國
立
交
通
大
學
分子醫學與生物工程研究所
碩 士 論 文
Gefitinib 誘發細胞凋亡經由表皮生長因子接受器
非依賴性路徑的分子機制
Molecular mechanism of gefitinib-induced apoptosis
through epidermal growth factor receptor
independent pathway
研 究 生:余勝壹
指導教授:趙瑞益
Gefitinib 誘發細胞凋亡經由表皮生長因子接受器
非依賴性路徑的分子機制
Molecular mechanism of gefitinib-induced apoptosis
through epidermal growth factor receptor
independent pathway
研 究 生:余勝壹 Student:Sheng-Yi Yu
指導教授:趙瑞益 Advisor:Jui-I Chao
國 立 交 通 大 學
分 子 醫 學 與 生 物 工 程 研 究 所
碩 士 論 文
A Thesis
Submitted to Institute of Molecular Medicine and Bioengineering
National Chiao Tung University
in Partial Fulfillment of the Requirements
for the Degree of
Master
in
Molecular Medicine and Bioengineering
July 2010
Hsinchu, Taiwan, Republic of China
摘 要
表皮生長因子接受器屬於酪胺酸激酶受體的ErbB/HER 家族之成員,活化後會 促進腫瘤的生長。Gefitinib (Iressa™, ZD1839)是一種小分子的酪胺酸激酶抑制劑, 已經被證實具有抗癌的活性,主要經由標靶到表皮生長因子接受器的催化部位與 ATP 去競爭鍵結位置。然而,gefitinib 在癌細胞中誘發表皮生長因子接受器非依賴 性的細胞凋亡機制仍然不清楚。在本篇研究中,我們主要探討由gefitinib 誘發的表 皮生長因子接受器非依賴性之癌細胞凋亡訊息傳遞路徑。Gefitinib 會在 RKO (大腸 癌)、A549 (肺癌)、BFTC905 (膀胱癌)、MCF7 (乳癌)及 A375 (皮膚癌)細胞中造成 細胞毒性,以10-60 μM gefitinib 處理 24 小時後,細胞毒性的敏感度依序為 A375 > MCF7 > BFTC905 > A549 > RKO 細胞。有趣地,A375、MCF7 以及 RKO 細胞表 現非常低量的磷酸化表皮生長因子接受器與總量表皮生長因子接受器蛋白;相反 地,BFTC905 與 A549 細胞則高度的表現。再者,A375 與 BFTC905 細胞在 gefitinib 處理後都能誘發細胞凋亡的產生。Securin 又稱作腦下垂體腫瘤轉形基因,大量地 表現在許多的癌症,可以促進腫瘤的形成。我們觀察到癌細胞處理gefitinib 後,明 顯降低securin 蛋白的表達。進一步研究發現,缺乏 securin 的細胞會比功能正常的 細胞對gefitinib 誘發的細胞毒性更為敏感,相反地若轉殖大量表現 securin 的質體 到癌細胞,則對gefitinib 誘發的細胞毒性產生抗性。ATF3 是一種轉錄因子,可藉 由調控細胞存活與死亡的訊息平衡影響癌症的形成。在我們的研究中發現,gefitinib 處理後可以誘發 ATF3 蛋白的表達與轉移到細胞核。綜合以上結果,我們推測 gefitinib 所誘發的癌細胞凋亡可透過表皮生長因子接受器非依賴性的途徑,而釐清 securin 與 ATF3 在 gefitinib 誘發的癌細胞死亡訊息的調控機轉,可提供未來癌症 治療的新策略。Abstract
The epidermal growth factor receptor (EGFR) belongs to the ErbB/HER family of tyrosine kinase receptors. The activation of EGFR is important for promoting tumor growth. Gefitinib (Iressa™, ZD1839), a small molecule tyrosine kinase inhibitor, has been demonstrated the promising antitumor activity. It targets the catalytic domain of EGFR to compete with the ATP binding site. However, the precise EGFR-independent apoptotic mechanisms by gefitinib remain incompletely clear. In this study, we investigated the effects of gefitinib on the EGFR-independent cell death signaling pathways in human cancer cells. Direct cytotoxicity was observed in RKO (colon cancer), A549 (lung cancer), BFTC905 (bladder cancer), MCF7 (breast cancer) and A375 (skin cancer) cells by gefitinib. The order of cytotoxic sensitivity was A375 > MCF7 > BFTC905 > A549 > RKO cells following treatment with 10-60 μM gefitinib for 24 h. Interestingly, A375, MCF7 and RKO cells expressed very low protein level of the phosphorylated-EGFR and total EGFR; in contrast, BFTC905 and A549 cells expressed the high level. Moreover, treatment with gefitinib induced apoptosis in A375 and BFTC905 cells. Securin, also known as pituitary tumor-transforming gene (PTTG), overexpresses in a variety of tumors and promotes tumorigenesis. We also observed that gefitinib inhibited the securin protein expression in cancer cells. Furthermore, loss of securin enhanced the gefitinib-induced cell death; conversely, an overexpression of securin resisted the gefitinib-induced cell death. Activating transcription factor 3 (ATF3), a transcription factor, may regulate the delicate balance between proliferative and apoptotic signals that control the development of cancer. We found that gefitinib induced ATF3 protein expression and translocated to nuclei. As a whole, we suggest that gefitinib can induce apoptosis that may be through the EGFR-independent pathways in cancer cells. Understanding the mechanisms which securin and ATF3 signal transduction on the regulation of apoptosis following treatment with gefitinib may contribute to the novel therapeutic strategies in cancers.
誌 謝
時值驪歌聲響的畢業季節,研究生的生涯也到了尾聲的階段。兩年的實驗學 習即將成為過去,不過一本論文的完成,要感謝的人實在太多了。 首先,由衷地感謝指導教授趙瑞益老師給予了學生這個學習的機會,讓我能 進入抗癌研究的領域鑽研相關知識。冗長的碩士生涯裡,無論是相關課程的學習、 實驗研究的討論、抑或是生活上的各種關心,一切都很感謝老師兩年來的指導與 照顧。而著作論文期間,也承蒙老師不厭其煩的校閱與改正。另外還要感謝王雲 銘教授與陳清漂教授對於論文的審閱以及口試報告上諸多寶告的意見,使學生的 論文能更加的完善。 而學習的開始除了老師的指導外,能遇到優秀學長姊的帶領是一件幸福的 事。真的非常感謝實驗室大學姊惠芳學姊以及大學長光凱學長兩年來一切大大小 小對於實驗上細心的指導,也感謝剛進實驗室時真宜學姊與瑞良學長認真的帶領 著我學習實驗,並感謝和欽學長在實驗問題上的討論。再者,作研究的過程最怕 的就是孤單一個人,很高興我能有靜怡、厚巽、繼慶這些同學的陪伴,大家一起 上課、趕報告、做實驗,度過這兩年來辛苦的碩士班生活。實驗室一切與大家生 活的點滴,終將成為美好的回憶。 同時,我要謝謝老同學志原、國政,總是在我學業或生活上遇到不順遂時聆 聽我的抱怨並給我安慰。另外,非常謝謝我的摯友政冠,在我碩士生涯最艱困難 熬的時刻不斷給予我支持與鼓勵,真的非常謝謝你。還要感謝葉爸爸、葉媽媽在 我求學期間對予我的關懷,也感謝曉君學姊、亞萱學姊時常給予我生活上的關心。 漫長的研究生生活裡,除了一群關心我的師長與朋友外,最重要的就是家人 的陪伴。一路走來,最感謝的就是親愛的媽媽支持我完成學業,另外還有外公、 外婆、哥哥、弟弟的關懷。家人的支持與鼓勵,便是我努力完成學業的最大動力。 最後,以此論文獻給所有關心我的師長與親友。 余勝壹 謹誌於 分子抗癌研究室 2010 年 7 月Contents
Abstract (Chinese)...i Abstract (English)...ii Acknowledgements...iii Contents...iv List of figures...vi List of appendixes...vii Abbreviations...viii 1. Introduction...11.1 Molecular targeting strategies in cancer therapies………..1
1.2 EGFR family………...………1
1.3 EGFR targeting cancer threapies...2
1.4 Gefitinib...3
1.5 Apoptosis...3
1.6 Securin and cancer...4
1.7 ATF3 and cancer...5
1.8 The purpose of the study...6
2. Materials and methods...7
2.1 Chemicals and reagents...7
2.2 Antibodies...7
2.3 Cell lines and cell culture...7
2.4 Cytotoxicity assay...8
2.5 Cell cycle analysis...8
2.6 Annexin V and PI assay...9
2.7 Observation of living cell image………...9
2.9. Western blot...10
2.10 Immunofluorescence staining and confocal microscopy...10
2.11 Transfection...11
2.12 Statistical analysis...11
3. Results...13
3.1 Gefitinib elicits the cytotoxicity in both low level EGFR and high level EGFR 3.1 cancer cells………....…13
3.2 Gefitinib induces apoptosis in both A375 and BFTC905 cancer cells…..13
3.3 Gefitinib promotes the hyperpolarization of mitochondria and elevates the 3.3. activation of caspase-3 and the cleavage of PARP...14
3.4 Gefitinib inhibits the securin protein expression……….14
3.5. Loss of securin enhances the gefitinib-induced cell death and overexpression of 3.5 securin resists the gefitinib-induced cell death……….….15
3.6 Gefitinib increases the ATF3 protein expression………15
3.7 Gefitinib analogue-1 raises higher cytotoxicity than gefitinib in the low level . EGFR colon cancer cells………...……16
4. Discussion...17
5. Conclusion...20
References...21
Figures...30
List of figures
Fig. 1. ..The effect of gefitinib on the cell viability in a variety of human cancer cell
Fig. 1. .lines...30
Fig. 2. .The protein level of phospho-EGFR and total EGFR in a variety of human Fig. 2. ..cancer cell lines...31
Fig. 3...The effect of gefitinib on the cell cycle progression and sub-G1 formation in ..A375 and BFTC905 cells...32
Fig. 4.. The effect of gefitinib on apoptosis in A375 and BFTC905 cells...33
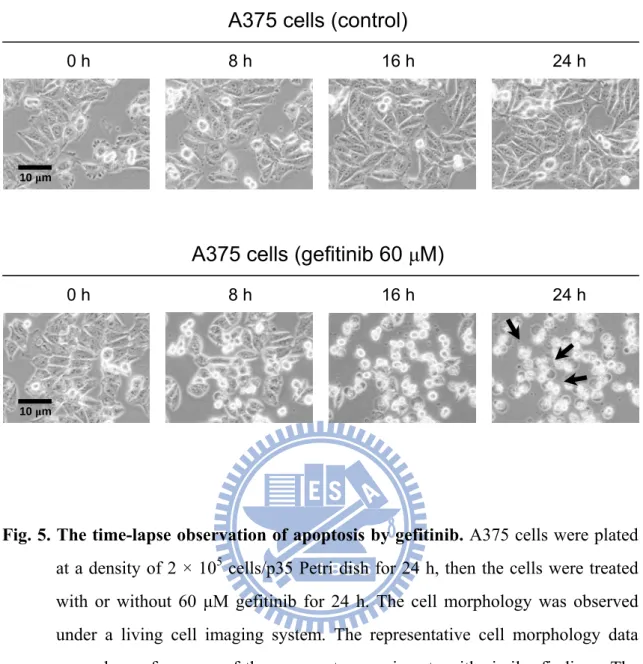
Fig. 5.. The time-lapse observation of apoptosis by gefitinib...34
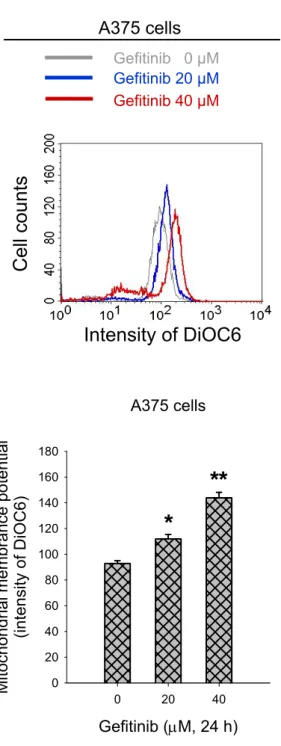
Fig. 6.. The effect of gefitinib on the hyperpolarization of mitochondria...35
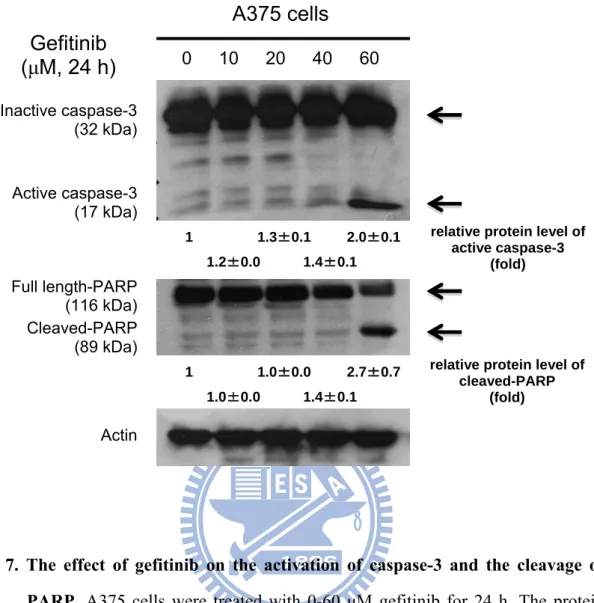
Fig. 7.. The effect of gefitinib on the activation of caspase-3 and the cleavage of Fig. 7.. PARP...36
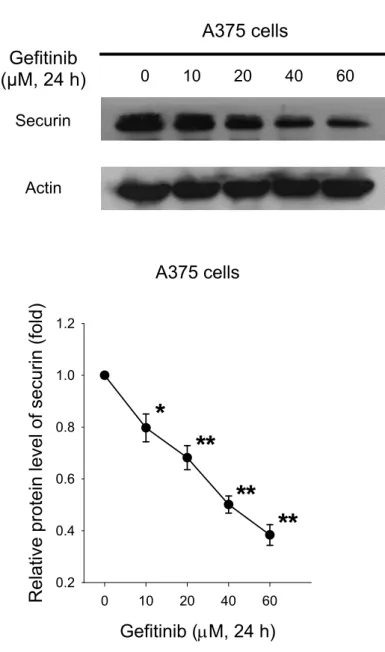
Fig. 8.. The effect of gefitinib on the securin protein expression...37
Fig. 9.. The effect of gefitinib on the expression and location of securin...38
Fig. 10. The comparison of cytotoxicity between the securin-wild type and -null HCT 116 cancer cells following gefitinib treatment……….…………..…….39
Fig. 11. The effect of the overexpression of securin on the gefitinib-induced cytotoxi- Fig. 11. city...40
Fig. 12. The effect of gefitinib on the ATF3 protein expression...41
Fig. 13. The effect of gefitinib on the expression and location of ATF3...42
Fig. 14. The comparison of cytotoxicity between the gefitinib and its analogues in a . variety of human cancer cell lines...43
Fig. 15. The schematic diagram of antitumor activity by gefitinib...44
List of appendixes
Abbreviations
AIF: apoptosis-inducing factor
ATF3: activating transcription factor 3 bZIP: basic-region leucine zipper
caspase: cysteine aspartic acid specific protease CREB: cyclic AMP response element-binding DiOC6: 3,3’-dihexiloxadicarbocyanine
EGFR: epidermal growth factor receptor ERK: extracellular signal-regulated kinase FBS: fetal bovine serum
FITC: fluorescein isothiocyanate
MAPK: mitogen-activated protein kinase mTOR: mammalian target of rapamycin
MTT: 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide PARP: poly(ADP-ribose) polymerase
PBS: phosphate-buffered saline PI: propidium iodide
PI3K: phosphoinositide 3-kinase
PTTG1: pituitary tumor-transforming gene 1 RTK: receptor tyrosine kinase
SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis TGF-α: transforming growth factor-α
TKI: tyrosine kinase inhibitor
1. Introduction
1.1 Molecular targeting strategies in cancer therapies
Cancers are leading cause of mortality in the world. The statistical data demonstrated that the malignancy has been situated the first place of the causes of death in Taiwan from 1982 (Department of Health, Executive Yuan, R.O.C., 1982). Last year, cancer still has been listed as the first cause of death in Taiwan. Despite improvements in surgical and diagnostic techniques, the outcome for people with cancer remains poor. Conventional chemotherapeutical agents act by creating toxic effects on all dividing cells, frequently resulting in severe damage of normal tissues leading to side effects like myelosuppression, alopecia, or gastrointestinal problems. The optimum goal is to find a treatment modality that specifically kills malignant cells and causes little or no side effects.
Cancer is the result of a multistep process in which genetic alterations in proto-oncogenes and/or tumor suppressor genes can eventually lead to the evolution of cells [1]. Advances in the understanding of cancer biology and more specifically of cell signalling pathways have led to the identification of several potential molecular targets and to the development of new agents directed against these targets. Targeted therapies were developed to target key elements that play a role in tumor development and tumor growth. Recently developed anticancer drugs target these molecular mechanisms and good results have been reported for various cancer types [2]. The anticancer target drugs have numerous good qualities. First, in many tumor types they tend to stabilize tumor progression and may create a chronic disease state which is no longer immediately life threatening. Second, side effects are minimal when compared to conventional chemotherapeutic agents. Third, synergistic effects are seen in vitro when anticancer target drugs are combined with radiotherapy and/or conventional chemotherapeutic agents [3].
Normal cell growth is tightly regulated through activation of cellular signal transduction pathways. Growth factors and their receptors play a fundamental role in the communication between outside the cell surface and the inside compartments. The epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases, also known as the HER or ErbB family, include four known members: EGFR (HER1/ ErbB1), HER2/ErbB2, HER3/ErbB3, and HER4/ErbB4 [4]. The EGFR is composed of an extracellular ligand-binding domain, a transmembrane domain and an intracellular tyrosine kinase domain [5]. The ErbB tyrosine kinases and the EGF-like peptides form a complex system [6]. Binding with ligands, such as epidermal growth factor (EGF) or transforming growth factor (TGF)-α, EGFR will be activated by dimerization and autophosphorylation of tyrosine kinase domain with subsequent initiation of downstream signaling molecules [7]. EGFR activates two major downstream intracellular signaling pathways: (1) Ras-Raf-mitogen-activated protein kinase kinase (MEK)-mitogen-activated protein kinase (MAPK) and (2) the phosphoinositide 3-kinase (PI3K)-Akt/protein 3-kinase B-mammalian target of rapamycin (mTOR) cascades [8; 9]. The Ras-Raf-MEK-MAPK pathway modulates several cellular processes including gene transcription, G1/S cell-cycle progression, and cellular proliferation. The PI3K pathway regulates anti-apoptotic and prosurvival signal cascades [10].
1.3 EGFR targeting cancer therapies
Tumor cells are able to synthesize and to respond to a number of different peptide growth factors [11]. Dysregulation of EGFR pathways by overexpression or constitutive activation can promote tumor processes including angiogenesis and metastasis [12]. This is associated with poor prognosis in many human malignancies [13]. Two major classes of EGFR-targeted therapies have been developed: the anti-ErbB monoclonal antibodies (MAb) and ErbB-specific tyrosine kinase inhibitors (TKIs) [14]. Monoclonal antibodies and TKIs clearly differ in their mode of action at target level. Anti-EGFR MAb such as cetuximab and panitumumab bind to the extracellular domain of EGFR on the surface of tumor cells, thus preventing EGFR ligands from interacting and activating the receptor, as well as receptor-ligand internalization [15]. By contrast, TKIs such as gefitinib and erlotinib block the binding of adenosine triphosphate to the intracellular
signaling [16].
1.4 Gefitinib
A quinazoline-derived agents that are developing by specific ATP competitors of EGFR tyrosine kinase, one representative of which is gefitinib [17]. Gefitinib was the first commercially available EGFR TKI. It has now been approved for the treatment of advanced colorectal cancer, squamous cell carcinoma of the head and neck, advanced non-small-cell lung cancer, as well as pancreatic and breast cancer [18]. The recommended dose for use was established at 250 mg/day [19]. Gefitinib shows antiproliferative activity in various human cancer cell types. It inhibits the production of pro-angiogenic factors and induces apoptosis in numerous in vitro and xenograftmodels
[20]. This sensitivity to gefitinib is associated with dependence on both Akt and ERK1⁄ 2 pathways [21; 22]. Moreover, tumor growth inhibition by gefitinib is potentiated by combination with a variety of cytotoxic anticancer agents [23].
Anti-EGFR therapy has shown significant efficacy in some cancers; however, no therapeutic response was seen in the majority of cancer patients [24]. In addition, patients initially responsive to anti-EGFR therapy develop resistance over time of treatment. Potential mechanisms of resistance to EGFR-targeted therapy might rely on activation of alternative RTKs which bypass the EGFR pathway or constitutive activation of signaling pathways downstream EGFR, as well as EGFR gene amplification and receptor mutations [25].
1.5 Apoptosis
The maintenance of cellular homeostasis is fundamental for tissue integrity in multicellular organisms. Programmed cell death (apoptosis) is a highly conserved mechanism that has evolved to maintain cell numbers and cellular positioning within tissues comprised of different cell compartments [26]. It is activated in response to diverse signals during normal tissue development, homeostasis, and disease pathogenesis [27; 28]. Apoptosis was first described in 1972 by Currie and colleagues [29]. Mammals have two distinct, but ultimately converging, apoptosis signaling
pathways: the extrinsic (also called ‘death receptor’) pathway, which is activated by death receptors; and the intrinsic (also called ‘mitochondrial’ or ‘Bcl-2-regulated’) pathway [30]. Apoptosis requires controlled degradation of cellular macromolecules by hydrolytic enzymes, and therefore proteases play a central role in this process [31; 32; 33; 34]. These proteases were collectively referred to as caspases (cysteine aspartic acid specific proteases) [35]. Since activated caspases are demonstrated in apoptotic cells, these proteases could presumably cleave their target proteins anywhere in the cell and at different times to complete the execution phase of apoptosis [36]. The activation of caspase-3 is a final parameter of caspase family on apoptosis. Poly(ADP-ribose) polymerase (PARP), a nuclear protein implicated in DNA repair, is one of the earliest proteins targeted for a specific cleavage to the signature 89-kDa fragment during apoptosis [37]. PARP is one of the prime target proteins for caspase-3 [38]. Failure of this apoptosis regulation results in pathological conditions such as developmental defects, autoimmune diseases, neurodegeneration or cancer [39]. On the other hand, apoptosis-regulating proteins also provide targets for drug discovery and new approaches to the treatment of cancer [40].
Mitochondria functions can control cellular life and death [41]. Mitochondria play important roles in cell death through the release of pro-apoptotic factors such as cytochrome c and apoptosis-inducing factor (AIF), which activate caspase-dependent and caspase-independent cell death [42]. On apoptosis, cytochrome c and AIF release to the cytosol, mitochondrial fragmentation as well as mitochondrial hyperpolarisation followed by an oxidative burst, and breakdown of mitochondrial membrane potential [43].
1.6 Securin and cancer
Pituitary tumor-transforming gene (PTTG) was isolated from rat pituitary tumor cells in 1997 [44]. It consists of a homologous family of proteins expressed in different species that includes Cut2 in fission yeast, Pds1 in budding yeast, Pimples in Drosophila, and securin in human [45; 46; 47; 48]. Securin acts as an anaphase inhibitory protein that plays an important role in preventing abnormal chromosome segregation during mitosis [47]. The activity of separase is inhibited by securin in
metaphase [49]. At the metaphase-anaphase transition, securin is degraded with subsequent release of separase to mediate the separation of sister chromatids by cleavage of the chromosomal cohesin [50]. In normal condition, securin also maintains genomic stability [51]. Recently, it has been shown that securin regulates DNA repair following UV and X-ray damages [52].
Securin overexpression has been reported in a variety of endocrine-related tumors, especially pituitary, thyroid, breast, ovarian, and uterine tumors, as well as nonendocrine-related cancers involving the central nervous system, pulmonary system, and gastrointestinal system [53; 54; 55; 56; 57]. It has been shown that securin can promote the cell proliferation and tumorigenesis [58; 59]. Securin levels correlate with tumor invasiveness, and it has been identified as a key signature gene associated with tumor metastasis [60].
1.7 ATF3 and cancer
Activating transcription factor 3 (ATF3) is a member of the ATF/cyclic AMP response element-binding (CREB) family of transcription factors [61; 62]. The common feature that these proteins share is the basic-region leucine zipper (bZIP) element [63]. This domain is responsible for specific DNA binding, while the leucine zipper region is responsible for forming homodimers or heterodimers with other bZIP-containing proteins [64]. ATF/CREB proteins were initially identified for their binding to the cyclic AMP response element (CRE) in various promoters, which has the consensus sequence TGACGTCA [65].
ATF3 is an adaptive-response gene that participates in cellular processes to adapt to extra- and/or intracellular changes, where it transduces signals from various receptors to activate or repress gene expression [66; 67]. Emerging evidence suggests that ATF3 may play another critical function in host defence by regulating the delicate balance between proliferative and apoptotic signals that contribute to the development of cancer [68]. Interestingly, ATF3 has been demonstrated to play differing roles in cancer development depending on the cell type and context [69]. There are many studies that support an oncogenic role of ATF3 [70]. ATF3 expression contributes to the successful propagation of human cancer [71]. In contrast to the studies above, some evidence
suggests that ATF3 may be able to inhibit tumourigenesis [72]. The loss of ATF3 function results in loss of tumour suppression [73].
1.8 The purpose of the study
The induction of apoptosis has been considered to be the major mechanism for these gefitinib mediated anticancer effects. However, the precise downstream signaling molecules of EGFR-independent have not yet been elucidated. In this study, we investigated the effects of gefitinib on the EGFR-independent cell death signaling pathways in human cancer cells. We also studied the role and regulation between securin and ATF3 on the cell viability following gefitinib treatment.
2. Materials and methods
2.1 Chemicals and reagents
Gefitinib was purchased from proteinkinase.de (Biaffin GmbH & Co KG, Kassel, Germany) and dissolved in DMSO. Gefitinib analogues were kindly provided by Dr. C. Chen (National Dong-Hwa University, Hualien, Taiwan) and dissolved in DMSO. 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), propidium iodide (PI), the Cy3-labeled mouse anti-β-tubulin (c-4585) and Hoechst 33258 were purchased from Sigma Chemical Co. (St. Louis, MO, USA). 3,3’-dihexiloxadicarbocyanine (DiOC6) was purchased from Calbiochem (San Diego, CA, USA). BODIPY FL phallacidin (B-607) was purchased from Invitrogen (Carlsbad, CA, USA). A431 cell lysate (12-301) were purchased from MILLIPORE (Temecula, CA, USA).
2.2 Antibodies
Anti-phospho-EGFR (05-1128) and anti-EGFR (05-104) antibodies were purchased from MILLIPORE (Temecula, CA, USA). Anti-caspase-3 (3004-100) was purchased from BioVision Research Products (Mountain View, CA, USA). Anti-PARP (#9542) antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). The anti-securin (ab-3305) was purchased from Abcam (Cambridgeshire, UK). Anti-ATF3 (sc-188) and the FITC (fluorescein isothiocyanate)-labeled goat anti-mouse IgG (sc-2010) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The Cy3-labeled goat anti-rabbit IgG was purchased from Amersham Pharmacia Biotech (Little Chalfont Buckinghamshire, UK). Anti-actin (MAB1501) antibodies were purchased from CHEMICON International, Inc. (Temecula, CA, USA).
2.3 Cell lines and cell culture
The A549 cell line was derived from lung carcinoma that contained the wild type p53. BFTC905 cells were derived from bladder carcinomas of Chinese patients. The MCF7 cell line was derived from breast adenocarcinoma of 69 years adult female. A375 cells were derived from skin carcinomas of malignant melanoma. The securinwild type and -null HCT116 colorectal carcinoma cell lines were kindly provided by Dr. B. Vogelstein of Johns Hopkins University (Baltimore, MD). RKO and A375 cells were maintained in DMEM medium (Gibco, Life Technologies, Grand Island, NY). A549, BFTC905 and MCF7 cells were cultured in RPMI-1640 medium (Gibco, Life Technologies, Grand Island, NY, USA). The securin-wild type and -null HCT116 cells were cultured in McCoy’s 5A medium (Sigma Chemical). The complete medium was supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin and sodium bicarbonate. These cells were maintained at 37°C and 5% CO2 in a
humidified incubator (310/Thermo, Forma Scientific, Inc., Marietta, OH).
2.4 Cytotoxicity assay
The cells were plated in 96-well plates at a density of 1 × 104 cells/well for
overnight. Following gefitinib of its analogues treatment for 24 h, the cells were washed with phosphate-buffered saline (PBS) and were replaced fresh medium for cultured 2 days. Thereafter, the medium was replaced and the cells were incubated with 0.5 mg/ml of MTT in complete medium for 4 h. The surviving cells converted MTT to formazan that generates a blue-purple color when dissolved in dimethyl sulfoxide. The intensity of formazan was measured at 565 nm using a plate reader (Molecular Devices, VERSAmax). The relative percentage of cell viability was calculated by dividing the absorbance of treated cells by that of the control in each experiment.
2.5 Cell cycle analysis
The cell cycle progression after treatment with gefitinib was measured by flow cytometer. The cells were plated at a density of 1 × 106 cells per 60-mm Petri dish in complete medium for overnight. Then the cells were treated with 0-60 μM gefitinib for 24 h. At the end of treatment, the cells were collected and fixed with ice-cold 70%
ethanol overnight at −20 °C. After centrifugation, the cell pellets were treated with 4 μg/ml PI solution containing 1% Triton X-100 and 100 μg/ml RNase at 37 °C for 30 min (in the dark). After re-centrifugation, the cells resuspended in 1 ml ice-cold PBS. To avoid cell aggregation, the cell solutions were filtrated through nylon membrane (Becton-Dickinson, San Jose, CA). Subsequently, the samples were analyzed by flow cytometer. A minimum of ten thousand cells was analyzed for DNA content, and the percentage of cell cycle phases was quantified by a ModFit LT software (Ver. 2.0, Becton-Dickinson).
2.6 Annexin V and PI assay
The level of apoptosis was determined by annexin V-PI staining analysis. The annexin V-PI staining kit (BioVision, Mountain View, CA) was used to examine the cells by incubated with fluorescein isothiocyanate (FITC)-conjugated-annexin V and PI according to the manufacturer's instruction. The cells were cultured in 60-mm Petri dish at a density of 1 × 106 cells for overnight. After treatment with or without gefitinib for
24 h, the cells were washed with PBS. The cells were trypsinized and collected by centrifugation at 1500 rpm for 5 min. Thereafter, the cells were incubated with 500 μL of annexin V-PI labeling solution (containing 5 μL of annexin V-FITC and 5 μL of PI) at 25 °C in the dark for 5 min. Finally, the samples were analyzed by flow cytometer using CellQuest software (FACScan, Becton-Dickinson, San Jose, CA). The cells showed annexin V(+)/PI(−) and annexin V(+)/PI(+), which indicated at early and late apoptosis, respectively.
2.7 Observation of living cell image
To examine the effect of gefitinib on cell death. After the cells were plated at a density of 2 × 105 cells/p35 Petri dish for 24 h, the cells were exposed to 0 or 60 μM gefitinib. And then the cell morphology was observed under an imaging system of living cell observation in inverted microscope (OLYMPUS, IX71, Japan) for 24 h.
Mitochondrial function was evaluated by the cells stained with the mitochondrial sensitive probe DiOC6. Lipophilic cation DiOC6 accumulated in the mitochondrial matrix driven by the electrochemical gradient. The cells were cultured in 60-mm Petri dish at a density of 1 × 106 cells for overnight. After treatment with or without gefitinib, the cells were washed with ice-cold PBS. The cells were trypsinized and collected by centrifugation. Then cell pellets were incubated with 50 nM DiOC6 in complete medium at 37 °C for 30 min (in the dark). Finally, the cell pellets were collected by centrifugation and resuspended in 1 ml ice-cold PBS and analyzed by flow cytometer (FACScan, Becton Dickinson, San Jose, CA).
2.9 Western blot
The cells were plated at a density of 6 × 106 cells per p100 Petri dish in complete medium for overnight. Then the cells were treated with 0-60 μM gefitinib for 24 h. At the end of treatment, the cells were lysed in the icecold cell extract buffer (pH 7.6) containing 0.5 mM DTT, 0.2 mM EDTA, 20 mM HEPES, 2.5 mM MgCl2, 75mM NaCl,
0.1 mM Na3VO4, 50 mM NaF, 0.1% Triton X-100. The protease inhibitors including 1
μg/ml aprotinin, 0.5 μg/ml leupeptin, and 100 μg/ml 4-(2-aminoethyl) benzenesulfonyl fluoride were added to the cell suspension. The protein concentrations were determined by the BCA protein assay kit (Pierce, Rockford, IL). The total cellular protein extracts were prepared. And they were separated on 6-12% sodium dodecyl sulfate-polyacrylamide gels, and electrophoretic transfer of proteins onto polyvinylidene difluoride membranes. The membranes were sequentially hybridized with primary antibody and followed with a horseradish peroxidase-conjugated secondary antibody. The protein bands were visualized on the X-ray film using the enhanced chemiluminescence detection system (PerkinElmer Life and Analytical Sciences, Boston, MA). Western analyses of various phospho-EGFR, total EGFR, caspsae-3, PARP, securin, ATF3 and actin were performed using specific antibodies. To verify equal protein loading and transfer, actin was used as the protein loading control. A gel digitizing software, Un-Scan-It gel (Ver. 5.1, Silk Scientific, Inc.), was used to analyze the intensity of bands on X-ray film by semi-quantification.
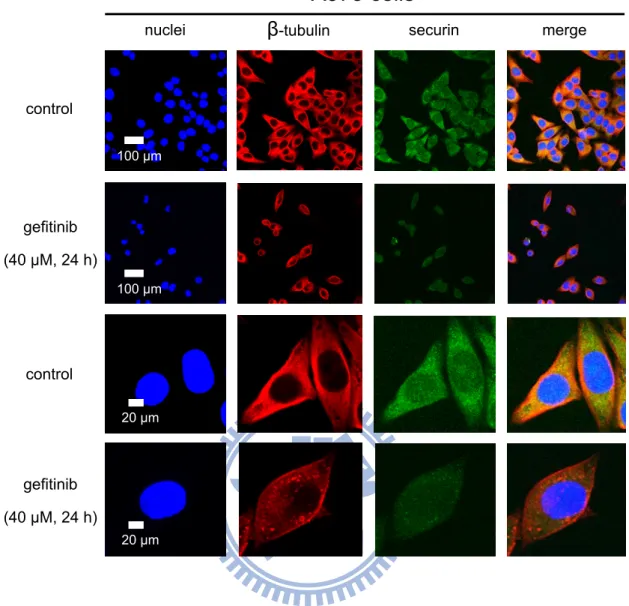
To view the localization and expression of proteins after gefitinib treatment, the cells were subjected to immunofluorescence staining and confocal microscopy. The cells were cultured on coverslips, which were kept in 6-well plates at a density of 2 × 105 per well for overnight before treatment. After treatment with or without 40 μM gefitinib for 24 h, the cells were washed with isotonic PBS (pH 7.4). Then fixation with 4% paraformaldehyde solution for 1 h at 37 °C, the cells were washed three times with PBS. And non-specific binding sites were blocked in PBS containing 10% FBS and 0.25% Triton X-100 for 1 h at 37 °C, and washed three times with 0.25% Triton X-100 in PBS. Thereafter, the cells were incubated with mouse anti-securin (1:120) or rabbit anti-ATF3 (1:200) antibodies in PBS containing 10% FBS and 0.25% Triton X-100 overnight at 4 °C, and washed three times with 0.25% Triton X-100 in PBS. Then the cells were incubated with goat mouse FITC-labeled IgG (1:120) and goat anti-rabbit Cy3-labeled IgG (1:200) in PBS containing 10% FBS and 0.25% Triton X-100 for 2.5 h at 37 °C, and washed three times with 0.25% Triton X-100 in PBS. The β-tubulin, F-actin, and nuclei were stained with the Cy3-labeled anti-β-β-tubulin, BODIPY FL phallacidin and Hoechst 33258, respectively. Finally, the samples were stored in the dark until examined under a confocal microscope (OLYMPUS, Japan) that equipped with an UV laser (405 nm), an Ar laser (488 nm), and a HeNe laser (543 nm).
2.11 Transfection
The pCT-GFP2 and pCT-GFP-sec2 were employed for transfection using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer's recommendations. The cells were plated in 96-well plates at a density of 8 × 103 per well in complete medium for overnight, then were transfected with 50 μg/mL of control or securin-expressed vectors in 50 μL/well serum-free medium for 6 h at 37 °C in a CO2 incubator
according to the manufacturer’s recommendations. Then, the equal amount medium with 20% fetal bovine serum was added without removing the transfection mixture, and incubation proceeded for an additional 24 h. After transfection, the cells were subjected to cytotoxicity analysis as described above.
Each experimentwas repeated at least three times. Data from the population of cells treated with different conditions were analyzed using paired Student’s t-test. In a comparison of multiple groups, data were analyzed by one-way or two-way analysis of variance (ANOVA), and further post Tukey’s tests using the statistic software of GraphPad Prism 5 (GraphPad software, Inc. San Diego, CA). A p value of < 0.05 was considered as statistically significant in each experiment.
3. Results
3.1 Gefitinib elicits the cytotoxicity in both low level EGFR and high
3.1 .
level EGFR cancer cells
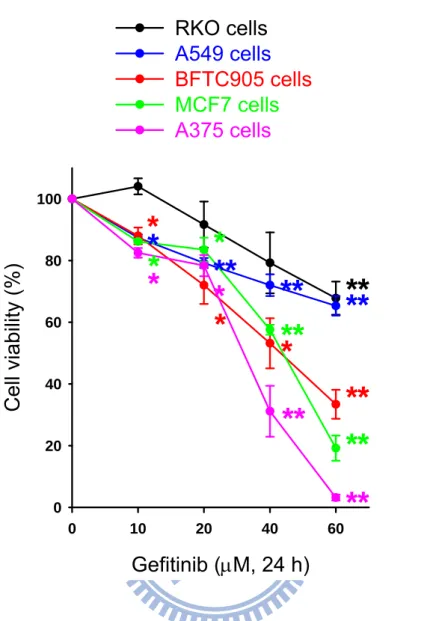
We examined the cytotoxicity following treatment with gefitinib in a variety of human cancer cell lines including RKO (colon cancer), A549 (lung cancer), BFTC905 (bladder cancer), MCF7 (breast cancer) and A375 (skin cancer) cells. Gefitinib significantly induced cancer cell death via a concentration-dependent manner in these cancer cell lines (Fig. 1). The order of cytotoxic sensitivity was A375 > MCF7 > BFTC905 > A549 > RKO cells by exposure to 10-60 μM gefitinib for 24 h. The IC50
value of gefitinib toward cultured human cancer cells lines was 31.45 μM in A375 cells, 40.82 μM in MCF7 cells, 43.49 μM in BFTC905 cells, 83.16 μM in A549 cells, and 91.37 μM in RKO cells.
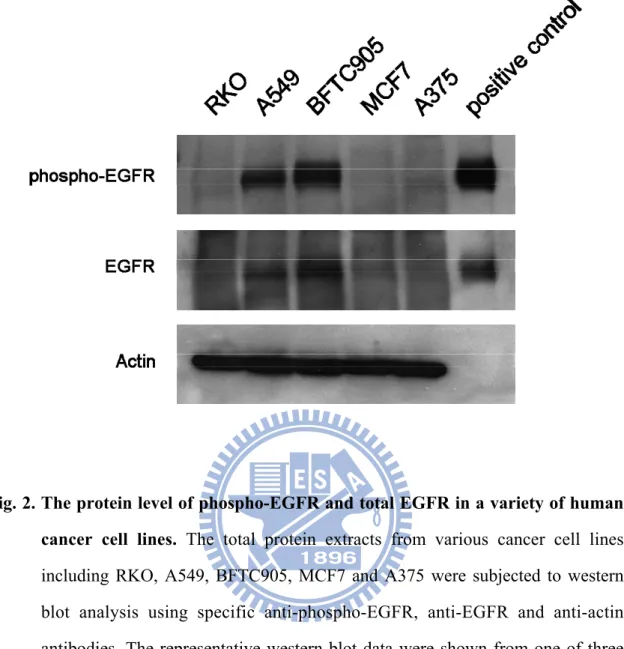
We have further determined the protein expression of phospho-EGFR and total EGFR in these cencer cell lines. The total protein extracts from various cancer cell lines including RKO, A549, BFTC905, MCF7 and A375 were subjected to western blot analysis. The immunoblot analysis indicated that various human cancer cell lines contained different the protein level of phospho-EGFR and total EGFR (Fig. 2). A375, MCF7 and RKO cells did not significantly express the proteins of the phosphorylated-EGFR and total phosphorylated-EGFR; in contrast, BFTC905 and A549 cells expressed high level of EGFR. Actin was used as a loading control protein. Positive control of phospo-EGFR and EGFR were purified from proteins of A431 cell extracts.
3.2 Gefitinib induces apoptosis in both A375 and BFTC905 cancer cells
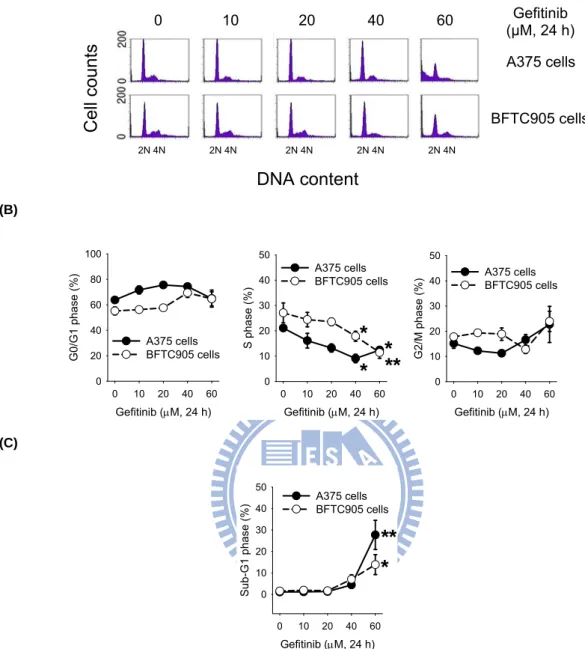
Subsequently, we determined the possible involvement of gefitinib in the regulation of cell cycle progression, the effect of gefitinib on A375 and BFTC905 cells was analyzed by flow cytometry (Fig. 3A). Gefitinib did not significantly alter the fractions of G0/G1 and G2/M phases; however, it markedly decreased the fractions of S
phase in both A375 and BFTC905 cells (Fig. 3B). Meanwhile, gefitinib increased the fraction of sub-G1 phase. Treatment with 60 μM gefitinib for 24 h increased apoptosis by 26.6% and 12.3% in A375 and BFTC905 cells, respectively (Fig. 3C).
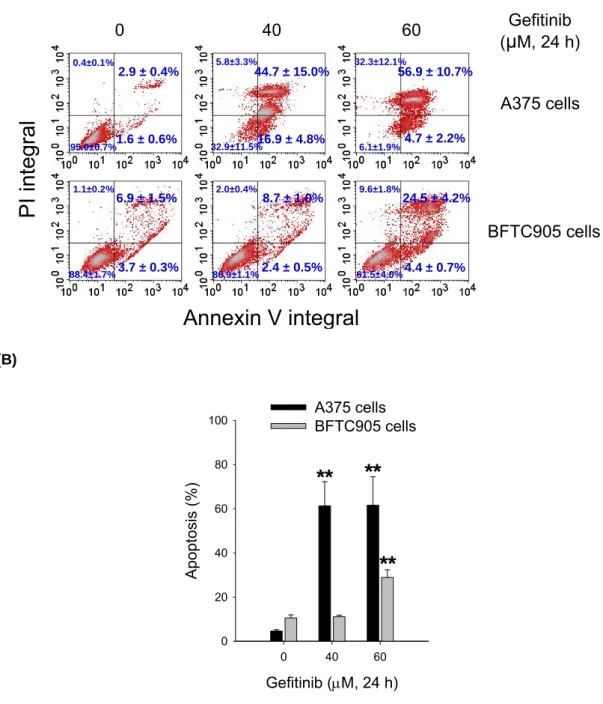
We further assessed apoptosis from the cells that had been exposed to gefitinib by annexin V and PI staining analysis. The control cells were not significantly stained with fluorochromes; however, the annexin V (+)/PI (−) cells (early apoptosis) and annexin V (+)/PI (+) cells (late apoptosis) were increased by treatment with gefitinib 60 μM for 24 h in both A375 and BFTC905 cells (Fig. 4A). The percentages of apoptosis populations (early and late stages) were quantified. Gefitinib at 60 μM induced about 61.6% of apoptosis in A375 cells and 28.9% of apoptosis in BFTC905 cells (Fig. 4B).
3.3 Gefitinib promotes the hyperpolarization of mitochondria and
3.3 .
elevates the activation of caspase-3 and the cleavage of PARP
The time-lapse observation of cell death was found by gefitinib treatment. Treatment with gefitinib significantly induced the cell disruption in A375 cells; in contrast, the control group did not induce the cell death (Fig. 5) .
To examine the induction of apoptosis pathway following gefitinib treatment, the cells were subjected to mitochondrial functional assay. Gefitinib induced the hyperpolarization of mitochondrial membrane potential (Fig. 6A). The quantified data showed that treatment with gefitinib for 24 h significantly increased the levels of DiOC6 intensities in a concentration-dependent manner (Fig. 6B). In A375 cells, gefitinib increased the fluorescence intensity of DiOC6 by about 19.1% and 51.0% at 20 μM and 40 μM, respectively.
The apoptosis-regulated proteins were also analyzed by gefitinib. The active form of caspase-3 (17 kDa) protein was induced following treatment with gefitinib for 24 h; the cleaved form of PARP (89 kDa) protein was also elicited after treatment with gefitinib for 24 h. Actin was used as a loading control protein (Fig. 7).
To investigate the role of securin on the gefitinib-induced cancer cell death, the securin protein expression after gefitinib-treated A375 cells were analyzed by western blot. The securin protein expression was reduced by gefitinib in A375 cells (Fig. 8A). Actin was used as a loading control protein. After semi-quantification, gefitinib significantly reduced the protein level of securin in a concentration-dependent manner (Fig. 8B).
Consistently, we examined the effect of gefitinib on the securin protein expression by immunofluorescence staining and confocal microscopy. The β-tubulin and nuclei were stained with the Cy3-labeled anti-β-tubulin and Hoechst 33258, respectively. The intensities of green fluorescence (FITC) exhibited by securin proteins that were markedly decreased after treatment with 40 μM gefitinib for 24 h (Fig. 9).
3.5 Loss of securin enhances the gefitinib-induced cell death and
3.5 .
overexpression of securin resists the gefitinib-induced cell death
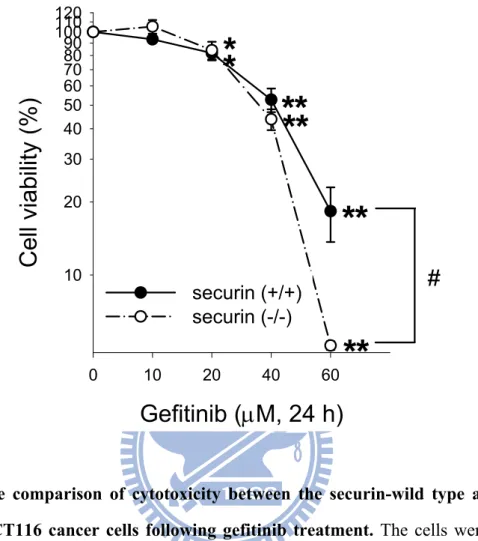
To determine the role of securin on the cell viability after gefitinib treatment, the securin-wild type and -null HCT116 cancer cells were compared. The cell viability was reduced in both the securin-wild type and -null HCT116 cancer cells following gefitinib treatment with. Treatment with 60 μM gefitinib for 24 h, the cell viability was 18.3% and 5.1% in securin-wild type and -null cells, respectively. At 60 μM gefitinib, the securin-null cells exhibited greater cytotoxicity than the securin-wild type cells (Fig. 10). To further determine the role of securin in regulating the gefitinib-induced apoptosis, we transfected a securin-expressed vector (pCT-GFP-sec2). The cell viability was 61.80% and 95.45% in control vector of transfection and securin vector of transfection by treatment with 40 μM gefitinib for 24 h, respectively (Fig. 11). Overexpression of securin by pCT-GFP-sec2 vector also increased the cell viability in cancer cells. Besides, the transfection of pCT-GFP-sec2 vector was more resistant to the gefitinib-induced cell death than control vector.
We had examined the effect of gefitinib on the ATF3 protein expression in cancer cells. Gefitinib increased ATF3 protein expression in A375 cells (Fig. 12A). Actin was used as a loading control protein. The semi-quantified data of western blot showed that gefitinib 40-60 μM for 24 h significantly elevated the protein levels of ATF3 in A375 cells (Fig. 12B).
The expression of ATF3 proteins after treatment with gefitinib were also analyzed by immunofluorescence staining and confocal microscopy. The cytoskeleton and nuclei were stained with BODIPY FL phallacidin and Hoechst 33258, respectively. The red fluorescence (Cy-3) intensity exhibited by ATF3 proteins was increased to translocate to nuclei after treatment with gefitinib 40 μM for 24 h (Fig. 13).
3.7 Gefitinib analogue-1 raises higher cytotoxicity than gefitinib in the
3.7 .
low level EGFR colon cancer cells
We examined the gefitinib and its analogues on the cytotoxicity in a variety of human cancer cell lines. The cell viability was reduced by treatment with 20 to 80 μM of gefitinib analogue-1 and gefitinib in all cancer cell lines including RKO, A549, BFTC905, MCF7 and A375 cells; however, gefitinib analogue-2 did not significantly induce cytotoxicity in these cancer cells (Fig. 14). Gefitinib analogue-1 was higher on the induction of colon cancer cell death than gefitinib. At the concentrations 60 and 80 μM gefitinib analogue-1 for 24 h in RKO cells, the cell viability was 39.1% and 5.9%, respectively; however, the cell viability for gefitinib at the concentrations 60 and 80 μM was 67.8% and 32.9%, respectively.
4. Discussion
EGFR as targets for cancer therapy have been investigated for over 20 years and the field of targeted therapy is evolving rapidly as new insights in receptor biology and signal transduction [18]. Gefitinib (Iressa™, ZD1839), a small molecule tyrosine kinase inhibitor, has been demonstrated the promising antitumor activity [74]. It targets the catalytic domain of EGFR to competewith the ATP binding site [75].The induction of apoptosis has been considered to be the major mechanism for these gefitinib mediated anticancer effects [76]. However, the precise downstream signaling molecules of EGFR-independent have not yet been elucidated. In this study, we investigated the effects of gefitinib on the EGFR-independent cell death signaling pathways in human cancer cells. Here, we found that gefitinib significantly induced cancer cell death in various human cancer cell lines. Interestingly, A375, MCF7 and RKO cells expressed very low protein level of the phosphorylated-EGFR and total EGFR; in contrast, BFTC905 and A549 cells expressed the high level.However, gefitinib more effectively induced cytotoxicity in A375 cells (low level EGFR) compared with BFTC905 cells (high level EGFR). In our data, treatment with 60 μM gefitinib for 24 h in cell cycle progression analysis increased apoptosis by about 26.6% and 12.3% in A375 and BFTC905 cells, respectively. And in annexin V and PI staining assay, gefitinib at 60 μM induced about 61.6% of apoptosis in A375 cells and 28.9% of apoptosis in BFTC905 cells.We suggest that gefitinib induces cancer cell death that may be through the EGFR-independent pathway (Fig. 15A). Although the underlined mechanism concerning the gefitinib-independent EGFR remains unknown, our analyses reveal a plausible hypothesis centered without EGFR, explaining how gefitinib elicits cell apoptosis (Fig. 15B).
Apoptosis plays an important role in development and maintenance of tissue homeostasis [77]. Caspases are central effectors of apoptosis [78]. Cells undergo apoptosis through two major pathways, the extrinsic pathway (death receptor pathway) or the intrinsic pathway (the mitochondrial pathway) [79]. PARP is one of the prime target proteins for caspase-3 [38]. Finally, the contents of dead cells are packaged into apoptotic bodies, which are recognized by neighboring cells or macrophages and cleared by phagocytosis [80]. In present study, we demonstrated that gefitinib induced
apoptosis mediated by the activation of caspase-3 induced and the protein cleavage of PARP.
Subsequently, it is the first time to prove that gefitinib inhibits the securin protein expression in human cancer cells. Treatment with anticancer agents including ultraviolet, doxorubicin, and bleomycin also decrease the securin expression in cancer cells [81; 82]. It had been demonstrated that oxaliplatin inhibits the securin protein expression via a p53-dependent pathway, and p53 and securin may modulate the oxaliplatin-induced cytotoxicity in human colorectal cancer cells [83]. And p38 MAPK may oppositely regulate securin protein expression and γ-H2AX formation in the oxaliplatin-induced apoptosis of human colorectal cancer cells [84]. As well as the blockage of survivin and securin expression increases the cytochalasin B-induced cell death and growth inhibition in human cancer cells [85]. Furthermore, we found that the securin null cells were more sensitive than the wild type cells on the induction of cytotoxicity and apoptosis following treatment with gefitinib. These findings indicate that the loss of securin in cancer cells may increase the cytotoxicity and apoptosis after gefitinib treatment. Vector-basedoverexpression experiments provide compelling evidence for a role of securin ingefitinib-induced tumor cell apoptosis. We provide the securin signal that plays an important role for gefitinib-induced apoptosis in human cancer cells (Fig. 16). Recently, securin has been shown to participate in the cellular DNA repair [52]. Moreover, securin prevents abnormal sister chromatid segregation during mitosis and maintains genomic stability, and its defects can result in chromosomal instability [51]. It also had been demonstrated that depletion of securin increases arsenite-induced chromosome instability and apoptosis via a p53-independent pathway [86]. In our study, gefitinib markedly decreased the fractions of S phase in cancer cells and increased the fraction of sub-G1 phase indicating apoptotic induction. Therefore, we propose that the loss of securin maybe decline cellular DNA repair ability and increases chromosome instability after treatment with gefitinib. Nevertheless, the precise mechanism and role of securin on the regulation of DNA repair in the gefitinib-exposed cells require further investigation.
ATF3 ia a transcription factor and has been shown to dimerise with ATF3 or other ATF/CREB proteins [87; 88; 89]. Depending on the promoter context, these homodimers or heterodimers can act as either repressors or activators of transcription [90]. Therefore, the role of ATF3 as a repressor or activator of transcription cannot be
generalised. ATF3 expression is maintained at low levels in quiescent cells [91]. In tumor, the role of ATF3 in the regulation of the cell cycle and apoptosis has important implications for understanding susceptibility to and progression of several cancers. In our study, we found that gefitinib increased ATF3 protein expression and it seemed to translocate to nuclei. According to literature reviews, the potential role of ATF3 is as an oncogene and a tumour suppressor gene. In tumorigenesis, ATF3 overexpression protected malignant MCF10CA1a human breast cancer cells from apoptosis and promoted their metastatic potential, associated with an upregulation of fibronectin-1, TWIST-1, and Slug transcripts, which are key regulators of cell-cell or cell- extracellular matrix interaction [69]. On the other hand, the potential role of ATF3 as a tumour suppressor is also supported by its defined role in transforming growth factor beta (TGFβ) signaling [92]. TGFβ is a potent tumour suppressor in epithelial cells, which signals via Smad3 activation to directly induce ATF3 [93]. In our data, if induction of ATF3 by treatment with gefitinib and translocate to nuclei that does indeed act as a tumour suppressor, it may be responsible in part for the action of anticancer.
5. Conclusion
In conclusion, we provide for the first time that gefitinib can induce cancer cell apoptosis through an EGFR-independent pathway. Understanding the mechanisms by which securin and ATF3 signal transduction pathways regulate apoptosis following treatmentwith gefitinib may contribute to the novel therapeuticstrategies in cancers.
References
[1] A.J. Levine, The tumor suppressor genes. Annu Rev Biochem 62 (1993) 623-651. [2] T. de La Motte Rouge, M.C. Petrella, J. Michels, C. Even, C. Balleyguier, J. Duclos,
R. Mazeron, P. Morice, P. Pautier, C. Lhomme, New drugs and targeted therapeutic agents in ovarian cancer. Bull Cancer 96 (2009) 1215-1224.
[3] N. Steeghs, J.W. Nortier, H. Gelderblom, Small molecule tyrosine kinase inhibitors in the treatment of solid tumors: an update of recent developments. Ann Surg Oncol 14 (2007) 942-953.
[4] M. Sibilia, R. Kroismayr, B.M. Lichtenberger, A. Natarajan, M. Hecking, M. Holcmann, The epidermal growth factor receptor: from development to tumorigenesis. Differentiation 75 (2007) 770-787.
[5] A. Citri, Y. Yarden, EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol 7 (2006) 505-516.
[6] D.J. Riese, E.D. Kim, K. Elenius, S. Buckley, M. Klagsbrun, G.D. Plowman, D.F. Stern, The epidermal growth factor receptor couples transforming growth factor-alpha, heparin-binding epidermal growth factor-like factor, and amphiregulin to Neu, ErbB-3, and ErbB-4. J Biol Chem 271 (1996) 20047-20052.
[7] M.F. Press, H.J. Lenz, EGFR, HER2 and VEGF pathways: validated targets for cancer treatment. Drugs 67 (2007) 2045-2075.
[8] R.S. Herbst, J.V. Heymach, S.M. Lippman, Lung cancer. N Engl J Med 359 (2008) 1367-1380.
[9] F. Ciardiello, G. Tortora, EGFR antagonists in cancer treatment. N Engl J Med 358 (2008) 1160-1174.
[10] C. Ho, J. Laskin, EGFR-directed therapies to treat non-small-cell lung cancer. Expert Opin Investig Drugs 18 (2009) 1133-1145.
[11] S.A. Aaronson, Growth factors and cancer. Science 254 (1991) 1146-1153.
[12] J.R. Grandis, J.C. Sok, Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol Ther 102 (2004) 37-46.
[13] A.W. Hemming, N.L. Davis, A. Kluftinger, B. Robinson, N.F. Quenville, B. Liseman, J. LeRiche, Prognostic markers of colorectal cancer: an evaluation of DNA content, epidermal growth factor receptor, and Ki-67. J Surg Oncol 51 (1992) 147-152.
[14] P.R. Dutta, A. Maity, Cellular responses to EGFR inhibitors and their relevance to cancer therapy. Cancer Lett 254 (2007) 165-177.
[15] P.M. Harari, Epidermal growth factor receptor inhibition strategies in oncology. Endocr Relat Cancer 11 (2004) 689-708.
[16] J. Baselga, Why the epidermal growth factor receptor? The rationale for cancer therapy. Oncologist 7 Suppl 4 (2002) 2-8.
[17] J. Baselga, S.D. Averbuch, ZD1839 ('Iressa') as an anticancer agent. Drugs 60 Suppl 1 (2000) 33-40; discussion 41-32.
[18] G. Lurje, H.J. Lenz, EGFR signaling and drug discovery. Oncology 77 (2009) 400- 410.
[19] T. Takano, Y. Ohe, M. Kusumoto, U. Tateishi, S. Yamamoto, H. Nokihara, N. Yamamoto, I. Sekine, H. Kunitoh, T. Tamura, T. Kodama, N. Saijo, Risk factors for interstitial lung disease and predictive factors for tumor response in patients with advanced non-small cell lung cancer treated with gefitinib. Lung Cancer 45 (2004) 93-104.
[20] F. Ciardiello, R. Caputo, R. Bianco, V. Damiano, G. Pomatico, S. De Placido, A.R. Bianco, G. Tortora, Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor- selective tyrosine kinase inhibitor. Clin Cancer Res 6 (2000) 2053-2063.
[21] J.G. Paez, P.A. Janne, J.C. Lee, S. Tracy, H. Greulich, S. Gabriel, P. Herman, F.J. Kaye, N. Lindeman, T.J. Boggon, K. Naoki, H. Sasaki, Y. Fujii, M.J. Eck, W.R. Sellers, B.E. Johnson, M. Meyerson, EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304 (2004) 1497- 1500.
[22] M. Ono, A. Hirata, T. Kometani, M. Miyagawa, S. Ueda, H. Kinoshita, T. Fujii, M. Kuwano, Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway
for proliferation. Mol Cancer Ther 3 (2004) 465-472.
[23] F.M. Sirotnak, M.F. Zakowski, V.A. Miller, H.I. Scher, M.G. Kris, Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin Cancer Res 6 (2000) 4885-4892.
[24] I. Okamoto, Epidermal growth factor receptor in relation to tumor development: EGFR-targeted anticancer therapy. FEBS J 277 (2010) 309-315.
[25] E.R. Camp, J. Summy, T.W. Bauer, W. Liu, G.E. Gallick, L.M. Ellis, Molecular mechanisms of resistance to therapies targeting the epidermal growth factor receptor. Clin Cancer Res 11 (2005) 397-405.
[26] M.D. Jacobson, M. Weil, M.C. Raff, Programmed cell death in animal development. Cell 88 (1997) 347-354.
[27] M.C. Raff, Social controls on cell survival and cell death. Nature 356 (1992) 397- 400.
[28] M.C. Raff, B.A. Barres, J.F. Burne, H.S. Coles, Y. Ishizaki, M.D. Jacobson, Programmed cell death and the control of cell survival. Philos Trans R Soc Lond B Biol Sci 345 (1994) 265-268.
[29] J.F. Kerr, A.H. Wyllie, A.R. Currie, Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26 (1972) 239- 257.
[30] D.R. Green, G. Kroemer, The pathophysiology of mitochondrial cell death. Science 305 (2004) 626-629.
[31] S.J. Martin, D.R. Green, Protease activation during apoptosis: death by a thousand cuts? Cell 82 (1995) 349-352.
[32] T. Patel, G.J. Gores, S.H. Kaufmann, The role of proteases during apoptosis. FASEB J 10 (1996) 587-597.
[33] M. Weil, M.D. Jacobson, H.S. Coles, T.J. Davies, R.L. Gardner, K.D. Raff, M.C. Raff, Constitutive expression of the machinery for programmed cell death. J Cell Biol 133 (1996) 1053-1059.
[34] A.M. Chinnaiyan, W.L. Hanna, K. Orth, H. Duan, G.G. Poirier, C.J. Froelich, V.M. Dixit, Cytotoxic T-cell-derived granzyme B activates the apoptotic protease ICE-LAP3. Curr Biol 6 (1996) 897-899.
[35] E.S. Alnemri, Mammalian cell death proteases: a family of highly conserved aspartate specific cysteine proteases. J Cell Biochem 64 (1997) 33-42.
[36] L.M. Martins, T. Kottke, P.W. Mesner, G.S. Basi, S. Sinha, N. Frigon, Jr., E. Tatar, J.S. Tung, K. Bryant, A. Takahashi, P.A. Svingen, B.J. Madden, D.J. McCormick, W.C. Earnshaw, S.H. Kaufmann, Activation of multiple interleukin-1beta converting enzyme homologues in cytosol and nuclei of HL-60 cells during etoposide-induced apoptosis. J Biol Chem 272 (1997) 7421-7430. [37] P.J. Duriez, G.M. Shah, Cleavage of poly(ADP-ribose) polymerase: a sensitive parameter to study cell death. Biochem Cell Biol 75 (1997) 337-349.
[38] D.K. Miller, The role of the Caspase family of cysteine proteases in apoptosis. Semin Immunol 9 (1997) 35-49.
[39] C.B. Thompson, Apoptosis in the pathogenesis and treatment of disease. Science 267 (1995) 1456-1462.
[40] K. Vermeulen, D.R. Van Bockstaele, Z.N. Berneman, Apoptosis: mechanisms and relevance in cancer. Ann Hematol 84 (2005) 627-639.
[41] K. Labedzka, A. Grzanka, M. Izdebska, [Mitochondria and cell death]. Postepy Hig Med Dosw (Online) 60 (2006) 439-446.
[42] S.J. Hong, T.M. Dawson, V.L. Dawson, Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci 25 (2004) 259- 264.
[43] T. Eisenberg, S. Buttner, G. Kroemer, F. Madeo, The mitochondrial pathway in yeast apoptosis. Apoptosis 12 (2007) 1011-1023.
[44] L. Pei, S. Melmed, Isolation and characterization of a pituitary tumor-transforming gene (PTTG). Mol Endocrinol 11 (1997) 433-441.
[45] H. Funabiki, H. Yamano, K. Kumada, K. Nagao, T. Hunt, M. Yanagida, Cut2 proteolysis required for sister-chromatid seperation in fission yeast. Nature 381 (1996) 438-441.
[46] A. Yamamoto, V. Guacci, D. Koshland, Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s). J Cell Biol 133 (1996) 99-110.
[47] H. Zou, T.J. McGarry, T. Bernal, M.W. Kirschner, Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and
tumorigenesis. Science 285 (1999) 418-422.
[48] A. Dominguez, F. Ramos-Morales, F. Romero, R.M. Rios, F. Dreyfus, M. Tortolero, J.A. Pintor-Toro, hpttg, a human homologue of rat pttg, is overexpressed in hematopoietic neoplasms. Evidence for a transcriptional activation function of hPTTG. Oncogene 17 (1998) 2187-2193.
[49] K. Nasmyth, Disseminating the genome: joining, resolving, and separating sister chromatids during mitosis and meiosis. Annu Rev Genet 35 (2001) 673-745. [50] O. Stemmann, H. Zou, S.A. Gerber, S.P. Gygi, M.W. Kirschner, Dual inhibition of sister chromatid separation at metaphase. Cell 107 (2001) 715-726.
[51] P.V. Jallepalli, I.C. Waizenegger, F. Bunz, S. Langer, M.R. Speicher, J.M. Peters, K.W. Kinzler, B. Vogelstein, C. Lengauer, Securin is required for chromosomal stability in human cells. Cell 105 (2001) 445-457.
[52] K. Nagao, Y. Adachi, M. Yanagida, Separase-mediated cleavage of cohesin at interphase is required for DNA repair. Nature 430 (2004) 1044-1048.
[53] X. Zhang, G.A. Horwitz, A.P. Heaney, M. Nakashima, T.R. Prezant, M.D. Bronstein, S. Melmed, Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J Clin Endocrinol Metab 84 (1999) 761-767.
[54] C. Solbach, M. Roller, C. Fellbaum, M. Nicoletti, M. Kaufmann, PTTG mRNA expression in primary breast cancer: a prognostic marker for lymph node invasion and tumor recurrence. Breast 13 (2004) 80-81.
[55] R. Puri, A. Tousson, L. Chen, S.S. Kakar, Molecular cloning of pituitary tumor transforming gene 1 from ovarian tumors and its expression in tumors. Cancer Lett 163 (2001) 131-139.
[56] S.S. Kakar, M.T. Malik, Suppression of lung cancer with siRNA targeting PTTG. Int J Oncol 29 (2006) 387-395.
[57] A.P. Heaney, R. Singson, C.J. McCabe, V. Nelson, M. Nakashima, S. Melmed, Expression of pituitary-tumour transforming gene in colorectal tumours. Lancet 355 (2000) 716-719.
[58] X. Zhang, G.A. Horwitz, T.R. Prezant, A. Valentini, M. Nakashima, M.D. Bronstein, S. Melmed, Structure, expression, and function of human pituitary tumor-transforming gene (PTTG). Mol Endocrinol 13 (1999) 156-166.
tumorigenesis in human embryonic kidney cells. Mol Cancer 4 (2005) 3.
[60] K. Boelaert, C.J. McCabe, L.A. Tannahill, N.J. Gittoes, R.L. Holder, J.C. Watkinson, A.R. Bradwell, M.C. Sheppard, J.A. Franklyn, Pituitary tumor transforming gene and fibroblast growth factor-2 expression: potential prognostic indicators in differentiated thyroid cancer. J Clin Endocrinol Metab 88 (2003) 2341-2347.
[61] G. Liang, C.D. Wolfgang, B.P. Chen, T.H. Chen, T. Hai, ATF3 gene. Genomic organization, promoter, and regulation. J Biol Chem 271 (1996) 1695-1701. [62] T. Hai, M.G. Hartman, The molecular biology and nomenclature of the activating
transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene 273 (2001) 1-11.
[63] D. Lu, J. Chen, T. Hai, The regulation of ATF3 gene expression by mitogen- activated protein kinases. Biochem J 401 (2007) 559-567.
[64] M.R. Montminy, L.M. Bilezikjian, Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature 328 (1987) 175-178.
[65] P.J. Deutsch, J.P. Hoeffler, J.L. Jameson, J.C. Lin, J.F. Habener, Structural determinants for transcriptional activation by cAMP-responsive DNA elements. J Biol Chem 263 (1988) 18466-18472.
[66] B.P. Chen, C.D. Wolfgang, T. Hai, Analysis of ATF3, a transcription factor induced by physiological stresses and modulated by gadd153/Chop10. Mol Cell Biol 16 (1996) 1157-1168.
[67] M.G. Hartman, D. Lu, M.L. Kim, G.J. Kociba, T. Shukri, J. Buteau, X. Wang, W.L. Frankel, D. Guttridge, M. Prentki, S.T. Grey, D. Ron, T. Hai, Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol Cell Biol 24 (2004) 5721-5732.
[68] M.R. Thompson, D. Xu, B.R. Williams, ATF3 transcription factor and its emerging roles in immunity and cancer. J Mol Med 87 (2009) 1053-1060.
[69] X. Yin, J.W. Dewille, T. Hai, A potential dichotomous role of ATF3, an adaptive- response gene, in cancer development. Oncogene 27 (2008) 2118-2127.
[70] A.E. Pelzer, J. Bektic, P. Haag, A.P. Berger, A. Pycha, G. Schafer, H. Rogatsch, W. Horninger, G. Bartsch, H. Klocker, The expression of transcription factor activating transcription factor 3 in the human prostate and its regulation by androgen in prostate cancer. J Urol 175 (2006) 1517-1522.
[71] S. Bandyopadhyay, Y. Wang, R. Zhan, S.K. Pai, M. Watabe, M. Iiizumi, E. Furuta, S. Mohinta, W. Liu, S. Hirota, S. Hosobe, T. Tsukada, K. Miura, Y. Takano, K. Saito, T. Commes, D. Piquemal, T. Hai, K. Watabe, The tumor metastasis suppressor gene Drg-1 down-regulates the expression of activating transcription factor 3 in prostate cancer. Cancer Res 66 (2006) 11983-11990.
[72] X. Huang, X. Li, B. Guo, KLF6 induces apoptosis in prostate cancer cells through up-regulation of ATF3. J Biol Chem 283 (2008) 29795-29801.
[73] F.G. Bottone, Jr., Y. Moon, J.S. Kim, B. Alston-Mills, M. Ishibashi, T.E. Eling, The anti-invasive activity of cyclooxygenase inhibitors is regulated by the transcription factor ATF3 (activating transcription factor 3). Mol Cancer Ther 4 (2005) 693-703.
[74] F. Ciardiello, G. Tortora, A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res 7 (2001) 2958-2970.
[75] S.K. Grant, Therapeutic protein kinase inhibitors. Cell Mol Life Sci 66 (2009) 1163-1177.
[76] A. Arora, E.M. Scholar, Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther 315 (2005) 971-979.
[77] P. Meier, A. Finch, G. Evan, Apoptosis in development. Nature 407 (2000) 796- 801.
[78] Y. Shi, Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell 9 (2002) 459-470.
[79] Q.A. Liu, M.O. Hengartner, The molecular mechanism of programmed cell death in C. elegans. Ann N Y Acad Sci 887 (1999) 92-104.
[80] A. Degterev, M. Boyce, J. Yuan, A decade of caspases. Oncogene 22 (2003) 8543- 8567.
[81] F. Romero, A.M. Gil-Bernabe, C. Saez, M.A. Japon, J.A. Pintor-Toro, M. Tortolero, Securin is a target of the UV response pathway in mammalian cells. Mol Cell Biol 24 (2004) 2720-2733.
[82] Y. Zhou, K.R. Mehta, A.P. Choi, S. Scolavino, X. Zhang, DNA damage-induced inhibition of securin expression is mediated by p53. J Biol Chem 278 (2003) 462-470.
[83] S.J. Chiu, T.S. Hsu, J.I. Chao, Opposing securin and p53 protein expression in the oxaliplatin-induced cytotoxicity of human colorectal cancer cells. Toxicol Lett 167 (2006) 122-130.
[84] S.J. Chiu, J.I. Chao, Y.J. Lee, T.S. Hsu, Regulation of gamma-H2AX and securin contribute to apoptosis by oxaliplatin via a p38 mitogen-activated protein kinase-dependent pathway in human colorectal cancer cells. Toxicol Lett 179 (2008) 63-70.
[85] J.I. Chao, H.F. Liu, The blockage of survivin and securin expression increases the cytochalasin B-induced cell death and growth inhibition in human cancer cells. Mol Pharmacol 69 (2006) 154-164.
[86] J.I. Chao, S.H. Hsu, T.C. Tsou, Depletion of securin increases arsenite-induced chromosome instability and apoptosis via a p53-independent pathway. Toxicol Sci 90 (2006) 73-86.
[87] T.W. Hai, F. Liu, W.J. Coukos, M.R. Green, Transcription factor ATF cDNA clones: an extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Dev 3 (1989) 2083-2090.
[88] T. Hai, T. Curran, Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci U S A 88 (1991) 3720-3724.
[89] J.C. Hsu, R. Bravo, R. Taub, Interactions among LRF-1, JunB, c-Jun, and c-Fos define a regulatory program in the G1 phase of liver regeneration. Mol Cell Biol 12 (1992) 4654-4665.
[90] T. Hai, C.D. Wolfgang, D.K. Marsee, A.E. Allen, U. Sivaprasad, ATF3 and stress responses. Gene Expr 7 (1999) 321-335.
[91] D. Lu, C.D. Wolfgang, T. Hai, Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J Biol Chem 281 (2006) 10473- 10481.
[92] Y. Kang, C.R. Chen, J. Massague, A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in
epithelial cells. Mol Cell 11 (2003) 915-926.
[93] M.T. Ling, X. Wang, X. Zhang, Y.C. Wong, The multiple roles of Id-1 in cancer progression. Differentiation 74 (2006) 481-487.
Gefitinib (μM, 24 h)
0 10 20 40 60Cell viability
(%)
0 20 40 60 80 100RKO cells
A549 cells
BFTC905 cells
MCF7 cells
A375 cells
**
**
**
**
*
**
*
*
*
**
**
*
*
**
**
*
*
Fig. 1. The effect of gefitinib on the cell viability in a variety of human cancer cell lines. The cells were treated with 0-60 μM gefitinib for 24 h. After drug
treatment, the cells were washed with PBS and incubated for 2 days. The cell viability was measured by MTT assay. The results were obtained from four to seven experiments and the bar represents the mean ± S.E.M. *p < 0.05 and **p < 0.01 indicate significant difference between control and gefitinib treated samples.
Fig. 2. The protein level of phospho-EGFR and total EGFR in a variety of human cancer cell lines. The total protein extracts from various cancer cell lines
including RKO, A549, BFTC905, MCF7 and A375 were subjected to western blot analysis using specific anti-phospho-EGFR, anti-EGFR and anti-actin antibodies. The representative western blot data were shown from one of three separate experiments with similar findings. Actin was a loading control. Positive
(A) (B) Gefitinib (μM, 24 h) 0 10 20 40 60 G0 /G 1 p has e (% ) 0 20 40 60 80 100 A375 cells BFTC905 cells Gefitinib (μM, 24 h) 0 10 20 40 60 S pha se (%) 0 10 20 30 40 50 A375 cells BFTC905 cells
*
*
* **
Gefitinib (μM, 24 h) 0 10 20 40 60 G2/M phase (%) 0 10 20 30 40 50 A375 cells BFTC905 cells (C) Gefitinib (μM, 24 h) 0 10 20 40 60 Su b-G1 ph ase (%) 0 10 20 30 40 50 A375 cells BFTC905 cells**
*
Fig. 3. The effect of gefitinib on the cell cycle progression and sub-G1 formation in A375 and BFTC905 cells. (A) The cells were treated with 0-60 μM gefitinib for
24 h. At the end of treatment, the cells were trypsinized and then subjected to flow cytometry analysis. The representative flow data were shown from one of three to four separate experiments with similar findings. (B) The percentage of G0/G1, S and G2/M fractions were quantified by ModFit LT software. (C) The percentage of sub-G1 fractions were quantified by CellQuest software. The results were obtained from three to four experiments and the bar represents the mean ± S.E.M. *p < 0.05 and **p < 0.01 indicate significant difference between
2N 4N 2N 4N 2N 4N 2N 4N 2N 4N 0 10 20 40 60 A375 cells Cell counts DNA content BFTC905 cells Gefitinib (μM, 24 h)
(A) (B) Gefitinib (μM, 24 h) 0 40 60 Apopt osis (%) 0 20 40 60 80
100 A375 cellsBFTC905 cells
**
**
**
Fig. 4. The effect of gefitinib on apoptosis in A375 and BFTC905 cells. (A) After
.treatment with or without gefitinib for 24 h, the cells collected and then
.subjected to annexin V and PI staining. The representative flow data were
.shown from one of three to four separate experiments with similar findings. The
.results represented the mean ± S.E.M. (B) The percentage of apoptosis
.populations (early and late stages) were quantified by CellQuest software. The
.results were obtained from three to four experiments and the bar represents the
.mean ± S.E.M. **p < 0.01 indicates significant difference between control and
Annexin V integral
PI integral
BFTC905 cells Gefitinib (μM, 24 h) A375 cells 0 40 60 2.9 ± 0.4% 44.7 ± 15.0% 56.9 ± 10.7% 6.9 ± 1.5% 8.7 ± 1.0% 24.5 ± 4.2% 4.4 ± 0.7% 2.4 ± 0.5% 3.7 ± 0.3% 4.7 ± 2.2% 16.9 ± 4.8% 1.6 ± 0.6% 32.3±12.1% 0.4±0.1% 5.8±3.3% 1.1±0.2% 2.0±0.4% 9.6±1.8% 95.0±0.7% 32.9±11.5% 6.1±1.9% 88.4±1.7% 86.9±1.1% 61.5±4.0%Fig. 5. The time-lapse observation of apoptosis by gefitinib. A375 cells were plated
at a density of 2 × 105 cells/p35 Petri dish for 24 h, then the cells were treated with or without 60 μM gefitinib for 24 h. The cell morphology was observed under a living cell imaging system. The representative cell morphology data were shown from one of three separate experiments with similar findings. The
arrows indicated the apoptotic cells.
0 h 8 h 16 h 24 h
A375 cells (gefitinib 60 μM)
0 h 8 h 16 h 24 h
A375 cells (control)
10 μm
![HPSH [ 分子間作用力 - 凡得瓦力 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)