲. 11D. L. Dexter, J. Chem. Phys. 21, 836共1953兲.
12T. Förster, Ann. Phys.共Leipzig兲 2, 55 共1948兲.
13R. McWeeny, Methods of Molecular Quantum Mechanics, 2nd ed.
共Aca-demic Press, London, 1992兲.
14C.-P. Hsu, G. R. Fleming, M. Head-Gordon, and T. Head-Gordon, J.
Chem. Phys. 114, 3065共2001兲.
15G. D. Scholes, Annu. Rev. Phys. Chem. 54, 57共2003兲.
16B. P. Krueger, G. D. Scholes, and G. R. Fleming, J. Phys. Chem. B 102,
5378共1998兲.
17C.-P. Hsu, P. J. Walla, M. Head-Gordon, and G. R. Fleming, J. Phys.
Chem. B 105, 11016共2001兲.
18S. Faure, C. Stern, R. Guilard, and P. D. Harvey, J. Am. Chem. Soc. 126,
1253共2004兲.
19J. A. Mondal, G. Ramakrishna, A. K. Singh, H. N. Ghosh, M. Mariappan,
B. G. Maiya, T. Muskherjee, and D. K. Palit, J. Phys. Chem. A 108, 7843 共2004兲.
20G. L. Closs, P. Piotrowiak, J. M. MacInnis, and G. R. Fleming, J. Am.
Chem. Soc. 110, 2652共1988兲.
21G. L. Closs, M. D. Johnson, J. R. Miller, and P. Piotrowiak, J. Am. Chem.
Soc. 111, 3751共1989兲.
22J.-P. Launay, Chem. Soc. Rev. 30, 386共2001兲.
23J. L. Katz, J. Jortner, S. I. Choi, and S. A. Rice, J. Chem. Phys. 39, 1897
共1963兲.
24J. Jortner, S.-I. Choi, J. L. Katz, and S. A. Rice, Phys. Rev. Lett. 11, 323
共1963兲.
25J. Jortner, S. A. Rice, J. L. Katz, and S. I. Choi, J. Chem. Phys. 42, 309
共1964兲.
26S.-T. Levy and S. Speiser, J. Chem. Phys. 96, 3585共1992兲.
27N. Koga, K. Sameshima, and K. Morokuma, J. Phys. Chem. 97, 13117
共1993兲.
28H. Nagae, T. Kakitani, T. Katoh, and M. Mimuro, J. Chem. Phys. 98,
8012共1993兲.
29A. Damjanovic, T. Ritz, and K. Schulten, Phys. Rev. E 59, 3293共1999兲. 30A. Aspuru-Guzik, O. E. Akramine, J. C. Grossman, and W. A. Lester, Jr.,
J. Chem. Phys. 120, 3049共2004兲.
31A. H. A. Clayton, G. D. Scholes, K. P. Ghiggino, and M. N.
Paddon-Row, J. Phys. Chem. 100, 10912共1996兲.
32K. Ohta, G. L. Closs, K. Morokuma, and N. J. Green, J. Am. Chem. Soc. 108, 1319共1986兲.
33A. Broo and S. Larsson, Chem. Phys. 148, 103共1990兲.
34A. Farazdel, M. Dupuis, E. Clementi, and A. Aviram, J. Am. Chem. Soc. 112, 4206共1990兲.
35L. Y. Zhang, R. A. Friesner, and R. B. Murphy, J. Chem. Phys. 107, 450
共1997兲.
36M. D. Newton and N. Sutin, Annu. Rev. Phys. Chem. 35, 437共1984兲. 37A. Szabo and N. S. Ostlund, Modern Quantum Chemistry: Introduction
to Advanced Electronic Structure Theory, 1st ed. 共McGraw-Hill, New
York, 1993兲.
38J. B. Foresman, M. Head-Gordon, J. A. Pople, and M. J. Frisch, J. Phys.
Chem. 96, 135共1992兲.
39E. Kreyszig, Advanced Engineering Mathematics, 8th ed.共Wiley, New
York, 1999兲.
40J. Kong, C. A. White, A. I. Krylov et al., J. Comput. Chem. 21, 1532
共2000兲.
41Z.-Q. You, Y. Shao, and C.-P. Hsu, Chem. Phys. Lett. 390, 116共2004兲. 42T. Clark, J. Chandrasekhar, G. W. Spitznagel, and P. v. R. Schleyer, J.
Comput. Chem. 4, 294共1983兲.
044506-9 Triplet-triplet energy-transfer coupling J. Chem. Phys. 124, 044506共2006兲
43R. A. Kendall, T. H. Dunning, Jr., and R. J. Harrison, J. Chem. Phys. 96,
6796共1992兲.
44M. Rust, J. Lappe, and R. J. Cave, J. Phys. Chem. A 106, 3930共2002兲. 45M. D. Newton, in Cluster Models for Surface and Bulk Phenomena,
NATO Advance Studies Institute, Series B: Physics, edited by G. Pac-chioni, P. S. Bagus, and F. Parmigiani共Plenum, New York, 1992兲, Vol. 283, p. 551.
46M. D. Newton, J. Phys. Chem. 92, 3049共1988兲.
47H.-C. Chen and C.-P. Hsu, J. Phys. Chem. A共in press兲.
48Z. Liu, H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An, and W.
Chang, Nature共London兲 428, 287 共2004兲.
49J. Standfuss, A. C. T. van Scheltinga, M. Lamborghini, and W.
Kühlbrandt, EMBO J. 24, 919共2005兲.
50A. A. Pascal, Z. Liu, K. Nroess, B. van Oort, H. van Amerongen, C.
Wang, P. Horton, B. Robert, W. Chang, and A. Ruban, Nature共London兲
436, 134共2005兲.