國
立
交
通
大
學
應用化學系分子科學研究所
碩 士 論 文
硝酸在二氧化鈦表面的吸附及反應情形
Adsorption Configurations and Reactions of Nitric Acid
on TiO
2Rutile (110) and Anatase (101) surface
研 究 生:張靜怡
指導教授:林明璋 院士
硝酸在二氧化鈦表面的吸附及反應情形
Adsorption Configurations and Reactions of Nitric Acid on TiO
2Rutile (110) and Anatase (101) surface
研 究 生:張靜怡 Student:Ching Yi Chang
指導教授:林明璋 院士 Advisor:M. C. Lin
國 立 交 通 大 學 應用化學系分子科學研究所 碩 士 論 文 A ThesisSubmitted to Department of Applied Chemistry and Institute of Molecular Science National Chiao Tung University
in partial Fulfillment of the Requirements for the Degree of Master
in
Applied Chemistry July 2008 Hsinchu, Taiwan
硝酸在二氧化鈦表面的吸附及反應情形
研 究 生:張靜怡 指導教授:林明璋 院士
國立交通大學應用化學系分子科學研究所
摘 要
本論文藉由VASP(Vienna Ab-initio Simulation Package) 計算軟體中 的理論計算方法,電子密度泛函理論(Density Functional Theory)及超軟贋 位勢近似法(Ultrasoft pseudopotential approximation, US-PP)來探討單分子 硝酸及雙分子硝酸分別在二氧化鈦金紅石(rutile)(110)表面及銳鈦礦 (anatase)(101)表面吸附結構及可能的反應路徑。 從我們的計算結果中可知,單分子硝酸中的氧原子和二氧化鈦金紅 石表面上的 Ti5c形成單一鍵結,而且硝酸分子中的 H 原子與表面的 O2c 形成H 鍵是最穩定的結構,其吸附能為 6.7 kcal/mol。此硝酸分子的 H 原 子可以解離至表面上最鄰近的O2c上,此反應步驟幾乎不需要任何的能障
就可以生成NO3(a) + H(a)的結構 。然後,NO3分子可以旋轉Ti5c-O 鍵而
形成Ti5c-ON(O)-Ti5c,H-O2c(a)的結構,但需要跨越 12.2 kcal/mol 的能障,
Ti5c-ON(O)-Ti5c,H-O2c(a)的吸附能為 16.5 kcal/mol。
在雙分子硝酸吸附的型態中,最穩定的結構是以兩個最穩定單分子 的結構組成2(Ti5c_ON(O)OH…O2c(a)),其吸附能為 12.8 kcal/mol。從
2(Ti5c_ON(O)OH…O2c(a)) 形成 N2O5 分子需要跨越 46.2 kcal/mol 的能
障,由此可知聚合反應難以發生。單分子及雙分子硝酸在二氧化鈦銳鈦 礦表面的吸附及反應路徑和在金紅石表面相似。
演一個重要的角色,特別是對 NO3自由基。NO3可能成為一個有效的連
接分子以連接半導體量子點系統,例如:氮化銦(InN)半導體量子點,和二 氧化鈦表面。此外,我們也進一步計算以 NO3連接氮化銦團簇(InN)X及
Adsorption Configurations and Reactions of Nitric Acid on TiO
2Rutile (110) and Anatase (101) surfaces
Student:Ching Yi Chang Advisor:M. C. Lin
Institute of Molecular Science National Chiao Tung University
Abstract
The adsorption and reactions of the monomer and dimer of nitric acid on TiO2 rutile (110) and anatase (101) surfaces have been studied by
first-principles calculations based on the density functional theory in conjunction with ultrasoft pseudopotential approximation implemented in the Vienna Ab-initio Simulation Package (VASP).
The most stable configuration of HNO3 on the rutile surface is a
molecular monodentate adsorbed on the 5-fold coordinated Ti atom with the hydrogen bonded to a neighboring surface bridging oxygen with the adsorption energy of 6.7 kcal/mol. It can dissociate its H atom to a nearest bridged oxygen with approximately no barrier to produce NO3(a) + H(a). The
rotation of NO3 requires a barrier of 12.2 kcal/mol to form the by didentate
configuration Ti5c-ON(O)-Ti5c,H-O2c(a), which adsorbs on two 5-fold
coordinated Ti atoms with the adsorption energy of 16.5 kcal/mol.
In the case of the adsorption of 2HNO3 molecules, the most stable
configuration, 2(Ti5c_ON(O)OH…O2c(a)), has a structure similar to two single
energy of 12.8 kcal/mol, which is about twice that of the single HNO3 molecule.
The result suggests that the interaction of the two planar HNO3 adsorbates is
negligible. The dehydration from 2(Ti5c_ON(O)OH…O2c(a)) forming N2O5
requires an energy barrier of 46.2 kcal/mol, indicating that the dimerization of the two HNO3(a) is hard to occur. Similar adsorption phenomena appear on the
anatase (101) surface.
The result of our calculations shows that the co-adsorption of hydrogen plays a significant role in the adsorption energies of adsorbates, especially for the NO3 radical, which may be employed as a linker between semiconductor
quantum dots such as InN with the TiO2 surface. Furthermore, we calculate
the charge distribution of the NO3 group connecting (InN)x clusters with
Acknowledgements
這兩年對我而言是很特別的一段日子,沒有念大學的主科—物理,反 而念了一樣是基礎科學的化學,有時候有點迷惘,有時候又覺得選一條有 挑戰的路也是挺有意思的。 在這些日子裡,很感謝我的指導老師—林明璋院士,與老師的談話 中,總可以讓我得到一些課本上學不到的東西,帶我看到不一樣的世界; 學問方面也謝謝老師的教導,總能有不同的見解。從老師身上看到的態度 是遠遠比這兩年中學到的知識要重要得多,這些態度對我有一定程度的影 響! “Where there is a will, there is a way"對嗎?其次感謝的就是朱超原、王念夏教授和林聖賢院士,在碩一上當朱超 原老師的助教時,了解原來做學問要抱持著很嚴謹的態度,如何扎扎實實 的做學問,以及老師在碩一下計算化學課程的教導,讓我從門外漢探進門 內,看到這屬於學問的美。感謝王念夏老師,總能給我一些協助,並且可 以把問題都簡單化,讓我了解原來可以以輕鬆的態度面對困難。也謝謝林 聖賢院士能在碩士班口試時當我的口試委員,謝謝院士當天的笑容與鼓 勵,口試那一天對我而言是難忘的! 在碩士論文中,也感謝陳欣聰博士的指導,對於我這個愛問問題的學 生,總是能給予一定程度的解答。也謝謝秀琴,禎翰學長,雯妃學姐,老 王學長,佩儀學妹,實驗室的大家及在交大的老伴琇雅和春慧。 這篇致謝也同時與疼我的外婆、放心不下我的老媽、超信任我的老 爸、適時關心我的老哥分享與遠在台南的廷璁男友分享。台北的家和中壢 的外婆家始終是我的避風港,謝謝你們無怨無悔的包容與支持! 謝謝大家!
Table of Contents
page Chinese abstract ...I English abstract ... Ⅲ Acknowledgements... Ⅴ Table of contents ... Ⅵ List of Figures ... Ⅸ List of Tables...ⅩIChapter One Introduction... 1
1-1 Preface ... 1
1-2 Discussion on TiO2 research field……..………..1
1-3 Reference ... 4
Chapter Two Computational methods ... 6
2-1 Introduction... 6
2-2 Density Functional Theory (DFT) ... 6
2-3 Reduced Density Matrix methods ... 7
2-4 Kohn-Sham Theory... 8
2-4-1 In general form ... 9
2-4-2 In ground state form ... 9
2-5 Exchange-correlation energy ... 11
2-5-2 General Gradient Approximation (GGA)... 12
2-5-3 Meta-GGA Methods... 12
2-6 Methods for finding reaction pathways between two stable states ... 12
2-6-1 Self-Penalty Walk (SPW) methods... 13
2-6-2 Nudged Elastic Band (NEB) method ... 13
2-7 Reference ... 15
Chapter Three Results and Discussion ... 16
3-1 Verification ... 16
3-1-1 TiO2 rutile(110) and anatase(101) surface... 17
3-1-2 Testing the Model with H2O adsorption... 19
3-2 Adsorption and reaction of HNO3 and HNO3 dimer on TiO2 surface and reaction mechanism ... 20
3-2-1 Adsorption of HNO3 and NO3 on TiO2 (110) rutile ... 20
3-2-2 Adsorption of HNO3 and NO3on TiO2 (101) anatase ... 21
3-2-3 Reaction path for the adsorption and dissociation of HNO3 on the rutile(110) surface ... 21
3-2-4 Reaction path for the adsorption and dissociation of HNO3 on the anatase (101) surface... 22
3-2-5 Reaction path for the adsorption and dissociation of HNO3 dimeron the rutile(110)surface ... 23
3-2-6 Reaction path for the adsorption and dissociation of HNO3 dimer on the anatase (101) surface ... 25
3-4 Bader atomic charges... 28
3-5 (InN)x, x=1, 2, 3, 6, 10 ... 29
3-6 Bader Charge analysis of (InN)x, x=1, 2, 3, 6, 10... 30
3-8 Reference ... 31
List of Figures
Figure 3-1-1(a) TiO2 rutile (110) surface 1x2x2 super cell... 36
3-1-1(b) TiO2 rutile (110) surface 1x4x1 super cell ... 36
Figure 3-1-1(c) TiO2 anatase (101) surface 2x1x2 super cell... 37
3-1-1(d) TiO2 anatase (101) surface 3x1x2 super cell ... 37
Figure 3-1-1(e) Calculated geometry of HNO3 molecule... 38
3-1-1(f) Calculated geometry of NO3 molecule... 38
Figure 3-1-2(a) Optimized geometries of adsorbed H2O on TiO2 rutile (110) surface... 39
Figure 3-1-2(b) Optimized geometries of adsorbed H2O on TiO2 anatase (101) surface ... 40
Figure 3-2-1(a) Optimized geometries of adsorbed of HNO3 monomer on TiO2 rutile (110) surface ... 41
Figure 3-2-1(b) Optimized geometries of adsorbed of HNO3 dimer on TiO2 rutile (110) surface ... 42
Figure 3-2-2(a) Optimized geometries of adsorbed of HNO3 monomer on TiO2 anatase (101) surface... 43
Figure 3-2-2(b) Optimized geometries of adsorbed of HNO3 dimer on TiO2 anatase (101) surface... 44
Figure 3-2-3 Potential energy surface for the HNO3 monomeron TiO2 rutile (110) surface... 45 Figure 3-2-4 Potential energy surface for the HNO3 monomer on TiO2 anatase
(101) surface ... 46 Figure 3-2-5 Potential energy surface for the HNO3 dimeron TiO2 rutile
(110) surface... 47 Figure 3-2-6 Potential energy surface for the HNO3 dimeron TiO2 anatase
(101) surface ... 48 Figure 3-2-7 Geometrical of illustrations of LM1 and TS1 TS2 TS3 TS4... 49 Figure 3-4 Bader Charge Analyses ... 50 Figure 3-5-1(a) Optimized geometries of (InN)x, x=1, 3 adsorbed on TiO2
rutile (110) surface... 51 Figure 3-5-2(a) Optimized geometries of (InN)x, x=1, 3 adsorbed on TiO2
anatase (101) surface... 52 Figure 3-5-1(b) Optimized geometries of (InN)x, x=6, 10 adsorbed on TiO2
rutile (110) surface... 52 Figure 3-5-2(b) Optimized geometries of (InN)x, x=6, 10 adsorbed on TiO2
anatase (101) surface... 53
List of Tables
Table 3-1-2(a) Optimized adsorption energies for H2O on two size of TiO2 (110)
surface ...54 Table 3-1-2(b) Optimized bond lengths (Å) and adsorption energies for H2O on
TiO2 (110) rutile surface ...54
Table 3-1-2(c) Optimized bond lengths (Å) and adsorption energies for H2O on
TiO2 (101) anatase surface ...54
Table 3-2-1(a) Optimized bond lengths (Å) and adsorption energies for HNO3 on
TiO2 (110) surface ...55
Table 3-2-1(b) Optimized bond lengths (Å) and adsorption energies for HNO3
dimer and its fragments on TiO2 (110) surface ...55
Table 3-2-2(a) Optimized bond lengths (Å) and adsorption energies for HNO3
and its fragments on TiO2 (101) surface...55
Table 3-2-2(b) Optimized bond lengths (Å) and adsorption energies for HNO3
dimer and its fragments on TiO2 (101) anatase...56
Table 3-2-3 Optimized bond lengths (Å) for transition state and intermediate on TiO2 surface...56
Table 3-3(a) Optimized bond lengths (Å) and adsorption energies for HNO3 with
H atoms co-adsorbed on bridged oxygen on TiO2 (110) surface...57
Table 3-3(b) Optimized bond lengths (Å) and adsorption energies for HNO3 with
H atoms co-adsorbed on bridged oxygen on TiO2 (101) surface...57
PW91 Level...58 Table 3-5(a) Optimized bond lengths (Å) and adsorption energies for (InN)x,
x=1, 3, 6, 10 on TiO2 (110) rutile surface...58
Table 3-5(b) Optimized bond lengths (Å) and adsorption energies for (InN)x, x=1,
3, 6, 10 on TiO2 (101) anatase surface ...59
Table 3-6(a) Bader Charge Analyses for (InN)x-ON(O)O-TiO2 rutile surface...59
Chapter One
Introduction
1-1 Preface
In the 21th century, we face the crisis in energy resources. We will run out of petroleum within forty years. We are also facing the detrimental effect of greenhouse gases, CO2 in particular, produced from fossil fuel combustion. It is
vital for us to search for environmentally clean alternative energy resources. A renewable energy such as solar radiation is ideal to meet the projected demand but requires new strategies to harvest incident photons with a higher efficiency, for example, by employing nano-structured semiconductors and molecular assemblies.
1-2 Discussion on TiO2 research field
In 1991, Grätzel et al.1 invented the dye-sensitized solar cells (DSSC) system using TiO2 nano particle films. Organic dye molecules were attached on
the porous TiO2 structures as absorbate which provided an alternative
photovoltaic device for electricity generation. The overall energy conversion (light to electricity) was typically 7.1%~7.9% in 1991. In DSSC system2, charges on the TiO2 interface go from photo-excited dye to the conduction
band of the semiconductor. The sunlight in the range between the wavelengths from the UV to the near IR region has a broad spectral adsorption for energy conversion. The efficiency may reach over 10%. However, dyes (N3 dye) are relatively expensive, and organic compounds have shorter lifetimes and are less stable than inorganic materials. Many researchers, therefore, prefer to employ semiconductor quantum dots (QD) instead of dyes for solar cells.
Moreover, TiO2 has been studied broadly for its physical and chemical
characteristic and applications , such as in heterogeneous photocatalysis on surfaces3,4, the nature of photovoltaic action5,6 and nanomaterial7 properties. All the previous information provides a rich scientific basis on its potential solar cell development.
Furthermore, investigating all the rich properties of titanium dioxide not only depend on experiments, but also on theoretical studies. Experiments and theoretical calculations are complementary to each other. We can know the reaction mechanisms in detail by theoretical calculations. The advantage of theoretical calculations is that it can reveal the mechanism of a reaction which we can not obtain through experiment and these save more expensive cost in experiment. Theoretical results contain rich and many-sided views on various effects such as doping (N-doped8,9, C-doped10 and S-doped11) and vacancy(O-vac12) of titanium dioxide surfaces. Recently more and more studies on semiconductor quantum dots13 (GaN14, InN15-17, AlN18, InP19) have been made by theoretical calculations. Further studies of quantum-dot sensitized TiO2 solar cells (QDSSC), by which their optical properties can maximize solar
photon adsorption, have been carried out, for example, Wang et al.20 reported that the absorption spectrum of InN/TiO2 films showed a pronounced broad
band absorption in the UV-Vis range, covering 390-800 nm, similar to Grätzel’s black dye. TiO2 solar cell with InAs21 quantum dots has also been studied. In
these systems, in order to achieve higher efficiencies, it is essential to find a good linker between quantum dots (InN, InAs…) and the TiO2 surface.
In our lab, we have studied the HN3/TMIn22,23 reaction to produce InN
quantum dots on TiO2 fabricated films and have analyzed adsorption/reaction
work, we want to investigate the adsorption and reaction of nitric acid (HNO3)
on TiO2 surfaces and compare the chemistry with the Boric acid (H3BO3)/TiO2
system24. We regard HNO3 as a potential linker group27 to connect InN clusters
1-3 Reference
(1) O'Regan, B.; Grätzel, M. Nature 1991, 353, 737.
(2) Grätzel, M. Journal of Photochemistry & Photobiology, C: Photochemistry Reviews 2003, 4, 145.
(3) Linsebigler, A. L.; Lu, G.; Yates Jr, J. T. Chemical Reviews 1995, 95, 735.
(4) Mills, A.; LeHunte, S. Journal of Photochemistry and Photobiology a-Chemistry
1997, 108, 1.
(5) Cahen, D.; Hodes, G.; Grazel, M.; Guillemoles, J. F.; Riess, I. J. Phys. Chem. B
2000, 104, 2053.
(6) Durrant, J. R.; Haque, S. A.; Palomares, E. Chemical Communications 2006,
2006, 3279.
(7) Chen, X.; Mao, S. S. Chem Rev 2007, 107, 2891.
(8) Asahi, R.; Morikawa, T.; Ohwaki, T.; Aoki, K.; Taga, Y. Science 2001, 293, 269. (9) Di Valentin, C.; Pacchioni, G.; Selloni, A. Physical Review B 2004, 70, 85116. (10) Di Valentin, C.; Pacchioni, G.; Selloni, A. Chem. Mater 2005, 17, 6656. (11) Tian, F.; Liu, C. J. Phys. Chem. B 2006, 110, 17866.
(12) Mutombo, P.; Kiss, A. M.; Berk, A.; Cháb, V. Modelling Simul. Mater. Sci. Eng
2008, 16, 025007.
(13) Loss, D.; DiVincenzo, D. P. Physical Review A 1998, 57, 120.
(14) Yeo, Y. C.; Chong, T. C.; Li, M. F. Journal of Applied Physics 1998, 83, 1429. (15) Persson, C.; Ferreira da Silva, A. Journal of Physics: Condensed Matter 2001, 13,
8945.
(16) Ahmed, R.; Akbarzadeh, H. Physica B: Physics of Condensed Matter 2005, 370, 52.
(17) Bagayoko, D.; Franklin, L. Journal of Applied Physics 2005, 97, 123708.
(18) Kandalam, A. K.; Pandey, R.; Blanco, M. A.; Costales, A.; Recio, J. M.; Newsam, J. M. J. Phys. Chem. B 2000, 104, 4361.
(19) Blackburn, J.; Ellingson, R.; Micic, O.; Nozik, A. J. Phys. Chem. B 2003, 107, 102.
(20) Wang, J. H.; Lin, M. C. ChemPhysChem 2004, 5, 1615.
(21) Yu, P.; Zhu, K.; Norman, A. G.; Ferrere, S.; Frank, A. J.; Nozik, A. J. J. Phys.
Chem. B 2006, 110, 25451.
(22) Wang, J. H.; Lin, M. C. Journal of Physical Chemistry B 2006, 110, 2263. (23) Tzeng, Y. R.; Raghunath, P.; Chen, S. C.; Lin, M. C. Journal of Physical
Chemistry A 2007, 111, 6781.
(24) Raghunath, P.; Lin, M. C. J. Phys. Chem. C 2008, 112, 8276.
(25) Huang, W. F.; Chen, H. T.; Lin, M. C. Chem Phys Lett 2008, communicated. (26) Huang, W. F.; Raghunath, P.; Lin, M. C. J. Comput. Chem 2008, communicated.
(27) Lee, H. J.; Kim, D. Y.; Yoo, J. S.; Bang, J.; Kim, S.; Park, S. M. Bulletin of the
Chapter Two
Computational methods
2-1 Introduction:
VASP (Vienna ab-initio Simulation Package)1 computational program is based on Density Functional Theory (DFT) which can be used to calculate the electronic ground state of a chemical system including gases and solids.
In this thesis, all the geometrical structures are calculated by VASP on the basis of first-principles2 with the electron-correlation methods such as LDA (local density approximation and GGA3 (generalized gradient approximation) at 0K. According to the VASP guide, the interaction between ions and electrons is described by ultra-soft Vanderbilt pseudopotentials (US-PP) or by the projector-augmented wave (PAW) method with a plane basis set. We select US-PP as pseudopotentials in the following calculations, except the Bader Charge which is done with the PAW method.
The following is the introduction of the DFT, Kohn-Sham Theory and NEB methods.
2-2 Density Functional Theory (DFT)4:
From a single particle Schrödinger equation eq.(1),ψ(r)K is a particle wave function ,ψ (r)∗ K is a particle conjugate wave function.=is called Dirac constant,
which is Plank constant divided by 2π. E is the total energy of the particle including kinetic energy and potential energy. V(r) is the potential energy of the particle. T is the kinetic energy of the particle.
( ) ( )
Hψ rK =Eψ rK ... (1)
2 2 ( ) ( ) ( ) ( ) 2m ψ r V rψ r Eψ r − = ∇ K + K = K ... (3) 2 2 ( ) ( ) ( ) ( ) 2m ψ r V rψ r Eψ r ∗ ∗ ∗ − = ∇ K + K = K ... (4) Density defined as the probability of wave function
2
( )K = ∗( ) ( )K K = ( )K
r r r r
ρ ψ ψ ψ ... (5) With Laplacian operator ∇2 on eq.(5)
2[ ( ) ( )] 2[ ( )] ( ) ( )[ 2 ( )] 2 ( ) ( ) r r r r r r r r ψ∗ ψ ψ∗ ψ ψ∗ ψ ψ∗ ψ ∇ K K = ∇ K K + K ∇ K + ∇ K ⋅∇ K = −22m2 [E V r− ( )] ( ) 2ρ rK + ∇ψ∗( )rK ⋅∇ψ( )rK = ... (6) (6)× 2 4m − = 2 2 2 ( ) ( ) ( ) ( ) ( ) ( ) 4m ρ r V r ρ r Eρ r 2m ψ r ψ r ∗ − = ∇ K + K = K − = ∇ K ⋅∇ K ... (7) Another polar coordinate expression for ψ(r)K and ψ (r)∗ K in eq.(8) and eq.(9)
( ) ( ) ( ) i r r r eθ ψ K = ρ K K ... (8) ( ) ( ) ( ) i r r r e θ ψ∗ K = ρ K − K ... (9)
Combine eq.(8), eq.(9) into eq.(7)
2 2 1 2 ( ) ( ) [ ( )] ( )[ ( )] [ ] 4 r r r r r ψ ψ ρ ρ θ ρ ρ ∗ ∇ K ⋅∇ K = ∇ K + K ∇ K ≈ ∇ ... (10) 2 2 2 2 ( ) ( ) ( ) ( ) [ ( )] 4m ρ r V r ρ r Eρ r 8mρ( )r ρ r − = ∇ K + K ≈ K − = K ∇ K ... (11) If we can do ( ) n ( ) n n r f
θ K =
∑
λ ρ , we have exact equation equal to solving density equation as solving wave function.2-3 Reduced Density Matrix methods:
The exact solution for wave function of N-electron system, each electron (ri
K
A wave function ( , , ..., )1 2 3
K K K K N
r r r r
ψ for N electron systems contains 4N variables. In reduced density matrix methods, is ignored electron spin.
1 2 3 1 2 3
( , , ..., ) ( , , ..., )
e N N
Hψ r r rK K K rK =Eψ r r rK K K rK ... (12)
Define the first-order density matrix
1( , )r r1 1 N ( , , ..., ) ( , , ..., )r r r1 2 3 rN r r r1 2 3 r dr drN 2 3...drN
γ K K′ =
∫
ψ∗ K K K′ K ψ K K K K K K K ... (13) Define the second-order density matrix2( , , , )r r r r1 2 1 2 N N( 1) ( , , ..., ) ( , , ..., ) ...r r r1 2 3 rN r r r1 2 3 r drN 3 drN
γ ′ ′ = − ψ∗ ′ ′ ψ
∫
K K K K K K K K K K K K K K ... (14)
Diagonal matrix element of the first-order density matrix is electron density When 1
K
r=rK1′,rK2=rK2′ eq.(13) turns into eq.(16)
1 1 1 1 1 2 3 1 2 3 2 3 ( )r ( , )r r N ( , , ..., ) ( , , ..., )r r r rN r r r r dr drN ...drN ρ =γ = ψ∗ ψ
∫
K K K K K K K K K K K K K K ... (15) 2( , )r r1 2 2( , , , )r r r r1 2 1 2 N N( 1) ( , , ..., ) ( , , ..., ) ...r r r1 2 3 rN r r r1 2 3 r drN 3 drN ρ =γ = − ψ∗ ψ∫
K K K K K K K K K K K K K K K K (16) Eq.(16) operate on∫
drK2 2( , )r r dr1 2 2 (N 1) ( )1 r1 ρ = − ρ∫
K K K K ... (17) 2-4 Kohn-Sham theory:The basis for Density Functional Theory (DFT) is proved by Hohenberg and Kohn theorem5, which is determined the ground-state electronic energy completely by the one electron densityρ(r)K . There exists one-to-one correspond between the electron density of a system and the energy.6 The goal of DFT is to connect the electron density with the energy.7,8
electron kinetic energyT, nuclear-electron attraction Vneand electron-electron
repulsion Vee.In eq.(18), the nuclear-nuclear repulsion is a constant.
e ne ee
E= ψ H ψ = +T V +V ... (18)
1( )r1 ( )r
ρ K ≡ρ K ... (19) DFT is based on the ground state energy in eq.(20)
[ ]
e DFT
E= ψ H ψ →E ρ ... (20)
Following are three terms in total energy. Respectively,T ,Vneand Vee. In
eq.(18), the nuclear-electron attraction is a sum of terms, each depending on one-electron coordinate. The electron-electron repulsion depends on two electron coordinates. 2-4-1 In general form4 Kinetic energy 1 1 1 2 1 2 1 1 ( , ) 2 r r r T = −
∫
∇ γ r rK K′ = ′drK ... (21)Nucleus-electron Coulomb attraction energy
1 1 1 1 1 ( ) n N ne Z r V dr r R α α α ρ = = − −
∑∫
K KK K ... (22) Electron-electron repulsion energy, which will divide into two parts in Hartree-Fock theory, Coulomb and exchange parts,J[ ]ρ andK[ ]ρ .Veecombinestwo terms as correlation parts.
2 1 2 1 2 1 2 ( , ) 1 2 ee r r V dr dr r r ρ = −
∫
K KK K K K ... (23)When energy level in ground state, eq.(21) will lead to eq.(24), eq.(22) will lead to eq.(27) and so on eq.(23) will lead to eq.(29). Ts[ ]ρ is the exact kinetic
energy by assuming non-interacting, and Ts[ ]ρ is in Slater determinant
composed of molecular orbital ,φ . a
Kinetic energy 1 1 1 2 1 2 1 1 ( , ) [ ] [ ] ( [ ] [ ]) 2 r r r s s T = −
∫
∇ γ r rK K′ = ′drK →T ρ =T ρ + T ρ −T ρ ... (24) 2 1 2 N s a a T α φ φ =∑
− ∇ ... (25) 2 1 2 3 1 2 3 1 2 1 1 [ ] ( , , ,..., )( )( ) ( , , ,..., ) ... 2 N s s N i s N N i T ρ ψ∗ r r r r ψ r r r r dr dr dr = =∫
K K K K −∑
∫
∇ K K K K K K K 2 1 ( ) ( ) 2 a r a r dr α φ ∗ φ = −∑∫
K ∇ K K ... (26) Where T[ρ] T [ρ]− s implies the kinetic correlation energy.Nucleus-electron Coulomb attraction energy
1 1 1 1 1 1 ( ) ( ) [ ] n n N N ne ne Z r Z r V dr E dr r R r R α α α α α α ρ ρ ρ = = = − → = − − −
∑
∫
K KK K∑
∫
K KK K ... (27) [ ] ( ) ( ) ne ext E ρ =∫
v rK ρ r drK K ... (28) 1 ( ) n N ext Z v r r R α α= α = − −∑
K K K ... (29) ( ) ext ext v rK =ε ... (30) extε implies the external potential per electron.
2 1 2 1 2 1 2 ( , ) 1 [ ] [ ] ( [ ] [ ]) 2 ee ee ee r r V dr dr E J E J r r ρ ρ ρ ρ ρ = → = + − −
∫
K KK K K K ... (31)Electron-electron Coulomb interaction energy
1 ( ) ( ) [ ] 2 r r J drdr r r ρ ρ ρ = ′ ′ ′ −
∫
K KK K K K ... (32) (Eee[ ]ρ −J[ ])ρ =Exc[ ]ρ is the electron exchange-correlation energy which is arather small fraction of the total energy and the only unknown terms. In order to solve the exchange-correlation term needs lots of approximations.
[ ] [ ] [ ] [ ] [ ]
DFT s ne xc
E ρ =T ρ +E ρ +J ρ +E ρ ... (33)
In eq.(33), Exc[ ]ρ is the unknown part, the other terms are already known. [ ] ( [ ] [ ]) ( [ ] [ ]) [ ] ( )
xc s ee xc
E ρ = T ρ −T ρ + E ρ −J ρ ≡
∫
ε ρ ρ r drK K ... (34)[ ] xc
E ρ is the exchange-correlation energy;ε ρ is the exchange-correlation xc[ ]
energy per electron.( [ ]T ρ −Ts[ ])ρ is considered as the kinetic correlation
energy.Eee[ ]ρ −J[ ]ρ contains potential correlation and exchange energy.
Furthermore, [ ]= [ ]+ [ ]=
∫
( ) [ ( )]K K K+∫
( ) [ ( )]K K K xc x c x c E ρ E ρ E ρ ρ ε ρr r dr ρ ε ρr r dr ... (35) [ ] xE ρ is a pure exchange part,Ec[ ]ρ is a correlation part.Exc[ ]ρ has physical
meaning.
2-5 Exchange-correlation energy
( ) ( ) ( ) ( ) ( ) ′ ′ ′ = + ′
∫
K K K K K K xc xc xc r v r r r dr r δε ε ρ δρ ... (36) When eq.(35) is only considered first term, eq.(36) it is assumed the density locally as slowly varying function.( ) ( )
xc xc
v rK =ε rK ... (37)
The exchange energy for a uniform electron gas is given by the Dirac formula
4 3 ( )= −
∫
( ) LDA x x E ρ C ρ r dr ... (38) 1 3 = − LDA x Cx ε ρ ... (39)2-5-2 General Gradient Approximation (GGA)3:
When eq.(35) is considered about higher correlation, the exchange and correlation energies depend on both electron density and derivatives of the density. ( ) ( , ) ( ) ′ ≈ ∇ ′ K K xc r f r δε ρ ρ δρ ... (40)
2-5-3 Meta-GGA methods (meta-GGA):
When eq.(37) is considered about higher order gradient correlation, it is with the Laplacian (∇2ρ ) as the second order term.
2 ( ) ( , , ) ( ) ′ ≈ ∇ ∇ ′ K K xc r f r δε ρ ρ ρ δρ ... (41)
2-6 Methods for finding reaction pathways between two stable states
The following methods (SPW and NEB) connect the reactant and product in the reaction of the images or structures and lead to the saddle point and to an approximation of the whole reaction path. The reaction path is based on reaction coordinate acting from reactant to product along a minimum energy
path (MEP).
2-6-1 Self-Penalty Walk (SPW) methods
In this method, the approximation reaction path is minimized by the average energy along the path, which is given as a line integral between reactant (R) and product (P). The line integral is approximation as a finite sum of M points. M is in the order of 10~20.
1 1 1 ( , ) ( ) ( ) ( ) ( ) = =
∫
P ≈∑
M i Δ i R i T R P U x ds x U x s x L L ... (42) 1 1 ij 2 2 3 1 , 1 1 1 1 d 1 ( , , ,... , ) ( ) ( ) ( ) (- ) d − − − + = = > + =∑
M Δ + M∑
− + M∑
SPW M i i i i i i i j T R x x x P U x s x d d L γ ρ λ ... (43) = − ij i j d x x ... (44) 1 , 1 1 1 1 − + = = +∑
M i i i d d M ... (45)There are γ ,λand ρ parameters in theTSPW to find the suitable reaction
path .The transition state (TS) is the point with the highest energy after minimization of the function TSPW.
2-6-2 Nudged Elastic Band (NEB) methods:
The transition state is based on transition state theory at the maximum of MEP. Nudged Elastic Band (NEB) is modified versions of SPW. The Nudged Elastic Band method defines as the sum of energies and adds a penalty term having the purpose of distributing the points along the path.9A single spring constant kwill distribute images along the reaction path.
1 2 2 3 1 , 1 1 1 1 ( , , ,..., , ) ( ) ( ) 2 − − + = = =
∑
M + M∑
− NEB M i i i i i i T R x x x P U x k x x ... (46)When k is too large,TNEBwill lead to cutting corners; whenk is too
small,TNEB will lead to sliding down. In the Climbing Image (CI-NEB) version,
one of the images is allowed to move along the elastic band to become the exact saddle point.10 In our VASP calculation is constructed on CI-NEB methods. We have to decide how many middle points to set in( , ,...,x x2 3 xM−1)on
2-7 Reference
(1) Kresse, G.; Furthmuller, J. Vienna Ab-initio Simulation Package
VASP the guide, 2005.
(2) Segall, M. D.; Lindan, P. J. D.; Probert, M. J.; Pickard, C. J.; Hasnip, P. J.; Clark, S. J.; Payne, M. C. Journal of Physics-Condensed Matter 2002, 14, 2717.
(3) Perdew, J. P.; Burke, K.; Ernzerhof, M. Physical Review Letters 1996, 77, 3865. (4) Zhu, C. Lecture Note in Quantum Computational Chemistry, 2007.
(5) Hohenberg, P.; Kohn, W. Phys. Rev. B 1964, 136, 864. (6) Jensen Introduction to Computational Chemistry.
(7) Koch, W.; Holthausen, M. C. A Chemist's Guide to Density Functional Theory Wiley-VCH 2000.
(8) Koch, W.; Sham, L. J. Phys. Rev. A 1965, 140, 1133.
(9) Henkelman, G.; Jónsson, H. J. Chem. Phys. 2000, 113, 9978.
Chapter Three
Results and Discussion
3-1 Verification
In the following calculations, the slab model is applied, respectively, to study the interaction between nitric acid and the TiO2 rutile (110) surface as
well as nitric acid and the anatase (101)1 surface. The slab cell constructed for the TiO2 rutile and anatase calculations includes 16 TiO2 as a unit cell shown in
Figure 3-1-1(a), 3-1-1(b), 3-1-1(c) and 3-1-1(d). All slabs are separated by a vacuum space of greater than 13.0 Å is employed in the direction parallel to the <110> and <101> coordinate, respectively. This large separation is used to avoid the interaction force between the upper and lower slabs. The whole layers of the super cell are not fixed in the following calculations in order to simulate with good accuracy.
The parameters are set for the TiO2 rutile (110) and anatase (101) surfaces
with cut off energy (Ecut) equal to 600 eV to include kinetic energies ( 2 2
2
k m
=
) in the basis set. The cut-off energy is bigger than real kinetic energies in a convergent system. The Monkhorst-Pack k-points are based on the Brillouin zone which is chosen by 4x4x1 in rutile and 4x5x1 in anatase. The parameters set in the cut-off energy and k-point are useful to help the convergence of a system.
Similar parameters were set in gas molecule simulations, but we allow Monkhorst-Pack k-points to 1x1x1. Gas-phase atoms and molecules were separated by 15Å as vacuum space, which is simulated with a quantum cubic box with sides 15Å. The sufficiently large size vacuum space reduces interaction between neighboring systems.
The parameters set in transition state2 calculations respectively are ISPRING=-5, IBRION=1 and LCLIMB3=.True. IBRION=1 means to find local minimum, respectively. We utilize the climbing-image nudged-elastic band (CI-NEB)3 to achieve a bigger accuracy.
The adsorption energy, Eads is computed according to equation (46):
[
(
)]
ads total slab molecule
E
= −
E
−
E
+
E
... (46) Where Eslab indicates the energy of the clean slabs or surface, Emolecule is theenergy of a gas-phase molecule, Etotal is the total energy of adsorbed species on
the surface. The surface energy is considered for the TiO2 surface with a
hydrogen adsorbed on it, the adsorption energy is calculated as follows:
/ /
[
(
)]
ads total H slab H molecule
E
= −
E
−
E
+
E
... (47) Where Eslab/H indicates the energy of the slab with an H atom adsorbed on theslab, and Etotal/H is the total energy of the slab with the adsorbate and H
co-adsorbed on it. The spin-polarized effect is insignificant, because the difference in the adsorption energy is merely +0.05 kcal/mole with the spin-polarized parameter.
A positive value of Eads >0 suggests a stable adsorption. Analysis on
atomic charges of optimized structures is calculated by utilizing the Bader method with a program designed by Henkelman et al.4 With the program, we can analyze the data with the detail in the charge transfer between an adsorbate and the surface.
3-1-1 TiO2 rutile(110) and anatase(101) surface
cell for HNO3 monomer/TiO2 system shown in Figure 3-1-1(a) and 1x4x1 super
cell shown for HNO3 dimer/TiO2 system in Figure 3-1-1(b) with Ti16O32. The
surface sizes are 6.495 Å x 5.866 Å and 6.495 Å x 11.732 Å, respectively. We use two kinds of the TiO2 rutile surface to avoid the interaction of large-size
molecules with each other. We utilize two sizes of the anatase surface for the same reason.
For the anatase surface, the models used are the 2x1x2 super cell shown in Figure 3-1-1(c) for HNO3 monomer/TiO2 system and 3x1x2 super cell for
dimer HNO3/TiO2 system shown in Figure 3-1-1(d). In the 2x1x2 super cell
with Ti16O32, the size of the surface is 7.57 Å x 10.239 Å. In the 3x1x2 super
cell with Ti24O48 composition, the surface is 11.355 Å x 10.239 Å.
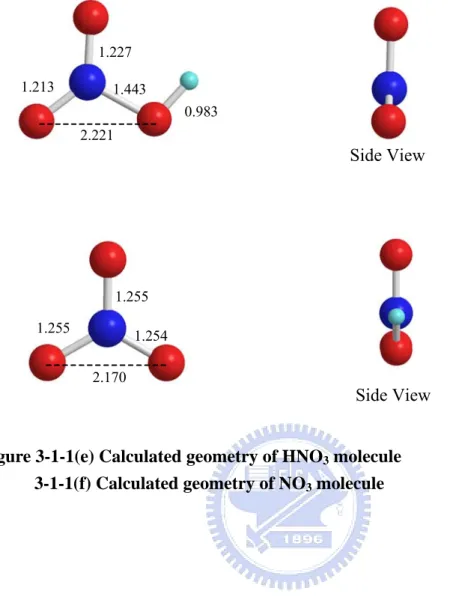
To ensure the reliability of the computational results, we first compared the calculated bulk lattice constants. The predicted lattice constants are a = 4.593 Å and c = 2.933 Å for rutile and a = 3.785 Å and c = 10.239 Å for anatase which are in good agreement with the experimental values of a = 4.594 Å and c = 2.958 Å for rutile5 and a = 3.782 Å and c = 9.502 Å for anatase6, respectively. The optimized geometries of HNO3 and NO3 are shown in Figure 3-1-1(e) and
Figure 3-1-1(f).
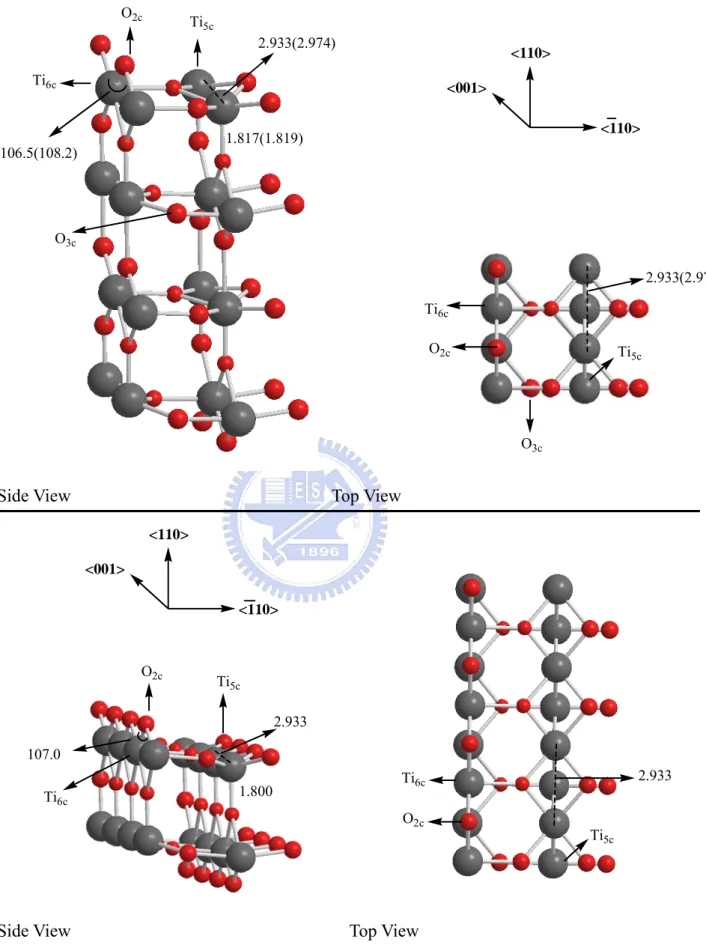
Figures 3-1-1(a) and 3-1-1(c) show different adsorption sites which have been labeled on the TiO2 surface. They are five-fold coordinated titanium,
six-fold coordinated titanium, two-fold bridging oxygen, and three-fold coordinated oxygen, corresponding to Ti5c, Ti6c, O2c(Ob), and O3c, respectively.
The twofold coordinated O atoms and fivefold coordinated Ti atoms are more active than the threefold O atoms and the sixfold coordinated Ti atom, respectively, due to their unsaturated coordinations. On rutile surface, the bond lengths of Ti5c-Ti5c, Ti5c-O3c are 2.933Å and 1.817Å, respectively. The angle of
Ti5c-O2c-Ti5c is 106.5°, which may be compared with those of Huang, et al.7, the
bond lengths of Ti5c-Ti5c, Ti5c-O3c are 2.974 Å, 1.819Å and the angle of
Ti5c-O2c-Ti5c is 108.2°, respectively.
3-1-2 Testing the Model with H2O adsorption
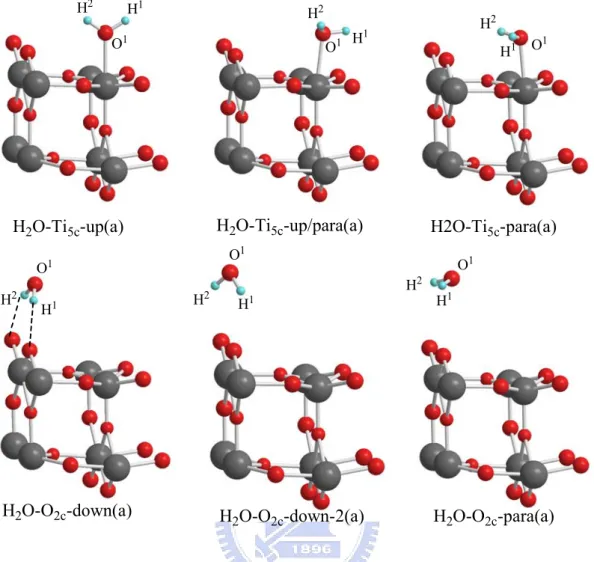
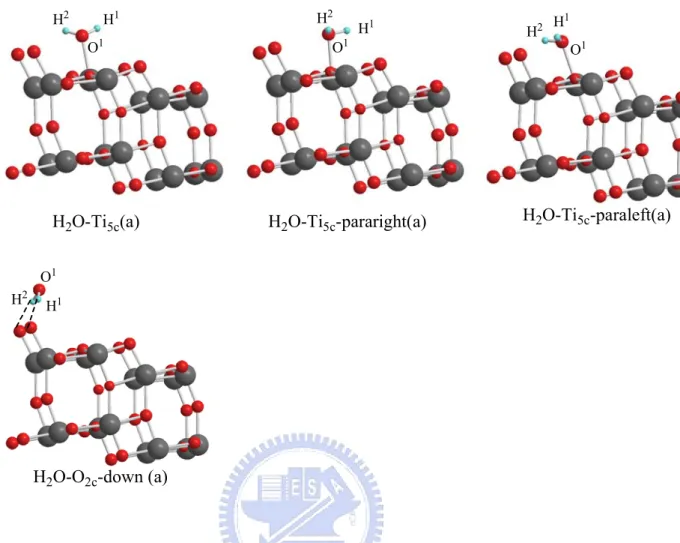
The test models have been compared with the adsorption energies of H2O8,9 on the surface. The H2O-rutile and H2O-anatase adsorption structures
are shown in Figures 3-1-2 (a), 3-1-2(b) and Table 3-1-2(a), 3-1-2(b), 3-1-2(c).
Adsorption of H2O:
As shown in the Figure 3-1-2 (a), the most stable structure H2O-Ti5c-up/para (a) for the H2O-TiO2 rutile system among the six kinds of
adsorption configurations is with one hydrogen bonding to the nearest bridged oxygen. The most stable structure is H2O-Ti5c-paraleft (a) for the H2O-TiO2
anatase system. The adsorption energies on the rutile and anatase surfaces are 16.6 kcal/mol and 15.7 kcal/mol, respectively, which are close to the result of another theoretical calculation 18.9 kcal/mole7 on the rutile and 19.2 kcal/mol8 on the anatase surfaces, indicating the adsorption of H2O on the Ti5c with anH
atom tilting forward to the bridged surface oxygen. Herman et al.10 in an experimental investigation show the adsorption of H2O on the rutile surface is
slightly higher than that on the anatase surface; the adsorption energies are from 0.74eV to 0.72 eV (17.1 kcal/mol to 16.6 kcal/mol) for the H2O adsorbed
on the anatase surface and from 0.74 eV to 0.64 eV (17.1 kcal/mol to 14.8 kcal/mol) for the H2O-ruilte system due to different water coverages. Our
calculated values are in very good with the experimental values10. Other structure (H2O-O2c-down (a)) with small adsorption energies of 2.3 kcal/mol on
without direct interactions with Ti5c atoms.
3-2 Adsorption and reaction mechanism of HNO3 and HNO3 dimer on TiO2
surface
3-2-1 Adsorption of HNO3 and NO3 on rutile TiO2 (110) Adsorption of HNO3:
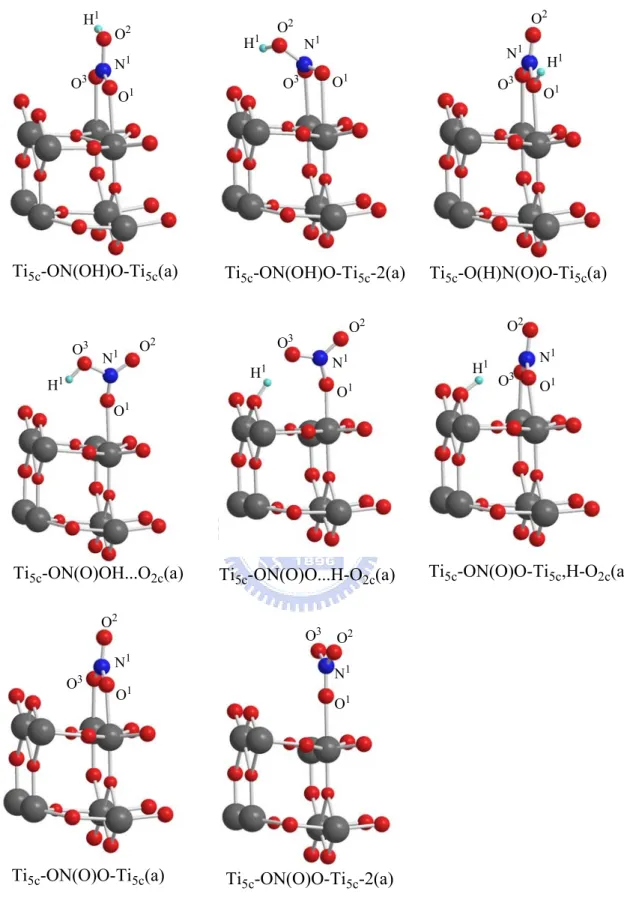
In our study, the adsorption of nitric acid was found to have four different
configurations on different sites of the metal oxide surface (Ti5c, O3c). These
adsorption configurations and their fragments on the TiO2 rutile (110) surface
are shown in Figure 3-2-1(a), and the associated bond lengths and adsorption energies are listed in Table 3-2-1(a).
In these four types of structures, the most stable one is a molecular configuration with an oxygen attached to the surface Ti5c atom and the
hydrogen formed a hydrogen bond with the nearest bridge oxygen on the surface indicated by Ti5c-ON(O)OH…O2c(a). Its adsorption energy is 6.7
kcal/mol and the Ti5c-O bond length is 2.428 Å and O3H1-O2c is 2.288 Å as
listed in Table 3-2-1(a). Although the adsorption energies of the exhibited four structures are not big enough to stabilize HNO3 for a long time,
Ti5c-ON(O)OH…O2c(a) shows clearly that the formation of a hydrogen bond
can increase the adsorption energy.
Adsorption of NO3:
We obtain two different configurations of ON(O)O on the rutile surface as shown in Figure 3-1-2(a); one is Ti5c-ON(O)O-Ti5c(a) with two oxygen atoms
bonding with Ti5c atoms on the surface and the other is Ti5c-ON(O)O-Ti5c-2(a)
with one oxygen on a Ti5c atom. The adsorption energy of the former is 12.0
kcal/mol, and that of the latter is 4.7 kcal/mol. Thus the formation of the O-Ti5c
3-2-2 Adsorption of HNO3 and NO3 on TiO2 (101) anatase
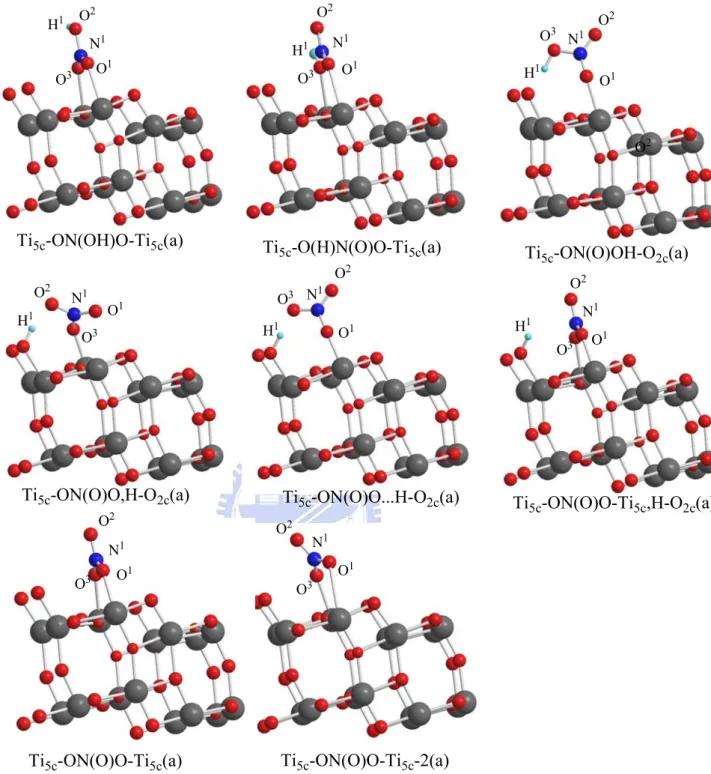
For the HNO3-anatase system, we utilize a 3x1x2 super cell with Ti16O32. Adsorption of HNO3:
We obtain three types of HNO3 adsorption configurations on the TiO2
anatase (101) surface as shown in Figure 3-2-1(b) and the bond length and adsorption energies are shown in Table 3-2-2(a). The most stable structure is similar to that on the rutile (110) surface with the configuration Ti5c-ON(O)OH-O2c(a), having a hydrogen bond involving the H atom and the
nearest neighboring oxygen site. Its adsorption energy is 13.3 kcal/mol. The other configurations of HNO3, with two oxygen atoms connecting to Ti5c atoms
on the surface are less stable than the hydrogen-bonded structure, the Ti5c-ON(O)OH-O2c(a).
Adsorption of NO3:
In Figure 3-2-2(a), we have two structures of NO3 adsorbed on the anatase
surface. The adsorption energy of Ti5c-ON(O)O-Ti5c(a) is 6.6 kcal/mol and that
of Ti5c-ON(O)O-Ti5c-2(a) is 5.6 kcal/mol. The bond lengths of Ti5c-O1 in
Ti5c-ON(O)O-Ti5c(a) and Ti5c-ON(O)O-Ti5c-2(a) are 2.259Å and 3.203Å,
respectively. The bond length of Ti5c-O is shorter, and the adsorption energy is
higher.
3-2-3 Reaction path for the adsorption and dissociation of HNO3 on the rutile
(110) surface
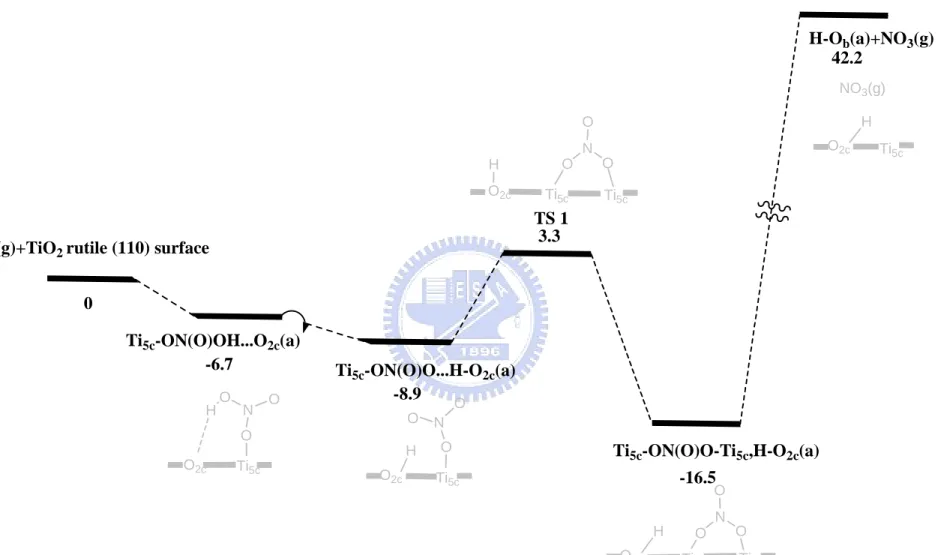
The potential energy surface of dissociative adsorption reactions of nitric acid on the clean rutile (110) surface is shown in Figure 3-2-3. The optimized structures are shown in Figure 3-2-1(a) and bond lengths are indicated in Table
considered. First, nitric acid on the clean rutile (110) surface is a monodentate structure with the hydrogen bond formed with the nearest neighboring bridging oxygen with an adsorption energy of 6.7 kcal/mol. In the dissociation process, the hydrogen atom can migrate to the neighboring O2c forming
Ti5c-ON(O)O...H-O2c(a); the reaction is slightly exothermic and occurs without
a well-defined transition state. Second, the adsorbed NO3 (a) can molecularly
rotate on the surface. The rotation of the adsorbate Ti5c-ON(O)O... H-O2c(a)
resulting in the formation of a covalently bond Ti5c-ON(O)O-Ti5c,H-O2c(a) as
shown in Figure 3-2-1(a). This rotation reaction has an activation barrier lying 3.3 kcal/mole above the reactants. The calculated activation barrier above the adsorbate is 12.2 kcal/mol. The isomerization process from Ti5c-ON(O)O...H-O2c(a) to Ti5c-ON(O)O-Ti5c,H-O2c(a) is calculated to be
exothermic by 7.6 kcal/mol. The transition state (TS1) of this process shown in Figure 3-2-7 and Table 3-2-3 corresponds to the formation of the Ti5c-O1 with a
bond length of 2.187 Å and the formation of the O2c-H1 with 0.969 Å.
According to the previous calculation, eliminating the NO3 gas molecule from
Ti5c-ON(O)O-Ti5c,H-O2c(a) to produce H-O2c(a) + NO3(g) requires 58.7
kcal/mol which implies that the adsorption process can not occur spontaneously.
On the other hands, NO3 (a) can more easily split into NO2 (a) + O (a) by
breaking one of the N-O bonds, which is endothermic by about 22.1 kcal/mol.
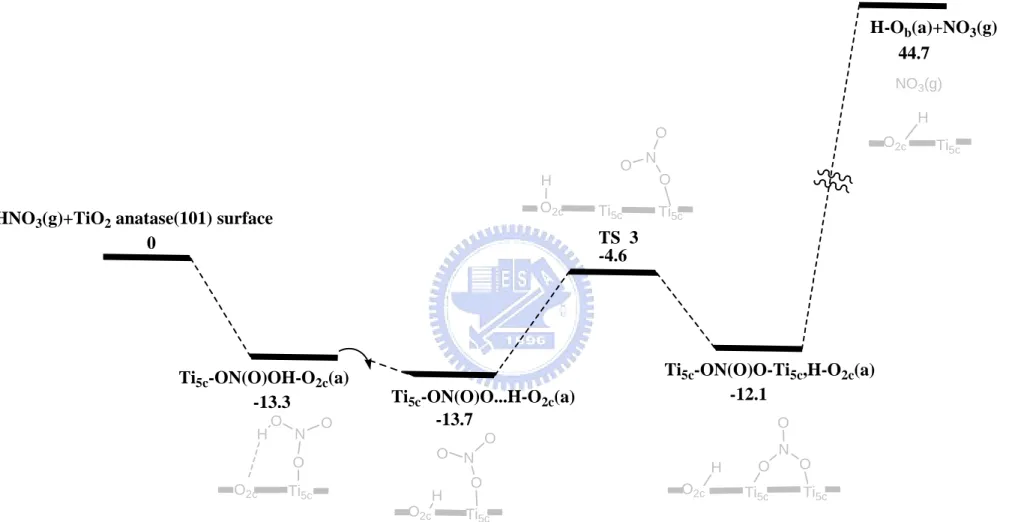
3-2-4 Reaction path for the adsorption and dissociation of HNO3 on the
anatase (101) surface
The computed potential energy surface for the dissociative adsorption reaction of HNO3 on the clean anatase (101) surface is shown in Figure 3-2-4.
The energies are all referenced to the initial reactants, HNO3 + TiO2 anatase
(101) surface. The optimized structures with the surface model are depicted in Figure 3-2-2(a) and the selected bond lengths are presented in Table 3-2-2(a). First, we consider the most stable adsorption configuration, Ti5c-ON(O)OH-O2c(a). The hydrogen of the coordinating O3H1 group can
migrate to the neighboring O2c to form Ti5c-ON(O)O…H-O2c(a) which is
slightly exothermic by 0.4 kcal/mol. The phenomenon of hydrogen migrating is same as that in the rutile (110) surface. The bond length of Ti5c-O1 is 2.027 Å.
The adsorbed ON(O)O molecule can rotate on the surface to form Ti5c-ON(O)O-Ti5c,H-O2c(a). The rotation requires 1.6 kcal/mol energy and the
bond length between Ti-O is increased by 0.12 Å. The rotation of ON(O)O molecule in the adsorbate Ti5c-ON(O)O-Ti5c,H-O2c(a) can result in the
formation of two Ti-O bonds on two Ti5c sites with the bond lengths of Ti5c-O1
and Ti5c-O3, 2.143 Å and 2.159 Å, respectively. In the isomerization process
from Ti5c-ON(O)…H-O2c(a) to Ti5c-ON(O)O-Ti5c,H-O2c(a), it has to overcome
an activation barrier of 9.1 kcal/mol at TS3, which is below the reactant by 4.6 kcal/mol in energy. In the transition state (TS3) shown in Figure 3-2-7 and Table 3-2-3, the bond lengths of Ti5c-O1 and Ti5c-O3 are 2.002 Å and 3.027 Å,
respectively. The decomposition process producing H-O2c(a) + NO3(g) from
Ti5c-ON(O)O-Ti5c,H-O2c(a) requires 56.8 kcal/mol and the final product H-O2c
+ NO3(g) is 44.7 kcal/mol above the initial reactants.
3-2-5 Reaction path for the adsorption and dissociation of HNO3 dimeron the
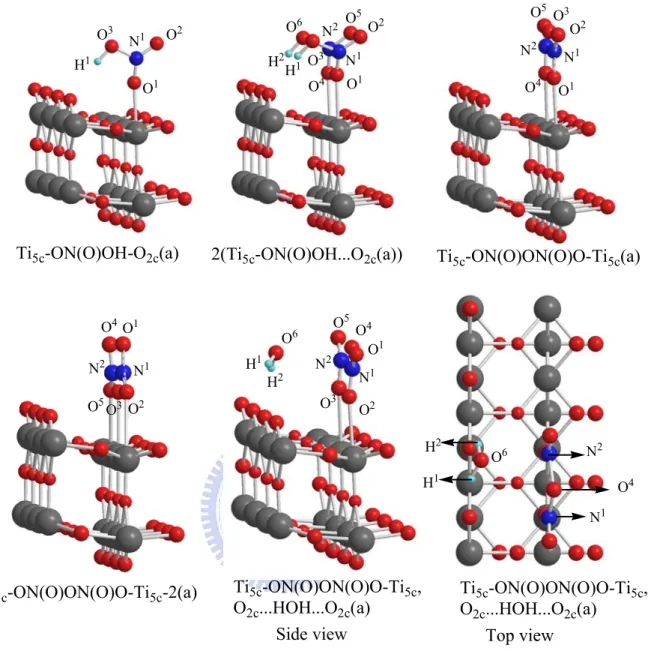
rutile(110)surface
Figure 3-2-5. The corresponding bond lengths and adsorption energies are listed in Table 3-2-1(b). In the dimer HNO3 adsorption on the rutile surface, we
use 1x4x1 super cell as our TiO2 rutile (110) model. The difference between
1x2x2 and 1x4x1 that is the latter has 2 times higher width with half of the height of the former.
In the reaction path, we only consider the most stable structure of HNO3
dimer as given for the monomer HNO3 adsorption with an adsorption energy of
12.8 kcal/mol, approximately twice that of the monomer. In the most stable conformation, two HNO3 molecules adsorbed parallel to each other on two
surface Ti5c atoms, having two hydrogen bonds formed with the closest
neighboring bridged oxygen O2c, depicted as 2(Ti5c-ON(O)OH-O2c(a)) in
Figure 3-2-2(b). Their bond lengths are Ti5c-O1=2.594 Å, Ti5c-O4=2.551 Å and
O3H1=2.266 Å, O3H1=2.163 Å.
Starting from the most stable adsorbate 2(Ti5c-ON(O)OH-O2c(a)), the
H1O3-N1 bond of one HNO3 molecule rotates and forms a hydrogen bond
with another HNO3 giving intermediate LM1, whose adsorption energy is
higher than that of 2(Ti5c-ON(O)OH…O2c(a)) by 5.1 kcal/mol. In the
intermediate LM1, the intermolecular hydrogen bond length is 2.203 Å between the H2 atom and the O3H1 of the first HNO3. This finding is
consistent with the results reported by M. J. Gillan11 who have showed the intermolecular hydrogen bonding can stabilize the configuration. From the intermediate, the H atom of the first HNO3 split to form H2O with the OH
group of the second HNO3 via TS2, producing a water complex
Ti5c-ON(O)ON(O)O-Ti5c,O2c…HOH…O2c(a). The process demands 46.2
kcal/mol for crossing the barrier. From the water complex, it can eliminate the H2O(g) from the surface to form a Ti5c-ON(O)ON(O)O-Ti5c(a) + H2O(g)
with 5.8 kcal/mol endothermicity. The adsorption energy of Ti5c-ON(O)ON(O)O-Ti5c(a) is not big enough to stabilize the structure on
TiO2.
Furthermore, the N2O5 molecule adsorbed on the rutile surface have
two different structures shown in Figure 3-2-1(b). The adsorption energies of Ti5c-ON(O)ON(O)O-Ti5c(a) and Ti5c-ON(O)ON(O)O-Ti5c-2(a) are 2.0
kcal/mol and 1.5 kcal/mol, respectively. These structures are not stable enough to form N2O5(a). Ti5c-ON(O)ON(O)O-Ti5c(a) easily splits into NO3
and NO2 molecules without a well-defined transition state. The final product
of NO3 and NO2 on the surface lies 3 kcal/mol below
Ti5c-ON(O)ON(O)O-Ti5c(a).
3-2-6 Reaction path for the adsorption and dissociation of HNO3 dimer on the
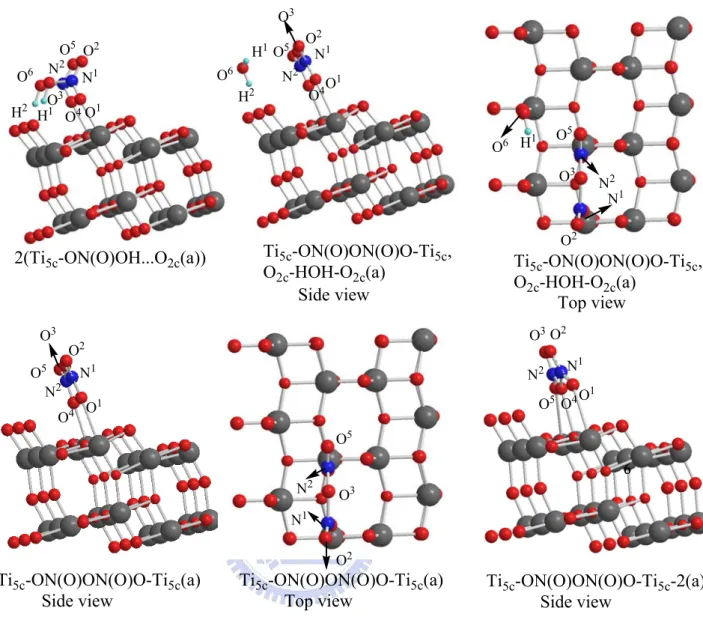
anatase (101) surface
Similarly, Figure 3-2-6 shows the potential energy diagrams and Figure 3-2-2(b) shows the related geometrical structures of HNO3 dimer on the TiO2
anatase (110) surface. For this process, we utilize 3x1x2 super cell with the 11.355Å x12.289 Å area on the TiO2 (101) anatase surface. For the HNO3
dimer adsorbed on a smaller 2x1x2 super cell, there is unavoidable interaction between the cells. The related bond lengths and the adsorption energies are presented in Table 3-2-2(b). We consider the most stable configuration Ti5c-ON(O)OH…O2c(a) given in the Figure 3-2-2(a) which has an adsorption
energy of 13.3 kcal/mol. Thus, the most stable HNO3 dimer is
2(Ti5c-ON(O)OH…O2c(a)) with the adsorption energy 28.3 kcal/mol, which is
Ti5c atoms on the surface and with the two hydrogen atoms forming hydrogen
bonds with the nearest two bridged oxygen atoms. Their bond lengths are Ti5c-O1 2.309 Å, Ti5c-O4 2.332 Å, O3H1-O2c 1.498 Å and O6H2-O2c 1.484 Å,
respectively.
From the most stable dimer adsorbate 2(Ti5c-ON(O)OH-O2c(a)), one of the
H atoms can react with the OH group of the second HNO311 to form H2O
giving Ti5c-ON(O)O-Ti5c,O2c…HOH…O2c(a) via TS4 as shown in Figure 3-2-7.
The transition state (TS4) is 40.0 kcal/mol higher than the initial reactants. The activation barrier for breaking the hydrogen bond requires 68.2 kcal/mol. The bond lengths of O3-H1 and O6-H1 are 1.873 Å and 3.120 Å. The Ti5c-ON(O)O-Ti5c,O2c…HOH…O2c(a) is 3.1 kcal/mol above the initial
reactants. From Ti5c-ON(O)O-Ti5c,O2c...HOH...O2c(a), it can eliminate H2O(g)
to produce Ti5c-ON(O)ON(O)O-Ti5c(a) + H2O (g). The dehydration reaction
requires 4.8 kcal/mol. Thus, Ti5c-ON(O)O-Ti5c,O2c…HOH…O2c(a) is not stable
and can not retain H2O on the surface.
Moreover, the N2O5 molecules can adsorb on the anatase surface with two
different structures as shown in Figure 3-2-2(b). The adsorption energies of Ti5c-ON(O)ON(O)O-Ti5c(a) and Ti5c-ON(O)ON(O)O-Ti5c-2(a) are 2.1 kcal/mol
and 0.2 kcal/mol, respectively; they are too small for the N2O5 molecules to
adsorb on the rutile surface.
3-3 Hydrogen effect on adsorbate structures and adsorption energies
In this section, we discuss the effect of H adsorbed on a neighboring oxygen (O2c) in the nitric acid system. The adsorption energies on rutile and
anatase are listed in Table 3-3(a) and Table 3-3(b), respectively. We will not take spin-polarization into consideration, because these are little change in
geometries and the difference in adsorption energies of nitric acid is no more than ± 0.1 kcal/mol. As discussed above, nitric acid adsorbed on clean TiO2
rutile (110) surface has four different of structures. The most stable structure is Ti5c-ON(O)OH…O2c(a) with an adsorption energy of 6.7 kcal/mol. For the
hydrogen bonding O2c atom, the bond lengths of Ti6c-O2c are 1.855 Å and 1.838
Å. Table 3-3(a) indicates the co-adsorption of H on a neighboring oxygen in Ti5c-ON(O)OH…O2c(a) to produce Ti5c-ON(O)O…H-O2c(a) has a higher
adsorption energy, 9.9 kcal/mol. The bond lengths of Ti6c-O2c are 2.070 Å and
1.838 Å. Moreover, the co-adsorption of two hydrogen atoms on the two neighboring O2c will increase adsorption energy to 11.7 kcal/mol. The bond
lengths of Ti6c-O2c are 2.087 Å and 2.008 Å.
Similarly, nitric acid adsorbed on clean TiO2 anatase has three different
structures. The most stable one is Ti5c-ON(O)OH…O2c(a) with 13.3 kcal/mol
adsorption energy. Comparing with one hydrogen adsorbed on a bridging oxygen, the adsorption energy is a bit lower than that of Ti5c-ON(O)OH-O2c(a),
10.5 kcal/mol. The bond lengths of Ti6c-O2c are 2.220 Å and 1.917 Å. So with
two hydrogen co-adsorbed on bridging oxygen atoms, the adsorption energy is slightly lower than the former, 8.9 kcal/mol. The bond lengths of Ti6c-O2c are
2.242 Å and 2.190 Å.
The effect of hydrogen absorbed on a bridging oxygen results in increasing adsorption energy on the TiO2 rutile (110) surface, but decreasing
the adsorption energy on the TiO2 anatase (101) surface. With the hydrogen on
bridging oxygen leads the increase in the bond length of Ti6c-O2c from 1.855 Å
to 2.087 Å in the rutile (110) surface and from 1.936 Å to 2.242 Å in the anatase (101) surface.
Ti5c-ON(O)O-Ti5c,H-O2c(a) on the rutile and anatase surfaces. The results are
shown in Figure 3-4 and Table 3-3(c). The adsorption energies with hydrogen attaching to the neighboring bridged oxygen on the rutile surface is 58.7 kcal/mol and that without hydrogen is 12.0 kcal/mol. On the anatase surface, the adsorption energy with and without hydrogen on the bridged oxygen are 56.7 kcal/mol and 6.6 kcal/mol, respectively. In order to explain the hydrogen effect on the adsorption energy, we analyze the Bader charges of Ti5c-ON(O)O-Ti5c(a) and Ti5c-ON(O)O-Ti5c,H-O2c(a) on both surfaces.
3-4 Bader atomic charges
We calculate the Bader charges for the adsorbate ON(O)O on the TiO2
rutile(110) and anatase (101) surfaces with and without an H atom co-adsorbated on the bridging oxygen as shown in Figure 3-4.
In the rutile (110) surface, H atom co-adsorption increases the charge of the ON(O)O adsorbate by 0.21 e, where e is the magnitude of the charge of the electron. In Figure 3-4, the charge of the bridged surface oxygen with an H atom is -1.45 e and the H atom is 1.00 e. Comparing with to the bridged surface oxygen without H, it is -0.82 e.
Similarly, in the anatase (101) surface, H atom adsorption on the bridged oxygen with the ON(O)O adsorbate increases the charge by 0.22 e. The charge of the bridged oxygen with H atom is -1.35 e and the H atom is 0.86 e. Without co-adsorbed H atom the charge is -0.84 e. This phenomenon suggests that an H atom on the bridged oxygen will distribute its atomic charge in the adsorbate and stabilize the configuration with a higher adsorption energy as listed in Table 3-3(c), in which the difference with and without a co-adsorbed H atom is 46.7 kcal/mol on the TiO2 rutile (110) surface, and 50.13 kcal/mol in
the TiO2 anatase (101) surface. The results of the Bader charge analysis on the
rutile surface are consistent with those on the anatase surface. The hydrogen effect on NO3 adsorbate is a significant difference in the adsorption energy due
to the charge transfer from surface to NO3.
3-5 (InN)x, x=1, 2, 3, 6, 10
The topic is about (InN)x clusters with nitric acid as a linker adsorbing on
TiO2 rutile(110) and anatase(101) surfaces. To understand the charge transfer
phenomenon and the adsorption energy of (InN)x, we computationally study
these systems with VASP.
The adsorption energies and bond lengths of (InN)x, x=1, 2, 3, 6, 10, each
connecting with an adsorbed NO3 are listed the Table 3-5(a) and Table 3-5(b)
for the rutile and anatase surfaces, respectively.
On the rutile surface, the (InN)x, x=1, 3 adsorbed on NO3 with an H atom
co-adsorbed on a bridged oxygen is shown in Figure 3-5-1(a). The (InN)x, x=1,
the In-N-O is linear and the In1-O2 bond length is 2.180 Å. The adsorption of (InN)-ON(O) on the rutile surface is merely 8.3 kcal/mol. When (InN)x, x=2,
the configuration of (InN)2-ON(O)O is not sufficiently stable to maintain the
(InN)2 structure on the TiO2 rutile(110) surface. (InN)2 easily emits N2 gas. For
(InN)x, x=3, the (InN)3 molecule has two kinds of hexagon structures, one with
In1-O2 bond length with 2.232 Å and the other with 2.347 Å. For the non-symmetric (InN)3 adsorbate, (InN)3-ON(O)O-Ti5c, its adsorption energy is
higher than that of the symmetric (InN)3-ON(O)O-Ti5c-2 by 9.8 kcal/mol. In the
case of (InN)x,x=6 and 10 with a wurtzite cluster structure, the adsorption
adsorption energy 14.5 kcal/mol for (InN)-ON(O)O and 45.4 kcal/mol and 34.6 kcal/mol for (InN)3-ON(O)O due to two different configurations as alluded to
above for the analogous with surface. The adsorption energies of (InN)6 and
(InN)10 on TiO2 anatase are 10.7 kcal/mol and 48.2 kcal/mol, respectively,
without the ON(O)O linker.
3-6 Bader Charge analysis of (InN)x, x=1, 2, 3, 6, 10
The analyses the charges of (InN)x-ON(O)O(a), x=1, 2, 3, 6, 10 with an H
atom co-adsorbed on TiO2 rutile(110) and anatase(101) are shown in Figures
3-5-1(a), 3-5-1(b) and 3-5-2(a), 3-5-2(b) and Table 3-6(a),3-6(b). The Ti5c
atoms on the surface without the ON(O)O linker are 2.46 e and 2.46 e on the rutile and anatase surface, respectively. The two Ti5c atoms attached with
ON(O)Omolecules will increase their charges to 2.51 e and 2.50 e, and 2.53 e and 2.50 e, on the rutile and anatase surface, respectively. The charges of Ti5c
atoms on both surfaces decrease to 2.48 e after (InN)x, adsorption.
With the ON(O)O molecule adsorbing on the TiO2 rutile and anatase
surfaces, the charges of ON(O)O are -0.78 e on both surfaces. Following the attachment of (InN)x, x=1, 2, 3, 6, 10, to ON(O)O, the charge distributions will
increase the electron density to near the ON(O)O molecule side. Because the ON(O)O molecule is an electron withdrawing radical, it can easily attract more electrons, the (InN)x cluster will thus become more positive. Therefore, we may
infer ON(O)O as a linker is not an enhancement in power conversion efficiency.
3-7 Reference
(1) Diebold, U. Surface Science Reports 2003, 48, 53.
(2) Eyring, H. J. Chem. Phys. 1935, 3, 107.
(3) Vasp TST Tools.
(4) Henkelman, G.; Arnaldsson, A.; Jónsson, H. Computational Materials Science
2006, 36, 354.
(5) Vinet, P.; Ferrante, J.; Smith, J.; Rose, J. J. Phys. C 1986, 19, L467.
(6) Burdett, J. K.; Hughbanks, T.; Miller, G. J.; Richardson Jr, J. W.; Smith, J. V.
Journal of the American Chemical Society 1987, 109, 3639.
(7) Huang, W. F.; Raghunath, P.; Lin, M. C. J. Comput. Chem 2008, communicated. (8) Raghunath, P.; Lin, M. C. J. Phys. Chem. C 2008, 112, 8276.
(9) Vittadini, A.; Selloni, A.; Rotzinger, F. P.; Gräzel, M. Physical Review Letters
1998, 81, 2954.
(10) Herman, G. S.; Dohnalek, Z.; Ruzycki, N.; Diebold, U. J. Phys. Chem. B 2003,
107, 2788.
(11) Lindan, P. J. D.; Harrison, N. M.; Gillan, M. J. Physical Review Letters 1998, 80, 762.
Chapter Four
Conclusions
4 Conclusions
In summary, we perform systematic investigations on the HNO3adsorbed
on the TiO2 rutile (110) and anatase (101) surfaces by DFT calculations. The
key conclusions are summarized as follows:
(1) The adsorption and reaction of the monomer nitric acid on the rutile (110) surface:
The most stable configuration for nitric acid on the clean rutile (110) surface is a monodentate structure with one hydrogen bonded to a neighboring bridging oxygen, its adsorption energy is 6.7 kcal/mol. The resulting Ti5c-ON(O)OH...O2c(a) can go over a small barrier for H atom migration to the
bonding bridged oxygen forming a more stable Ti5c-ON(O)O...H-O2c(a) with an
exothermicity of 8.9 kcal/mol. The rotation of ON(O)O adsorbate starting from Ti5c-ON(O)O...H-O2c(a) to form Ti5c-ON(O)O-Ti5c,H-O2c(a) requires a barrier
of 13.2 kcal/mol. Then, the elimination of NO3 from Ti5c-ON(O)O-Ti5c,H-O2c(a)
to produce H-O2c(a) + NO3(g) has an endothermicity of 58.7 kcal/mol, which
corresponds to the binding energy of NO3 with the H-covered rutile TiO2
surface.
(2) The adsorption and reaction of the dimer nitric acid on the rutile (110) surface:
In the case of HNO3 dimer, two equivalent monodentate structures nitric
acid 2(Ti5c-ON(O)OH…O2c(a)) absorbed on two 5-fold coordinated Ti atoms of
12.8 kcal/mol, which are slightly smaller than two times of the monodentate. The result indicates that the effect of adsorbate interaction is negligible. According to the PES shown in Figure 3-2-5, the intermediate LM1 is a local minimum with an exothermic energy of 17.8 kcal/mol. The dehydration process from LM1 to Ti5c-ON(O)ON(O)O-Ti5c,O2c...HOH...O2c(a) needs a high barrier
of 46.2 kcal/mol. The Ti5c-ON(O)ON(O)O-Ti5c,O2c...HOH...O2c(a) can
barrierlessly eliminate the H2O molecule dissociate to produce
Ti5c-ON(O)ON(O)O-Ti5c(a) + H2O(g) with a small endothermicity of 5.8
kcal/mol.
(3) The adsorption and reaction of the monomer nitric acid on the anatase (101) surface:
The most stable configuration for nitric acid on the clean anatase (101) surface is a monodentate structure Ti5c-ON(O)OH…O2c(a) with a hydrogen
bonded to a neighboring bridged oxygen, its adsorption energy is 13.3 kcal/mol. The Ti5c-ON(O)OH-O2c(a) can overcome a small barrier for H atom migration to
an adjacent bridged oxygen forming Ti5c-ON(O)O...H-O2c(a) with an
exothermicity of 0.4 kcal/mol. Similar to the reaction on the rutile (110) surface, for the rotation of ON(O)O molecule, from Ti5c-ON(O)O...H-O2c(a) to form
Ti5c-ON(O)O-Ti5c,H-O2c(a) has a barrier of 9.1 kcal/mol. The NO3 elimination
from Ti5c-ON(O)O-Ti5c,H-O2c(a) to generate the H-O2c(a) adsorbate and NO3(g)
has an endothermicity of 56.8 kcal/mol which represents the adsorption energy of NO3 on H-covered anatase TiO2 surface.
Similar to the rutile surface, the 2(Ti5c-ON(O)OH....O2c(a)) configuration
is the most stable one on the anatase surface with an adsorption energy of 28.9 kcal/mol, which are slightly bigger than two times of the monodentate 13.3 kcal/mol. No intermediate structure like LM1 on rutile can be formed as shown in Figure 3-2-6. To form the configuration Ti5c-ON(O)ON(O)O-Ti5c,O2c-HOH-O2c(a) requires a higher barrier of 68.2
kcal/mol than that on the rutile surface. Formation of Ti5c-ON(O)ON(O)O-Ti5c,O2c-HOH-O2c(a) can therefore not occur
spontaneously due to the high barrier. The elimination process from Ti5c-ON(O)ON(O)O-Ti5c,O2c-HOH-O2c(a) to the Ti5c-ON(O)ON(O)O-Ti5c(a) +
H2O(g) is only endothermic by 4.8 kcal/mol.
(5) Hydrogen effect
Hydrogen atom plays an important role in the adsorption energies of the adsorbates on TiO2 surface. We compare the adsorption energies of reveal
species with and without H atom on the bridged oxygen atom on rutile and anatase surfaces. According to our calculated results, the adsorption energies of NO3(g) adsorbed on the rutile and anatase surface increase by 46.7 and 50.1
kcal/mol, respectively (see in Table 3-3(c) ), when an H atom is adsorbed on the nearest bridged oxygen atom.
By analyzing the Bader charges, the charge of ON(O)O in Ti5c-ON(O)O-Ti5c,H-O2c(a) is -0.63 e and it is -0.42e in Ti5c-ON(O)O-Ti5c(a) on
the rutile. While the charge of ON(O)O is -0.65e in Ti5c-ON(O)O-Ti5c,H-O2c(a)
and it is -0.43 e in Ti5c-ON(O)O-Ti5c(a) on the anatase. It is found that there is a
charge transfer from TiO2 to NO3 in the presence of H atom co-adsorbed on a
(6) (InN)x, x=1, 2, 3, 6, 10 on ON(O)O-covered TiO2 surface
Our calculations show that the adsorption energies of (InN)x, are less than
50.2 kcal/mol on the rutile surface and 48.2 kcal/mol on the anatase surface covered with ON(O)O. By Bader charge analysis, we find that only a small electron density (0.36 e for rutile, 0.35 e for anatase) transfer from (InN)x, to the
ON(O)O linker. Therefore, the –ON(O)O- might not be a good linker between a semiconductor dot and the TiO2 surface.
<110> <110> <001> O2c Ti 5c Ti6c O3c 1.817(1.819) 2.933(2.974) 106.5(108.2) O2c Ti5c O3c Ti6c 2.933(2.974)
Side View Top View <110> <110> <001> O2c Ti 5c Ti6c 1.800 107.0 O2c Ti5c Ti6c 2.933 2.933 Side View Top View
Figure 3-1-1(a)TiO2 rutile (110) surface 1x2x2 super cell 3-1-1(b) TiO2 rutile (110) surface 1x4x1 super cell
<101> <101> <010> O2c O2c Ti5c Ti6c O2c O2c Ti5c Ti6c O3c O3c 1.858 1.809 3.788 3.801 Side View Top View
<101> <101> <010> O2c O2c Ti5c Ti6c O2c O2c Ti5c Ti6c O3c O3c 1.802 1.858 3.785 3.785
Side View Top View
Figure 3-1-1(c) TiO2 anatase (101) surface 2x1x2 super cell 3-1-1(d) TiO anatase (101) surface 3x1x2 super cell
1.227 1.213 1.443 0.983 1.255 1.255 1.254 Side View Side View 2.221 2.170
Figure 3-1-1(e) Calculated geometry of HNO3 molecule 3-1-1(f) Calculated geometry of NO3 molecule
H2O-Ti5c-up(a) H2O-Ti5c-up/para(a) H2O-Ti5c-para(a) H2O-O2c-down(a) O1 H1 O1 O1 O1 H1 H1 H1 H2 H2 H2 H2 H2O-O2c-para(a) H2O-O2c-down-2(a) H2 H 2 H1 H1 O1 O1
H2O-Ti5c(a) H2O-Ti5c-pararight(a) H2O-Ti5c-paraleft(a)
H2O-O2c-down (a)
O1 O1 O1 O1 H2 H2 H2 H2 H1 H1 H1 H1
Ti5c-ON(OH)O-Ti5c(a) Ti5c-O(H)N(O)O-Ti5c(a)
Ti5c-ON(O)OH...O2c(a)
Ti5c-ON(OH)O-Ti5c-2(a)
Ti5c-ON(O)O...H-O2c(a) Ti5c-ON(O)O-Ti5c,H-O2c(a)
Ti5c-ON(O)O-Ti5c(a) O2 N1 H1 O3 O1 O2 O3 O1 H1 N1 N1 N1 N1 N1 N1 N1 H1 H1 H1 O1 O1 O1 O1 O1 O1 O2 O2 O2 O2 O2 O2 O3 O3 O3 O3 H1 O3 O3 Ti5c-ON(O)O-Ti5c-2(a)
Figure 3-2-1(a) Optimized geometries of adsorbed of HNO3 monomer on TiO2 rutile (110) surface
Ti5c-ON(O)OH-O2c(a) 2(Ti5c-ON(O)OH...O2c(a)) Ti5c-ON(O)ON(O)O-Ti5c, O2c...HOH...O2c(a) Ti5c-ON(O)ON(O)O-Ti5c(a) Ti5c-ON(O)ON(O)O-Ti5c-2(a) O3 O2 H1 N1 O1 O1 O1 O1 O1 O2 O2 O2 O2 O3 N1 N1 N1 N1 H1 H1 O3 O3 O3 N2 N2 N2 N2 O6 O5 O4 O4 O4 O4 O5 O5 O5 H2 H2 Ti5c-ON(O)ON(O)O-Ti5c, O2c...HOH...O2c(a)
Side view Top view
H1 H2 O6 O6 N2 N1 O4
Figure 3-2-1(b)Optimized geometries of adsorbed of HNO3 dimer on TiO2 rutile (110) surface