行政院國家科學委員會專題研究計畫 期中進度報告
有機金屬化合物在催化反應研究(2/3)
期中進度報告(精簡版)
計 畫 類 別 : 個別型
計 畫 編 號 : NSC 95-2119-M-002-038-
執 行 期 間 : 95 年 08 月 01 日至 96 年 07 月 31 日
執 行 單 位 : 國立臺灣大學化學系暨研究所
計 畫 主 持 人 : 劉緒宗
處 理 方 式 : 期中報告不提供公開查詢
中 華 民 國 96 年 05 月 04 日
行政院國家科學委員會補助專題研究計畫
行政院國家科學委員會補助專題研究計畫
行政院國家科學委員會補助專題研究計畫
行政院國家科學委員會補助專題研究計畫 ]
期中進度報告
期中進度報告
期中進度報告
期中進度報告
立體阻障雙芽配基金屬化物之催化聚合應用
計畫類別:X 個別型計畫 □ 整合型計畫
計畫編號:NSC 96-2113-M-002-038
執行期間:95 年 8 月 1 日至 96 年 7 月 31 日
計畫主持人:劉緒宗
成果報告類型(依經費核定清單規定繳交):x 精簡報告 完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
出席國際學術會議心得報告及發表之論文各一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、列
管計畫及下列情形者外,得立即公開查詢
執行單位:台灣大學化學系
中 華 民 國 96 年 5 月 3 日
中英文摘要
中英文摘要
中英文摘要
中英文摘要
此計畫為三年期計畫,執行第二年之精簡報告。 設計有立體阻障碳烯的雙芽供體,並合成其過渡金屬化合物作為催化劑應用; 合成官能性離子液體分子; 水合陽離子之催化反應 關鍵詞: 多芽配位體,催化,離子液體,陽離子 Abstract:This is a progesss report based on a three-year research project. The research involves (1) the design of bulky carbene bidenates and their rhodium complexes, (2) synthesis of functionalized ionic liquid, (3) aqua metal complexes in catalysis.
Synthesis of Iridium Pyridinyl N-Hetreocyclic Carbene Complexes and Their Catalytic Activities on Reduction of Nitroarenes
In recent years, the use of N-heterocyclic carbenes as ligands for transition metal ions in catalysis has increased significantly due to the effect of their strong σ-donor nature on the stabilization of metal ions.1 Among the various metal ions, applications of the iridium carbene
complexes in homogenous catalysis have also been reported.2-9 It is known that N-heterocyclic
carbene iridium complexes can catalyze the Oppenauer-type oxidation,2 hydrogen transfer
reduction of carbonyl compounds,3 cyclization of alkynyl-carboxylic acids,4 hydrogenation of
olefins,5 hydrosilylation,6 hydroamination,7 C-H activation,8 and bornylation.8 However, to our
knowledge, the use of iridium-carbene complexes for catalytic reduction of nitro compounds has not been reported before.
Hydrogenation of functionalized nitrobenzenes produces anilines, which are important intermediates for the manufacture of a variety of agrochemicals, pharmaceuticals, dyes, and pigments. Reduction of nitro compounds can be carried out in the gas or liquid phase by using
metal catalysts.10 However, hydrogenation of aromatic nitro compounds poses problems,
particularly for those with other reducible groups in the reaction. Alternatively, the
hydrogen-transfer reduction offers a promising solution due to its selectivity.10Herein, we report
the preparation and characterization of a series of new pyridinyl-carbene (denoted as
pyN^C-R) iridium(I) complexes as well as their catalytic activities toward
hydrogen-transfer reduction of nitroarenes under mild reaction conditions.
N N N R pyN^C-R R = Mes, Me
Preparation of iridium complexes. The method for the preparation of iridium carbene
complexes is similar to that of the rhodium complexes prepared in our laboratory.11a In brief, the
pyridinyl-imidazolium salt 1, the precursor for the carbene ligand, was prepared by a simple substitution reaction of 2-bromomethyl-6-mesitylpyridine with the corresponding imidazole. Deprotonation of imidazolium salt 1 with a strong base, which was expected to produce the corresponding free carbene, unfortunately failed presumably due to the interference from the
deprotonation of benzylic methylene protons.12 Alternatively, the imidazolium salt 1 was
converted into its silver carbene complex (Scheme 1) via the reaction of 1 with excess of Ag2O in
characteristic shift for Ag-C(carbene) at δ 184.7, which is reasonably assigned to the
2C-imidazol-2-ylidene(carbene) carbon, whereas Ag-C(carbene) for complex 2b appears at δ 183.5,
and is in a similar range as that for Ag-C(carbene).13 This spectral data clearly illustrates the
formation of a silver carbene complex.
Br N Ar N N R 1a, R = Me 1b, R = Mes N Ar N N R Ag N Ir N Ar N R BF4 N Ar N N R Ir(COD)Cl (COD) 2a, R = Me
Ar = mesityl. (i) Ag2O, NaI, (ii) [Ir(COD)Cl]2 (iii) AgBF4
i ii iii 3a, R = Me 3b, R = Mes 4a, R = Me 4b, R = Mes Scheme 1 I 2
Further structural confirmation is given by x-ray analyses. ORTEP plot of 2a is shown in Figure 1, while the crystal structure of 2b has been reported previously.11a Unlike 2b, the molecule comprises two silver atoms bridged by iodides, while the ligand pyN^C-Me acts as monodentate from the carbene end. The four-member ring defined by Ag(1)-I(1)-Ag(1A)-I(1A) is in a planar
arrangement. The long distance of Ag-Ag (3.40 Å) indicates no metal-metal interaction.11b The
Ag–C bond length is 2.150(3) Å, similar to that of complex 2b [2.108(4) Å].
N Mes N N Mes Ag 2 I 2b
Treatment of [Ir(COD)Cl]2 with the silver carbene complexes 2 in dichloromethane at ambient
temperature gave the desired iridium complexes 3 as yellow solids in excellent yields. Characterization of these complexes, 3a and 3b, was performed by both spectroscopic and
elemental analyses. 13C{1H} NMR data for the coordinating carbene carbons appear at δ 179.8 for
3a and 179.6 for 3b, respectively, suggesting the formation of the iridium carbon bond. These
signals are all in the typical range for Ir-C(carbene) observed for the analogues.14 The 1H NMR shifts
corresponding to the proton of the pyrindinyl ring are essentially similar to those of the silver
complexes , indicating that the pyridinyl nitrogen donor remains un-coordinated. Furthermore, 1H
NMR spectra of both 3a and 3b display an AM type of splitting pattern for the methylene protons (Hd and He) respectively. The detailed coordination sphere around the metal center of 3b is confirmed by the x-ray crystal structural analysis.
The complete molecular structure of 3b is shown in Figure 2. Selected bond distances and bond angles are listed in Table 3. The structural determination shows that the molecular geometry around the iridium ion is in square planar arrangement with two coordination sites occupied by
carbene and chloride in cis fashion. The average distance of Ir-C(COD) trans to carbene donor [2.17
Å] appears to be longer than those in cis arrangement [2.10 Å], suggesting that the σ-donor nature of the diaminocarbene is stronger than that of chloride. No major deviation was observed in bond lengths (Table 3). It is noted that the imidazol-2-ylidene ring is bisected with the coordination plane by ca. 73.2o.
<< insert Figure 2>>
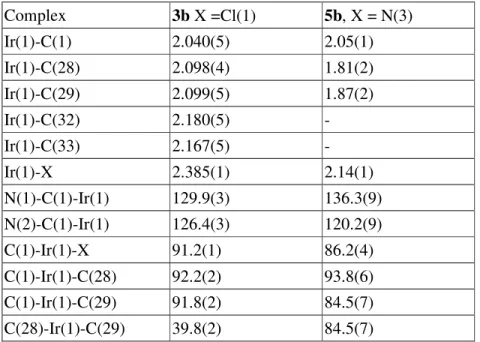
Table 3. Selected bond distances and bond angles of iridium carbene complexes 3b and 5b.
Complex 3b X =Cl(1) 5b, X = N(3) Ir(1)-C(1) 2.040(5) 2.05(1) Ir(1)-C(28) 2.098(4) 1.81(2) Ir(1)-C(29) 2.099(5) 1.87(2) Ir(1)-C(32) 2.180(5) - Ir(1)-C(33) 2.167(5) - Ir(1)-X 2.385(1) 2.14(1) N(1)-C(1)-Ir(1) 129.9(3) 136.3(9) N(2)-C(1)-Ir(1) 126.4(3) 120.2(9) C(1)-Ir(1)-X 91.2(1) 86.2(4) C(1)-Ir(1)-C(28) 92.2(2) 93.8(6) C(1)-Ir(1)-C(29) 91.8(2) 84.5(7) C(28)-Ir(1)-C(29) 39.8(2) 84.5(7)
The chelation of pyN^C-R toward the iridium center can be achieved by the treatment of 3 with
an equal molar amount of AgBF4, leading to the ligand substitution of chloride by the
pyridinyl-nitrogen. The appearance of δ 173.30 for 4a and 171.3 for 4b in the 13C{1H} NMR
spectra was assigned to the carbene carbons, which are essentially similar to those of 3a-b. All 1H
NMR signals of pyridinyl hydrogens in 4a-b are shifted downfield relative to those in silver
complexes and 3a-b, indicating the chelation of .pyN^C-R ligand. It is noticed that 1H NMR
spectra of 4a and 4b exhibit an AB type of splitting pattern for the methylene protons (Table 2). Besides the spectral data, elemental analyses are consistent with the proposed formula.
Under the atmospheric pressure of CO, a stirred solution of 4a-b gave the carbonyl substituted iridium complex 5a-b, respectively (Eq.1). IR spectra of these complexes show two carbonyl
stretching bands at 2064 and 2004 cm-1 for 5a as well as 2071 and 2004 cm-1 for 5b, characteristic
of the iridium dicarbonyl moiety. The coordination of the strong π-acid ligands around the metal
center causes the downfield shift of pyridinyl protons in the 1H NMR spectrum (Table 2). In
addition to the spectral data, an x-ray single crystal structure of 5b was determined to confirm the coordination environment of the complex.
N Ir N Ar' N R BF4 5a, R = Me 5b, R = Mes OC CO 4a-b CO (1)
An ORTEP diagram of 5b is represented in Figure 3, and selected bond distances and bond angles can be found in Table 3. As expected, the geometry around the metal center is square
planar, with the chelating pyN^C-Me [bite angle 86.2(4)o] and two carbonyl ligands. The
chelating ring is adopted into a boat conformation, which allows the two-methylene protons to be
in different environments, which is in agreement with the spectroscopic observation. The 1H NMR
shifts of –CH2- appear as two sets of doublets at δ 5.96 and 5.44 with the germinal coupling
constant ~14 Hz. All metal-carbon bond lengths, including the Ir-C(carbene) [2.05(1) Å] and
Ir-C(carbonyl) [1.81(2) and 1.87(2) Å], lie in the normal range, except the angle of N(1)-C(1)-Ir
[136.3(9)o]. The large deviation from 120o of this angle is presumably caused by the steric
interaction of mesityl group and the carbonyl ligand around the metal center. It is also noticed that the Ir-C(carbonyl) trans to the carbene moiety appears to be longer than that of cis orientaion by
about 0.06 Å, as anticipated, due to the trans influence.
Ligand Lability. N-donor ligands are, in general, more labile than those containing the more
strongly binding carbene donors. In solution, the pyridinyl-nitrogen donor of 4 undergoes dissociation with added acetonitrile and NMR experiments were applied to study the exchange.
Various amounts of acetonitrile were added to a CDCl3 solution of 4a (0.01 M) and the 1H NMR
of these samples were recorded at ambient temperature (Figure 4). As the concentration of acetonitrile increases, the signals of pyridinyl hydrogens and the methylene unit broaden. In addition, the resonances of the pyridinyl hydrogens shift upfield, whereas the splitting patterns
corresponding to the methylene unit are changed from the AB into the AM system. For the 1H
NMR of 4a in the presence of excess of acetonitrile, both the chemical shifts and the splitting
pattern are resemble to those of complex 3a in CDCl3, indicating that the coordinating pyridinyl
nitrogen has been replaced by the acetonitrile. Complex 4b behaves similarly.
-CH2
-pyridinyl-hydrogen CHCl
3
Figure 4. 1H NMR spectra of 4a in the presence of various amounts of acetonitrile in CDCl
Hydrogen transfer reduction. The hydrogen transfer reduction of a carbonyl function
catalyzed by transition metal complexes is well documented (Scheme 2).15 Likewise, the iridium
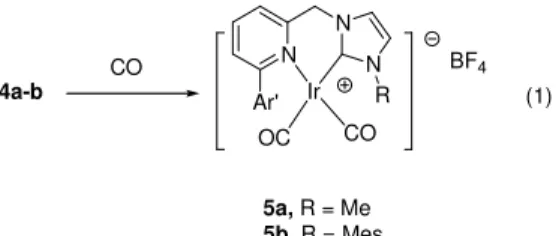
complexes 3-5, with the mixed donor carbene-pyridinyl nitrogen donors, are effective for the reduction of benzophenone. A mixture of substrate, iridium complex (0.1 mol %) and KOH (0.1M) in isopropyl alcohol was heated to reflux for 12h, and the product was obtained by a simple workup procedure. Results of the reduction catalyzed by 3-5 are compiled in Table 4.
As we can see from Table 4, complexes 3a and 3b with pyN^C-R ligand binding in a monodentate fashion show a slightly higher catalytic activity compared to their respect chelation complexes 4a, 4b and 5a, respectively. Nevertheless, these iridium complexes show decent activity toward the hydrogen transfer reduction of benzophenone. Under a similar condition, also noted is that from the TOF in 3a is on the same order of magnitude found by Hahn et al using a
five-coordinated iridium speces.16
Table 4. Catalytic transfer hydrogenation on reduction of benzophenone.a
Entry Catalyst (mol %) Yield (%) TOFb
1 3b (0.1) 93 78
2 3a (0.1) 90 75
3 4a (0.1) 50 42
4 4b (0.1) 53 44
5 5a (0.1) 60 50
a benzophenone (2.5 mmol), catalysts 2.5 x 10-3 mmol, KOH (0.1 M) in isopropanol (2 mL) under refluxing
temperature for 12h. b TOF = mol product/mol catalyst.h.
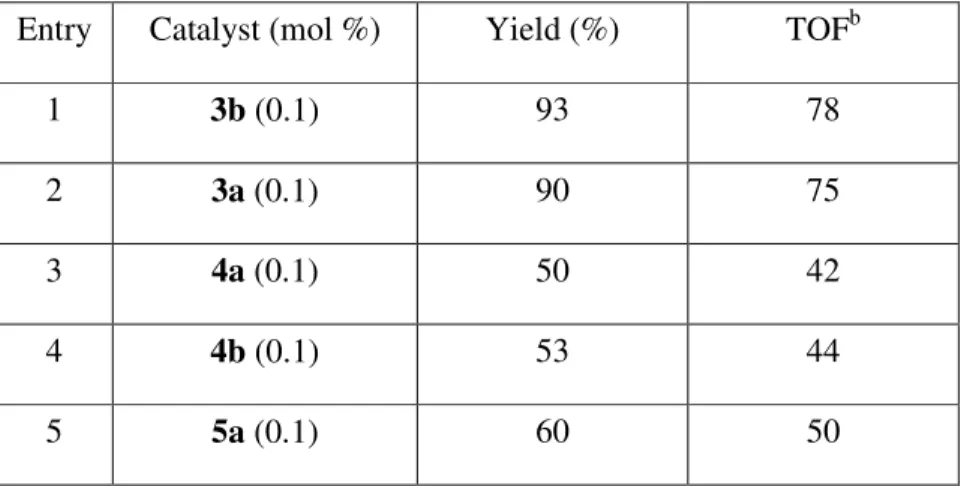
The promising result on the reduction of benzophenone encouraged us to further explore the catalytic capability toward the reduction of nitroarenes. In a typical case, p-bromonitrobenzene, iridium complex, solid KOH and isopropyl alcohol were placed in a round-bottomed flask and the mixture was heated under refluxing with stirring. The progress of the reaction was monitored by TLC. After completion, followed by the usual work-up procedures on the reaction mixture, aniline was produced, accompanied by the corresponding azo product (Eq 2). Accordingly a survey on a representative reduction of 4-bromonitrobenzene with various iridium complexes was carried out and the results are listed in Table 5.
NO2 NH2 + Ir complex KOH/i-PrOH (2) Br Br 6 7 N=N Br Br
As shown in the Table 5, the conversion is faster for the iridium carbene complexes 3-5, as
compared to [Ir(COD)Cl]2, with more than 80% conversion after 12 h under refluxing conditions.
There is no significant difference in activity among all iridium complexes under similar conditions. The concentration of base and catalysts, however, could affect the selectivity of products. Increasing the base concentration resulted in a higher amount of the reduction product 6. On the other hand, the amount of the azo product 7 increased with the use of a lower concentration of KOH. (see Table 5, entries 3-5). In principle, the catalytic path towards the reduction over the formation of azo compound 7 could be achieved if we carried the reaction under a higher dosage of catalyst (entry 6). When the reaction was carried out under a diluted concentration of the catalyst, the azo compound 7 was obtained predominately (Table 5, entry 7). This is probably attributable to the dominant metal-catalyzed hydrogen transfer reduction, namely that the higher concentration of catalyst favors the reduction path. Iridium complex 3a is less active when carrying out the reduction at room temperature, with only 69% conversion over 12 h, but with higher selectivity for providing solely the reduction product.
Table 5. Reduction of p-nitrobromobenzene using iridium carbene complexes.
yield
entry Cat.
concentration [KOH] temp time
Conv. %
6 7
1 [Ir(COD)Cl]2, 6.25 x 10-4 M 1.0 M reflux 24 h 24 30 10
2 [Ir(COD)Cl]2, 2.5 x 10-3 M 1.0 M reflux 12 h 50 49 trace
3 3a, 2.5 x 10-3 M 1.0 M reflux 12 h 88 72 13 4 3a, 2.5 x 10-3 M 0.5 M reflux 12 h 80 42 34 5 3a, 2.5 x 10-3 M 0.1 M reflux 12 h 100 18 73 6 3a, 5.0 x 10-3 M 1.0 M reflux 12 h 90 80 trace 7 3a, 6.25 x 10-4 M 1.0 M reflux 24 h 96 3 81 (76) 8 3a, 2.5 x 10-3 M 1.0 M r. t. 12 h 69 64 0 9 3b, 2.5 x 10-3 M 1.0 M reflux 12 h 60 14 40 10 4b, 2.5 x 10-3 M 1.0 M reflux 12 h 96 86 6 11 4a, 2.5 x 10-3 M 1.0 M reflux 12 h 98 89 (81) 3 12 5a, 2.5 x 10-3 M 1.0 M reflux 12 h 98 (73) trace
a Reaction conditions: nitrobenzene (0.1mmol), catalyst, KOH in isopropanol (4 mL); b NMR yield, isolated yields
given in parenthesis
The reduction steps of nitrobenzene leading to aniline are quite complex. A generally accepted mechanistic pathway is depicted in scheme 3, in which the aniline can be obtained via either the
direct reduction or the condensation followed by reduction;17 the latter route is favorable under the
basic conditions. In this work, the iridium carbene complexes are found to play key role in the reduction of aromatic nitro compounds via the catalytic transfer hydrogenation similar to that for the reduction of carbonyl functionality. The higher concentration of KOH increases the
concentration of isopropoxide, which further accelerates the ligand substitution on the metal center. The net result speeds up the β-elimination and the reduction process to yield the aniline product. Under a lower concentration of the catalyst, the base-catalyzed condensation of nitroso and hydroxyamine intermediates becomes the major pathway, yielding predominately the azo compound. (Table5, entry 7)
Under a similar reduction condition, we found that the iridium carbene complex catalyzes the reduction of azo compound to aniline in a very slow manner, with less than 10 % of azo compounds converted into aniline by the use of 3a, the result of which explains why the azo compound can be obtained as the major product.
NO2 [H] NO [H] NHOH [H] NH2 Disproportionation N N O -N N Azoxy Azo Aniline Condensation [H] [H] Scheme 3
Hydrogen transfer reductions of various nitroarenes in the presence of catalytic amount of iridium carbene complexes, were carried out under the same reaction conditions described for the reduction of p-bromonitrobenzene and the results are summarized. All instances provide good yields of the desired product except when p-nitrophenol and 2-chloropyridine were used. It is noticed that the iodo group is largely replaced by the hydrogen in the reduction of 2-iodo-4-nitroaniline. Catalytic hydrogen transfer reduction of 2-chloropyridine resulted in the formation of bis-(6-isopropoxypyridin-3-yl)diazene in poor yield (18 %), indicating that the chloride in the pyridine ring was replaced by isopropoxide.
1 NO2 Br 4a, 2.5 x 10-3 M 3a, 6.25 x 10-4 M [KOH] 1.0 M [KOH] 1.0 M 24 h 24 h p-BrC6H4NH2 p-BrC6H4N=NC6H4Br-p 89 % (81 %) 3 % 3 % 81 % (74%)
2 NO2 MeO 4a, 2.5 x 10-3 M 4a, 6.25 x 10-4 M [KOH] 2.0 M [KOH] 1.0 M 12 h 12 h p-MeOC6H4NH2 p-MeOC6H4N=NC6H4OMe-p 100% (97%) trace 89 %(82%)
a. Reaction conditions: substrate (1 mmol), catalyst and KOH in isopropanol (4 mL) under
refluxing temperature. b.NMR yield, isolated yields given in parenthesis
In summary, we have successfully prepared iridium pyridinyl N-hetreocyclic carbene complexes and studied their coordination behaviors. Compared to the carbene donor, the pyridinyl nitrogen donor is more labile and is readily replaced by acetonitrile. The prepared iridium complexes have shown salient catalytic activity on hydrogen transfer reductions of carbonyl and nitro functionalities. More importantly, these studies explore a great potential for selective reduction of nitroarene to aniline derivatives or azo compounds. The results should attract a broad spectrum of interest in the fields of organometallics and catalysis.
References
1. For a recent review see (a) Herrmann, W. A. Angew. Chem., Int. Ed., 2002, 41, 1290. (b) Crudden., C. M.; Allen, D. P. Coord. Chem. Rev. 2004, 248, 2274. (c) Cavell, K. J.; McGuinness,D. S. Coord. Chem. Rev. 2004, 248, 671. (d) Bourissou, D.;Guerret, O.; Gabbai, F. P.; Bertrand, G. Chem. Rev. 2000, 100, 39. (e) Peris, E.; Crabtree, R. H. Comptes Rendus Chim. 2003,
6, 33. (f) Arduengo, A. J. Acc. Chem. Res. 1999, 32, 913. (g) Cesar, V.; Bellemin-Laponnaz, S.;
Gade, L. H. Chem. Soc. Rev. 2004, 33, 619. (h) Reddy, R. K.; Liu, S.-T. Chem. Soc. Rev. 1999, 28, 315, and references cited therein.
2. Hanasaka, F.; Fujita, K.; Yamaguchi, R. Organometallics 2005, 25, 4643, and references therein.
3. (a) Albrecht, M.; Miecznikowski, J. R.; Samuel, A.; Faller, J. W.; Crabtree, R. H. Organometallics 2002, 21, 3596. (b) Miecznikowski, J. R.; Crabtree, R. H. Organometallics 2004,
23, 629. (c) Hahn, F. E.; Holtgrewe, C.; Pape, T.; Martin, M.; Sola, E.; Oro, L. A.
Organometallics 2005, 24, 2203. (d) Seo, H.; Kim, B. Y.; Lee, J. H.; Park, H.-J.; Son, S. U.; Chung, S. Y. K. Organometallics 2003, 22, 4783. (e) Herrmann, W. A.; Baskakov, D.; Herdtweck, E.; Hoffmann, S. D.; Bunlaksananusorn, T.; Rampf, F.; Rodefeld, L. Organometallics 2006, 25, 2449.
5. (a) Lee, H. M.; Jiang, T.; Stevens, E. D.; Nolan, S. P. Organometallics 2001, 20, 1255. (b) Vazquez-Serrano, L. D.; Owens, B. T.; Buriak, J. M. Chem. Commun. 2002, 2518. (c) Perry, M. G.; Cui, X.; Powell, M. T.; Hou, D.-R.; Reibenspies, J. H.; Burgess, K. J. Am. Chem. Soc. 2003,
125, 113. (d) Nanchen, S.; Pfaltz, A. Chem. Eur. J. 2006, 4550. (e) Baskakov, D.; Herrmann, W.
A.; Herdtweck, E. Hoffmann, S. D. Organometallics 2007, 26, 626, and references cited therein. 6. (a) Chianese, A. R.; Crabtree, R. H. Organometallics 2005, 24, 4432. (b) Mas-Marza, E.; Poyatos, M.; Sanau, M.; Peris, E. Inorg. Chem. 2004, 43, 2213. (c) Vicent, C.; Viciano, M.; Mas-Marzá, E.; Sanaú, M.; Peris, E. Organometallics 2006, 25, 3713.
7. Field, L. D.; Messerle, B. A.; Vuong, K. Q.; Turner, P. Organometallics 2005, 24, 4241. 8. (a) Corberan, R.; Sanau, M.; Peris, E. J. Am. Chem. Soc. 2006, 128, 3974. (b) Frey, G. D.;Rentzsch, C. F.; von Preysing, D.;Scherg, T.; Muehlhofer, M.;Herdtweck, E.;Herrmann, W. A. J. Organomet. Chem. 2006, 691, 5725.
9. Imlinger, N.; Mayr, M.; Wang, D.; Wurst, K.; Buchmeiser, M. R. Adv. Synth. Catal. 2004,
346, 1836.
10. Ragaini, F.; Cenini, S.; Gallo, E.; Caselli, A.; Fantauzzi, S. Curr. Org. Chem. 2006, 10,1479. 11. (a) Wang, C. Y.; Liu, Y. H.; Peng, S. M.; Liu, S. T. J. Organomet. Chem. 2006, 691, 4012. (b) Garrison, J. C.; Youngs, W. J. Chem Rev. 2005, 105, 3978.
12. McGuinness, D. S.; Cavell, K. J. Organometallics 2000, 19, 741. 13.Wang, H. M. J.; Lin, I. J. B. Organometallics 1998, 17, 972.
14.Chianese, A. R.; Li, X.; Janzen, M. C.; Faller, J. W.; Crabtree, R. H Organometallics 2003, 22, 1663.
15. Recent review: Saluzzo, C.; Lemaire, M. Adv. Synth. Catal. 2002, 344, 915, and references therein.
16. Hahn, F. E.; Holtgrewe, C.; Pape, T.; Martin, M.; Sola, E.; Oro, L. A. Organometallics 2005,
24, 2203.
Deprotection of Acetals and Ketals in A Colloidal Suspension Generated by Sodium Tetrakis(3,5-trifluoromethylphenyl)borate in Water.
Abstract: Deprotection of acetals and ketals can be achieved by using sodium
tetrakis(3,5-trifluoromethylphenyl)borate (NaBArF
4) as the catalyst in water at 30oC. For example,
a quantitative conversion of 2-phenyl-1,3-dioxolane into benzaldehyde was accomplished by using this sodium salt (0.1 mol %) in water within 5 min.
Protection/deprotection of carbonyl groups are of fundamental importance in the multistep organic synthesis. Although numerous methods for deprotection of acetals are known, chemists are still interested in developing processes of good efficiency under mild conditions. In recent years, deprotection of ketals and acetals under very smooth conditions such as CAN in neutral or mild
conditions,3 thiourea in EtOH/H
2O,4 SmCl3/ TMSCl,5 cyclodextrins in water,6 Bi(OTf)3 in
THF/H2O,7 Ce(OTf)3 in CH3NO2/H2O,8a TESOTf/pyridine/H2O8b and LiBF4 in CH3CN/H2O,9
have been reported. Quest for a mild and neutral approach particularly in water as reaction
medium which may affect the selective hydrolysis of acetals or ketals, is still of interest.10
In our previous investigation, we have found that sodium
tetrakis(3,5-trifluoromethylphenyl)borate dihydrate (NaBArF
4.2H2O) is able to render a proton to
initiate the polymerization of vinyl ether under anhydrous organic solvents (scheme 1).11 This sodium salt could also assist the hydrolysis of vinyl ethers in aqueous solution. Based on our preliminary finding, we have carried out a study on the deprotection of acetal or ketals by using this sodium salt in aqueous medium.
O R excess H2O H O R n CH3CHO + ROH CH2Cl2 Scheme 1 F3C F3C B Na(H2O)2 4 [Na(H2O)2BArF4]
Hydrolysis of 2-phenyl-1,3-dioxolane is selected as a model to examine the effectiveness of
NaBArF4 on this reaction. It was surprising to find out that 2-phenyl-1,3-dioxolane was efficiently
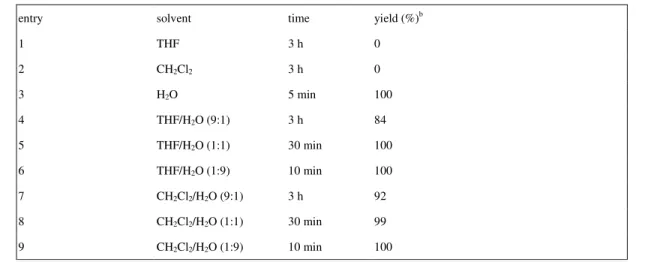
hydrolyzed into benzaldehyde in water as the solvent (entry 3, table 1). Besides water, we also studied the solvent effect for the reaction, because a colloidal dispersion of the reaction mixture occurred during the reaction. As shown in table 1, hydrolysis preceded smoothly in any aqueous/organic mixed solvent except in a pure organic solvent (entries 1~2, table 1). The use of organic/aqueous mixture for hydrolysis of acetals or ketals is a common choice due to the solubility reason. However, the larger portion of organic solvent was used, a longer reaction period was required for the completion. For example, in aqueous tetrahydrofuran only 84 % of deprotection product was collected after stirring for 3 h (entry 4, table 1), but 100 % conversion within 5 minutes in water. Of course, the colloidal dispersion disappeared with the use of a larger
quantity of organic portion. Nevertheless, the emulsion seemed to accelerate the hydrolysis process, suggesting that the tetrakis(3,5-trifluoromethylphenyl)borate anion might play as a role of surfactant.12
Table 1 Solvent Effect on Hydrolysis of 2-phenyl-1,3-dioxolane catalyzed by NaBArF4. a
entry solvent time yield (%)b
1 THF 3 h 0 2 CH2Cl2 3 h 0 3 H2O 5 min 100 4 THF/H2O (9:1) 3 h 84 5 THF/H2O (1:1) 30 min 100 6 THF/H2O (1:9) 10 min 100 7 CH2Cl2/H2O (9:1) 3 h 92 8 CH2Cl2/H2O (1:1) 30 min 99 9 CH2Cl2/H2O (1:9) 10 min 100
a Reaction conditions: 2-phenyl-1,3-dioxolane (5 mmol) and NaBArF
4 (0.005mmol) in solvent (10 mL) at 30 oC for 3h. b GC yield.
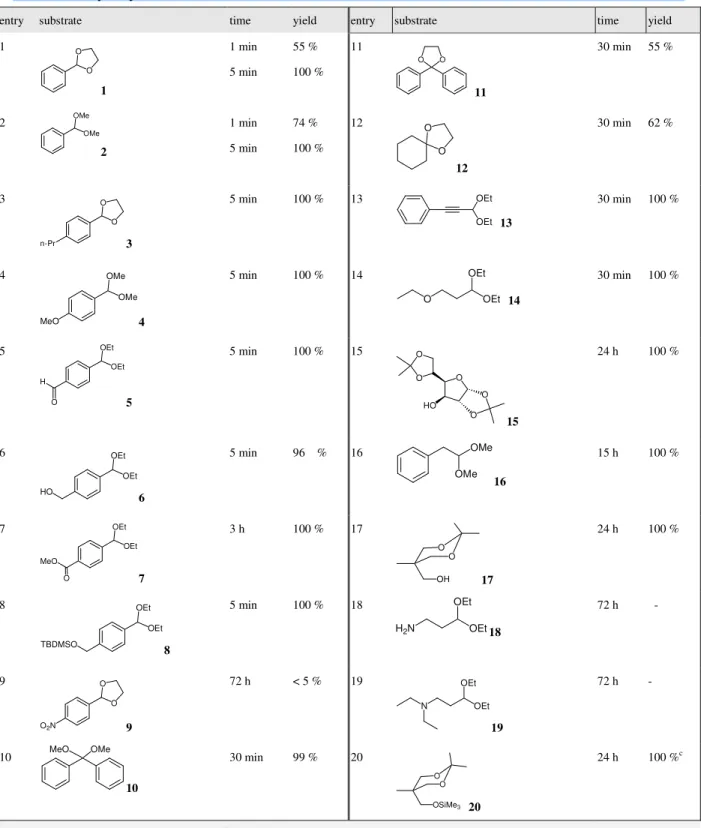
In view of the efficient hydrolysis of benzaldehyde acetal, the deprotection for various dialkyl, ethylene acetals and dioxolanes was investigated and the results are shown in Table 2. The experiment was simply conducted in a water solution of acetals or ketals in the presence of 0.1
mol% of NaBArF
4at 30 oC. Acetals of aromatic aldehydes underwent smooth hydrolysis in an
extremely fast rate to give the corresponding carbonyls in quantitative yields (entries 1-8, table 2). In most instances, the reactions were accomplished within 0.5 h, indicating the efficiency of this
approach as compared to other Lewis acid catalyzed methods.1-8 It was particularly noteworthy
that various functional group were tolerated under reaction conditions such as ester or
t-butyldimethylsilyl (TBDMS) groups. However, a competition on the hydrolysis of the
trimethylsilyloxy group versus the ketal was observed in compound 20. The process of
de-silyation appeared to be faster than that of hydrolysis. By on 1H nmr integration, about 50 % of
20 was converted into 17 (desilylation) within 3 h.
Previous studies showed that the benzaldehyde acetals with electron-withdrawing substituents
tended to be slow or resistant on the hydrolysis.13 Indeed, the deprotection of compound 7 (ester
substituted) took a longer period for completion, whereas the compound 9 remained almost intact for 3 days under the same conditions. Dialkyl aromatic acetals (or ketals) were more reactive toward the reagent than the corresponding cyclic acetals (or ketals) (entries 1 vs. 2, 10 vs. 11, table 2), whereas the hydrolysis of aromatic acetals seemed to be easier than those of the aliphatic ones (entries 12, 14, 16-17, table 2).
The beneficial effect of NaBArF
4 on the deprotection was also applicable to the hydrolysis of
acetonide of carbohydrate. When the 1,2:5,6-O-isopropylidene-α-D-glucofuranose 15 was subjected to this reaction conditions, a quantitative yield of 1,2-O-isopropylidene-α-D-gluco-
furanose was observed; no other hydrolyzed product was isolated even after 24 hours (entry 15, table 2). Such selectivity was presumably due to a favorable hydrolysis at the less steric hindered site.14
Table 2 Hydrolysis of acetals and ketals.a
entry substrate time yield entry substrate time yield
1 O O 1 1 min 5 min 55 % 100 % 11 O O 11 30 min 55 % 2 OMe OMe 2 1 min 5 min 74 % 100 % 12 O O 12 30 min 62 % 3 O O n-Pr 3 5 min 100 % 13 OEt OEt 13 30 min 100 % 4 OMe OMe MeO 4 5 min 100 % 14 O OEt OEt 14 30 min 100 % 5 OEt OEt H O 5 5 min 100 % 15 O O O HO O O 15 24 h 100 % 6 OEt OEt HO 6 5 min 96 % 16 OMe OMe 16 15 h 100 % 7 OEt OEt MeO O 7 3 h 100 % 17 O O OH 17 24 h 100 % 8 OEt OEt TBDMSO 8 5 min 100 % 18 H2N OEt OEt 18 72 h - 9 O O O2N 9 72 h < 5 % 19 N OEt OEt 19 72 h - 10 MeO OMe 10 30 min 99 % 20 O O OSiMe3 20 24 h 100 %c
a. Reaction conditions: substrate (5 mmol), NaBArF
4 (0.005 mmol) in water (10 mL) at 30 oC. b. Isolated yield. c. 1,1,1-Tris(hydroxymethyl)ethane as the obtained product.
The acetal molecules containing amino-functionality appear to be inert to these reaction
conditions as no reaction product was detected even after longer reaction time (72 h). The basicity of amines readily stopped the deprotection of acetals with all starting material recovered in both examples (entries 18 ~19, table 2). Presumably the coordination of amines toward the metal centers demolished the activity of the catalyst.
Lipshutz and coworkers have shown that acetals and ketals could be hydrolyzed by LiBF4 in
aqueous acetonitrile. It has been demonstrated that this cleavage might involve the participation of
HF and boron-containing Lewis acid.9 However, it is quite unlikely to generate such a species
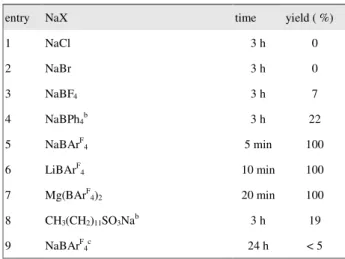
under the catalytic reaction conditions in our work. Thus the roles of cations and anions in this reaction were examined. Results of the treatment of 2-phenyl-1,3-dioxolane with various salts are summarized in table 3. Sodium salts of chloride, bromide and tetrafluoroborate, which are soluble in water, did not form colloidal particles in the presence of the organic substrates under the given conditions, thus the deprotection did not proceed (or insignificant). However, the benzaldehyde acetal underwent hydrolysis smoothly with the use of tetrakis(3,5-trifluoromethylphenyl)borate salts (entries 5~7, table 3), indicating the importance of -BArF4. The reactivty order of the borate
salts is Na+ > Li+ > Mg2+, which was consistent with the order of hydrolysis constant (pKh).15 It is
noticed that the sodium dodecylsulfonate could also act as a catalyst for the deprotection of acetals
in water, although slow. However, hydrolysis of actals catalyzed by NaBArF4 was completely
stopped, when 18-Crown-6 was added (entry 9, table 3).
Table 3 Hydrolysis of 2-phenyl-1,3-dioxolane by various salts.a
entry NaX time yield ( %)
1 NaCl 3 h 0 2 NaBr 3 h 0 3 NaBF4 3 h 7 4 NaBPh4b 3 h 22 5 NaBArF 4 5 min 100 6 LiBArF 4 10 min 100 7 Mg(BArF 4)2 20 min 100 8 CH3(CH2)11SO3Nab 3 h 19 9 NaBArF 4c 24 h < 5
a Reaction conditions: NaX (5 x 10-3 mmol), 2-phenyl-1,3-dioxolane (5 mmol) in H2O (10 mL) at 30 oC.
b from commercial source and use it without further purification. c. Addition of 18-C-6 (1 x 10-2 mmol).
It has been known that the formation of colloidal particles created by organic
substrates/surfactant/catalyst in water could accelerate chemical reactions.16 The hydrolysis of
acetals/ketals catalyzed by metal salts of tetrakis(3,5-trifluoromethylphenyl)borate makes another example. It is obvious that the trifluoromethyl subsitutent of the phenyl rings does have an
influence. In a previous report, we showed the existence of the coordination of Na toward F in the crystal structure of NaBArF4.2H2O.11 It is believed that such coordination might partially persist in
aqueous environment, and thus increases the Lewis acidity of metal ions. The formation of sodium crown ether complex however hinders the reaction (entry 9, table 3), showing that the interaction
of Na…F is readily interrupted by the addition of the crown ether. Further experiments and
theoretical consideration are necessary to confirm such phenomenon. By screening a series of
acid-base indicators, we found that the acidity of [Na(H2O)2]BArF4 in THF/H2O was estimated to
be in the range of pH = 3.6~4.5.
In summary, a new and chemoselective method for the cleavage of acetals and ketals in water under mild and neutral reaction conditions has been developed in this work. The advantages of this method are the general applicability in hydrolysis of alkyl and cyclic acetals (ketals) in high
yields, the observed selectivity, and a very mild reaction conditions such as 0.1 mol % NaBArF4 of
catalyst in water at 30oC. From the anion study, it is quite clear that the property of metal ions
changes dramatically by the surrounding anions. Furthermore, the activities of metal salts
associated with a tetrakis(3,5-trifluoromethylphenyl)borate anion on other reactions are currently investigated.
References
(1) Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis, 3rd ed.; John Wiley: New York, 1999.
(2) Recent reviews:(a) Kocienski, P. J. J. Chem. Soc., Perkin Trans. 1 2001, 2109. (b) Sartori, G.; Ballini, R.; Bigi, F.; Bosica, G.; Maggi, R.; Righi, P. Chem. Rev. 2004, 104¸ 199.
(3) Markó, I. E.; Ates, A.; Gautier, A.; Leroy, B.; Plancher, J. M.; Quesnel, Y.; Vanherck, J. C. Angew. Chem., Int. Ed. 1999, 38, 3207.
(4) Battacharjya, A.; Majumdar, S. J. Org. Chem. 1999, 64, 5682. (5) Yutaka, U.; Koumoto, N.; Fujisawa, T. Chem. Lett. 1989, 1623. (6) Ji, H. B. Eur. J. Org.Chem. 2003, 3659.
(7) Carrigan, M. D.; Sarapa, D.; Smith, R. C.; Wieland, L. C.; Mohan, R. S. J. Org. Chem. 2002, 67, 1027.
(8) (a) Dalpozzo, R.; De Nino, A.; Maiuolo, L.; Procopio, A.; Tagarelli, A.; Sindona, G.; Bartoli, G. J. Org. Chem. 2002, 67, 9093. (b) Fujioka, H.; Okitsu, T.; Sawama, Y.; Murata, N.; Li, R.; Kita, Y. J. Am. Chem. Soc. 2006, 128, 5930. (9) Lipshutz, B. H.; Harvey, D. F. Synth. Commun. 1982, 12, 267.
(10) Manabe, K.; Kobayashi, S. Org. Lett. 1999, 1, 1965.
(11) Chang, C.-T.; Chen, C.-L.; Liu, Y.-H.; Peng, S.-M.; Chou, P.-T.; Liu, S.-T. Inorg. Chem. 2006, 45, 7590. (12) Iwamoto, H. Tetrahedron Lett. 1983, 228, 369.
(13) Cappola, G. M. Synthesis 1984, 1021.
(14) Goud, P. M.; Venkatachalam, T. K.; Uckun, F. M. Synth. Commun. 2003, 33, 1185. (15) Kobayashi, S.; Nagayama, S.; Busujima, T. J. Am. Chem. Soc. 1998, 120, 8287.
(16) (a) Fendler, J. H.; Fendler, E. J. Catalysis in Micellar and Micromolecular Systems; Academic Press: London, 1975. (b) Holland, P. M., Rubingh, D. N. ACS Symposium Series, 1992, 501, 2.
(17) Kawasaki, M.; Goto, M.; Kawabata, S.; Kometani, T. Tetrahedton: Assymmetry 2001, 12, 585. (18) Barluenga, J.; del Pozo, C.; Olano, B. Synthesis 1995, 12, 1529.
(19) Mak, C. C.; Bampos, N; Darling, S. L.; Montalti, M.; Prodi, L.; Sanders, J. K. M. J. Org. Chem. 2001, 13, 4476. (20) Raffaelo, L.; Segio, B.; Paolo, P.; Francesco, T.; Piero, S. J. Organomet. Chem. 1985, 295, 371.
(21) Baillargeon, V. P.; Stille, J. K. J. Am. Chem. Soc. 1986, 108, 452. (22) Typical procedure for hydrolysis. A mixture of NaBArF
4.2H2O(5 x 10-3 mmol)11 and acetal (5.0 mmol) in water (10 mL)
was placed into a 25 mL flask. The resulted suspension solution was stirred vigorously at 30oC for a certain period as shown
in table 2.22 After the completion of the reaction, the reaction mixture was extracted with CH
2Cl2 (5 mL x 2). The extracts
were dried and filtered through a short column (silica gel) to remove the sodium salt. Upon concentration, the desired carbonyl products were obtained in pure form. For all products reported in table 2, full 1H and 13CNMR were compared
Mannich-type Reactions in a Colloidal Solution Formed by Sodium Tetrakis(3,5-trifluoro- methylphenyl)borate as the Catalyst in Water
Abstract—Sodium tetrakis(3,5-trifluoromethylphenyl)borate [NaBArF4] efficiently catalyzed the
one-pot, three-component Mannich reaction of ketones with aromatic aldehydes and different anilines in water at ambient temperature and afforded the corresponding β-amino carbonyl compounds in good to excellent yields.
Mannich reaction is one of the most important carbon-carbon bond forming reactions in organic
synthesis (Eq.1).1 This reaction provides the formation of β-amino carbonyl compounds, which
are important intermediates for construction of various nitrogen-containing natural products and
pharmaceutical.2 However, the drastic reaction conditions for the classical intermolecular
Mannich reaction limit its synthetic usefulness. Therefore, numerous modifications of this
reaction have been developed to overcome the drawbacks.1,3 Amongst, many metal complexes
have been used as Lewis acid catalysts to promote the reaction under anhydrous conditions,1-3 and
few water-compatible Lewis acids were reported.4
R1CHO + R2NH2 + O R4 R3 R1 R4 NH O R2 R3 (1) catalyst
In recent years, organic reactions in aqueous medium have received enormous attention, because the use of water has several advantages such as the easiest obtainable solvent, an environmentally
friendly substance and easy separation.5-7 In this context, researches focusing on the development
of new catalysts to promote organic reactions in water have become highly desirable. In an early report by Kobayashi and coworkers, they found that dodecylbenzenesulfonic acid could catalyze
the Mannich reaction at ambient temperature in water to give various β-amino ketones in good
yields.6 In these surfactant-aided systems, organic substrates form emulsion droplets that function
as reaction media in water. However, it is still required the presence of a Brønsted acid to carry out the reaction in most of instances. Here we offer an efficient method for the preparation of Mannich type products in water by using sodium tetrakis(3,5-trifluoromethyl-phenyl)borate
(NaBArF4) as a catalyst under mild and nearly neutral conditions.
F3C
F3C
B Na NaBArF4
4
Initially, the two-component Mannich reaction of N-benzylideneaniline (1.0 mmol) and various of
nucleophiles (2 mmol) was examined (table 1). As a preliminary study, we found that the NaBArF
4
did not catalyzethe reaction of acetophenone with benzylideneaniline in most of organic solvents
such as THF, dichloromethane or DMF. It was revealed that the present Mannich-type reaction proceeded only in aqueous media. Running the reaction of benzylideneaniline with ketone in water at ambient temperature gave the desired product in excellent yield (table 1, entry 4).
Encouraged by the results, we examined various ketones and silyl enolates to screen the scope of this new protocol. The silyl enolate as the nucleophile in the reaction appeared to be more reactive than the corresponding ketone compounds as evidenced by the shorter reaction time and better yields (table 1, entries 5~6).
Table 1. Results of Mannich reaction catalyzed by NaBArF4.a
entry Ketone or silyl enol ether solvent time yield
1 acetophenone CH2Cl2 96 h 0 % 2 acetophenone THF 96 h 0 % 3 acetophenone DMF 96 h trace 4 acetophenone water 96 h 96 % 5 C6H5C(OSiMe3)=CH2 water 30 min 1h 91 % 100 % 6 C6H5C(OSiMe3)=CHCH3 water 30 min 1h 77 % 100 % 7 C6H5C(O)CH2CH3 water 144 h 32 % 8 cyclohexone water 3 h 100 %
a Reaction conditions: NaBArF
4(0.005 mmol), ketone or silyl enol ether (2 mmol), N-benzylideneaniline (1 mmol) at 30oC.
With the success of the above coupling reaction, we examined the feasibility of the
three-component Mannich reaction. The reaction of benzaldehyde, aniline, and acetophenone in
the presence of NaBArF4 as a catalyst in water provided the desired compound in 81 % isolated
yield (table 2, entry 1). Again, this efficient catalyst was not observed in the reactions carried out in the organic solvents. Several of the substituted anilines underwent the reaction smoothly except
for the p-nitroaniline (table 2, entry 9). By using the 1H NMR to monitor the reaction, we realized
that the imine intermediate, (4-nitrobenzyl-idene)phenylamine, was not formed during the reaction.
Table 2. Results of three-component Mannich reaction.a
O CHO R1 NH2 R2 + + NH O R2 R1 H2O NaBArF4
entry R1 R2 time yield 1 H H 48 h 81 % 2 F H 48 h 51 % 3 Cl H 48 h 74 % 4 Br H 48 h 85 % 5 i-propyl H 48 h 53 % 6 CH3O H 48 h 21 % 7 H I 24 h 100 % 8 H Me 24 h 51 % 9 H NO2 48 h -
a Reaction conditions: NaBArF
4(0.005 mmol), acetophenone (2 mmol), aniline (1 mmol), aldehyde
(1 mmol) in H2O (2 mL) at 30oC.
The reactions proceeded not only for acetophenone but also for other dialkyl ketones in good
yields. As illustrated in Scheme 1, the NaBArF4 didcatalyzethe Mannich reaction of cyclohexone,
substituted benzaldehydes and anilines to yield the desired β-amino ketone products. The
stereoselectivity was determined by the 1H NMR spectroscopy and by comparison with known
compounds. The selectivity for anti-isomers is slightly favored. In addition, reaction of cyclohexanone and p-chloroaniline with 1-naphthalenecarbaldehyde yielded the coupling product, 2-[(4-Chlorophenylamino)naphthalen-1-ylmethyl]-cyclohexanone, in 65 % (anti:syn = 1:1). CHO Y O NH2 Z NH O Z Y Scheme 1 Y = -COOMe = -COOMe Z = I = Cl 63 % (anti : syn = 7:4) 57 % (anti : syn = 8:5) NaBArF4 H2O
For unsymmetric ketone, the regioselectivity of the less hindered site is observed. Thus, reaction
of 2-butanone, benzaldehyde and aniline in the presence of NaBArF4 in water provided the
Mannich products I and II (Eq. 2). The ratio of I:II is about 85:15, indicating that the reaction takes place at the less substituted carbon center.
Ph NH Ph O C6H5CHO + C6H5NH2 + O Ph NH Ph O + (2) I II
It should be noted that the addition of organic substrates to this sodium salt in water readily
found this salt did not completely dissociate into free ions in dichloromethane, with the formation
of aggregation instead.9 From the dynamic light scattering measurement, the average size of
aggregated particles of NaBArF4 and organic substrates is in the range of ~1 µm. Thus the addition
of organic substrates readily forms a colloidal dispersion, which is similar to that of a mixture of dodecylbenzenesulfonic acid (DBSA) and organic reagents in water reported by Kobayashi and
coworkers.6 Apparently, the formation of colloid particles plays an essential role in acceleration of
the coupling reaction. In our early study, the crystal structure of NaBArF4 reveals that the
fluorine atoms of trifluoromethyl groups do coordinate toward the metal center.9 We believe that
such coordination might partially persist in aqueous environment, which thus increases the Lewis acidity of metal ions.
In conclusion, this procedure offers several advantages for Mannich reaction such as low loading of catalyst, mild conditions, high yields, clean reactions, which make it a useful and attractive methodology for organic synthesis. Quite a number of products are solid and insoluble in water, which can be obtained by fitration and recrystallization. This simple workup procedure is also beneficial to this method. Further applications of this catalyst to other transformations are currently under investigation.