Synthesis and Characterization of Dendronized Polyfluorenes
Chia-Hung Chou and Ching-Fong Shu*
Department of Applied Chemistry, National Chiao Tung University, Hsin-Chu, Taiwan 30035, R. O. C.
Received July 1, 2002; Revised Manuscript Received October 9, 2002
ABSTRACT: A series of polyfluorenes containing pendant poly(benzyl ether) dendritic wedges of different generations was synthesized via the macromonomer approach, and then they were characterized by their structures and properties. The dendritic monomers were prepared by coupling benzyl bromide function-alized dendrons with 9,9-bis(4-hydroxyphenyl)-2,7-dibromofluorene and were subsequently copolymerized with a 9,9-dioctylfluorene diboronate derivative by the Suzuki coupling reaction to give the target alternating copolymers. Photophysical studies reveal that the photoluminescent properties of the dendronized polymers are greatly affected by the size of the dendritic side chains. Polymers appended with higher generation dendrons exhibit an enhanced photoluminescence efficiency and an improved luminescent stability. This is attributed to the shielding effect induced by the bulky dendrimer side chains, which prevent self-quenching and suppress the formation of aggregates/excimers.
Introduction
Since the discovery of polymer-based light-emitting diodes (PLEDs) in 1990,1considerable progress has been made in the development of new conjugated polymers and in the performance of related LEDs.2 Organic luminescent polymers are attractive due to the flex-ibility in fine-tuning their luminescent properties through the manipulation of chemical structures, along with the feasibility of utilizing spin-coating and printing pro-cesses for large area display devices. Polymers with large energy band gaps that emit blue light efficiently are of special interest; these materials are desired for full color display applications and also serve as energy-transfer donors in the presence of lower energy fluoro-phores.3 Due to their high photoluminescence and electroluminescence efficiencies, polyfluorenes are among the most promising candidates for blue-emitting poly-mers.4 However, a major problem with polyfluorenes concerns their tendency to form long-wavelength ag-gregates/excimers in the solid state upon heating.5This leads to issues of color instability of the light emitted from LEDs fabricated with polyfluorenes.
Dendritic macromolecules are characterized by a central polyfunctional core, from which monomeric units with branching points arise successively. This results in a nearly entanglement-free, highly branched poly-meric structure with a well-defined three-dimensional architecture.6 Dendronized polymers, consisting of a linear, polymeric core appended with dendrons or den-drimers, form another class of macromolecules which is presently being given considerable attention.7There are two principally different synthetic routes to den-dronized polymers. In the macromonomer route, the desired dendrons are attached to the monomer unit before polymerization.8 In the attach-to route, the polymers which become the core in the final product possess anchor groups to either convergently or diver-gently attach a dense sequence of dendrons.9 The dendronized approach has been applied to polymers with conjugated polymer backbones such as poly(p-phenylene),10poly(p-phenylenevinylene),11 poly(p-phen-yleneethynylene),12 and poly(thiophene).13 Recently, Mu¨ llen et al. reported polyfluorenes appended with
polyphenylene dendrons, anticipating that the incorpo-ration of the three-dimensional dendritic side chain to the light-emitting polymer would improve its lumines-cent activity.14 While this paper was in preparation, Carter et al. reported the synthesis of dendronized polyfluorenes containing “Fre´chet-type” dendrons, with their focal benzyl group directly bonded to the C-9 carbon of the fluorene units.15,16 Both groups demon-strated that the shielding effect provided by the den-dritic side chains on the conjugated polyfluorene back-bone suppresses the formation of aggregates/excimers.14,16 More recently, the Fujiki group reported an unsym-metrically substituted polyfluorene bearing a bulky poly(benzyl ether) dendron and a less bulky 3,6-dioxa-octyl group in the C-9 position and showed that this polymer gives a pure bluish photoluminescence with negligible excimer emission around 520 nm.17
The facile functionalization at the C-9 position of the fluorene unit provides the opportunity to improve both the solubility and processability of the resulting poly-mers, while offering the ability to control interchain interactions,14,16cross-linking,18and the charge-inject-ing or -transportcharge-inject-ing properties of the polymers.19,20 Herein, we report the synthesis and characterization of fluorene-based alternating copolymers containing poly-(benzyl ether) dendritic wedges as side chains, with their benzyl focal point attached through a 4-hydroxy-phenylene spacer to the C-9 carbon in every alternating fluorene unit. The extended 9,9-bis(4-hydroxyphenyl)-fluorenyl core is easily accessible, and will offer the following advantages. First, it avoids the direct attach-ment of benzyl groups to the fluorene unit. The benzyl linkages at the C-9 position of polyfluorenes are poten-tially susceptible to photooxidation, which may cause polymer degradation and device failure in polymer LEDs.2a,14 Second, the extended core reduces steric hindrance and offers easy access to the introduction of high generation dendrons with bulky frameworks into the fluorene unit.21In addition, the coupling of “Fre´chet-type” dendrons with the extended fluorenyl core is readily available via the Williamson ether synthesis to afford the desired macromonomers. Third, the insertion of a rigid phenylene spacer between the dendritic side chain and the polymer backbone may lead to a more 10.1021/ma0210271 CCC: $22.00 © 2002 American Chemical Society
efficient shielding effect on the polyfluorene main chain, while not blocking the reaction sites of the macromono-mer from the palladium-catalyzed polymacromono-merization reac-tion.
Results and Discussion
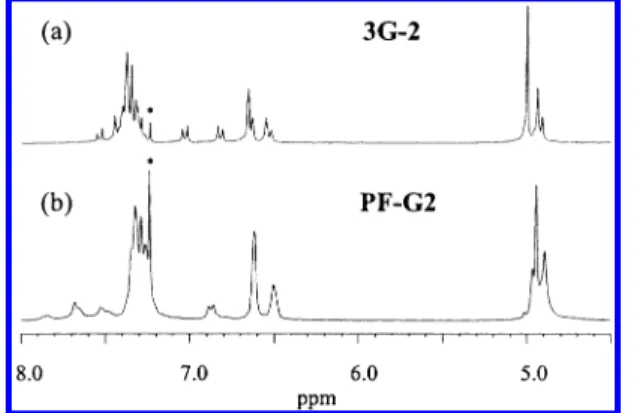
The general synthetic route to the dendritic mono-mers 3-GX (X ) 1-3) is illustrated in Scheme 1. The acid-catalyzed condensation reaction of 2,7-dibromo-9-fluorenone (1)22 with excess phenol gives 9,9-bis(4-hydroxyphenyl)-2,7-dibromofluorene (2). This method resembles that used very recently by Mu¨llen et al. to attach the triarylamine substituents to the C-9 position of fluorene.19 It provides a simple way to introduce hydroxyphenyl functionality to the C-9 position of fluorene. Subsequently, different generations of benzyl bromide-functionalized dendrons GX-Br (X ) 1-3), which were prepared according to well-known literature procedures by Hawker et al.,15 were coupled with compound 2 via the Williamson ether synthesis to afford the dendritic monomers 3-GX (X ) 1-3). The structures were verified by1H and 13C NMR analysis and mass spectrometry. Figure 1a shows the1H NMR spectrum of monomer 3-G2. Resonances of the benzyl protons occur in the region 4.90-5.02 ppm, and separated resonances are observed for each “layer” of benzylic units. Two distinct AB-type doublets are found at δ ) 6.82 and 7.03, respectively, and are attributed to the protons of the phenylene groups attached to the C-9 of the fluorene ring. As the macromonomer moves to the third generation, resonances corresponding to each “layer” of benzyl units overlap. The relative integrations of resonances of benzyl groups and phenylene rings confirm the structures and generation numbers of the dendritic monomers.
The macromonomers 3-GX (X ) 1-3) were then copolymerized, respectively, with the diboronate 4 (Scheme 2),
2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaboro-lan-2-yl)-9,9-dioctylfluorene,23 by the Suzuki coupling reaction.24The copolymerizations were carried out using Pd(PPh3)4 as catalyst in a mixture of toluene and aqueous potassium carbonate (2.0 M), in the presence of aliquate 336 as a phase-transfer reagent. At the end of polymerization, the end groups of the polymer chain were capped by refluxing the reaction mixture for a period of 6 h each with phenylboronic acid and bromo-benzene. The resulting polymer solutions were precipi-tated repeatedly into methanol-acetone and methanol several times to give alternating copolymers PF-GX (X ) 1-3), consisting of 9,9-dioctylfluorene and the corre-sponding dendronized fluorenes. The1H and13C NMR spectra of PF-GX (X ) 1-3) are consistent with the expected structure of the alternating copolymers. Figure 1b shows the1H NMR spectrum of PF-G2. The signals between δ ) 0.61 and 2.05 ppm are assigned to protons of the n-octyl chain (not shown in the figure). Reso-nances of benzyl protons and of aromatic protons occur in the regions δ ) 4.83-5.05 and 6.40-7.90 ppm, respectively. The resonance appearing at δ ) 6.87 corresponds to one of the doublets of the AB quartet of the phenylene rings; the other doublet of the AB quartet is obscured by other aromatic resonances. On the basis of the integration data of aliphatic, benzylic, and aromatic resonances, the structure of the polymer and the generation number of the dendron were confirmed. PF-GX (X ) 1-3) are readily soluble in common organic solvents, such as THF, chloroform, toluene, xylene, cyclohexanone, DMF, etc. The highly soluble nature of these copolymers is attributed to the presence of highly Scheme 1
Figure 1. 1H NMR spectra in the region of 4.5-8.0 ppm for (a) the macromonomer 3-G2 and (b) PF-G2 in CDCl3. / indicates a signal arising from CHCl3.
branched dendrons, along with flexible n-octyl chains in the fluorene units.
The molecular weights of PF-GX (X ) 1-3) were determined by gel permeation chromatography (GPC) with THF as the eluent, calibrated against polystyrene standards. GPC analysis showed that the weight-average molecular weights (Mw) and polydispersities (Mw/Mn) of the polymers are in the range of (3.8-6.5)× 104 and 1.9-2.5, respectively, as shown in Table 1. These values, however, should be taken as only indica-tive, since the backbone rigidity of the rigid, rod-type polyfluorenes studied may deviate strongly from that of the coillike polystyrene standards.25The data reveal that the degree of polymerization decreases as the size of the dendritic monomer increases. The same observa-tion was also reported in the synthesis of the similar dendronized polyfluorenes.16The alternating copolymer PF-G3, which contains the sterically crowded G3-dendron, has the lowest degree of polymerization, Dp∼ 4 (Mn) 15 300), corresponding to eight coupled fluorene units. A comparison of the molecular weights of PF-GX (X ) 1-3) with those of the similar polymers made by Carter et al. shows that our approach leads to dendronized polyfluorenes with higher molar masses.16 The thermal properties of PF-GX (X ) 1-3) were investigated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA), with the results presented in Table 1. The glass transition temperatures (Tg’s) of the dendronized polyfluorenes are in the range of 90-56 °C, declining with increasing size of the attached dendrons. All the polymers exhibit high ther-mal stability, and their 5% weight loss temperatures in nitrogen are up to approximately 350 °C. It was noted that PF-GX (X ) 1-3) exhibit higher Tg’s and similar decomposition temperatures, in comparison with the dendronized polyfluorenes, which have the focal benzyl group directly attached to the C-9 position of the fluorene units.16
The absorption and photoluminescence (PL) spectra of the dendronized, alternating copolymers in dilute solutions are presented in Figure 2. PF-G1 and PF-G2 in THF solutions exhibit an absorption at 388-392 nm, which resembles the absorption of a high-molecular weight poly-2,7-(9,9-dioctylfluorene) (POF) (λmax) 388 nm) and is assigned as a π-π* transition contributed from the polyfluorene backbone. On the other hand, the corresponding absorption of PF-G3 is blue-shifted to 380 nm, which is the same as the reported absorption (λmax ) 380 nm) for the fluorene oligomer with eight repeated units.26 This result is consistent with the degree of polymerization for PF-G3, Dp) 4, estimated from GPC measurements. All dendron-functionalized polymers display an additional absorption at 278 nm, at which the absorbance increases as the dendritic generation grows. This absorption is contributed from dendritic wedges, since the dendritic benzyl alcohol G3-OH in
THF have an absorption with λmax at 278 nm. As the emission maxima of fluorene oligomers remain virtually unchanged beyond six repeated units,26the PL spectra (λexc) 388 nm) of PF-GX (X ) 1-3), which represent the emission from the polyfluorene backbone, are almost identical and display a vibronic fine structure with two absorption bands at 417-419 and 439-443 nm. The value of the absorption and emission maxima for the dendronized polyfluorenes are similar to those for the alkyl- substituted polyfluorene, implying that the in-troduction of bulky dendritic wedges into the C-9 carbon of every alternating fluorene unit neither increased the torsion angle nor perturbed the conjugation of the polyfluorene backbone.
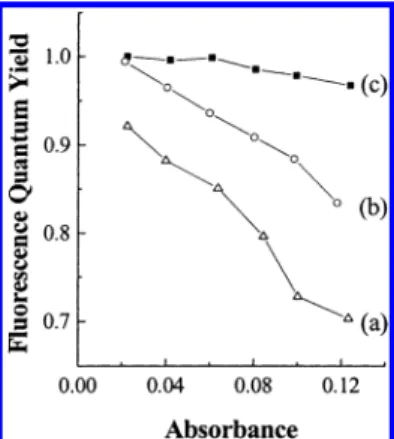
The fluorescence quantum yields (ΦFL) of PF-GX (X ) 1-3) in THF solution (λexc) 365 nm) were estimated by comparison with the standard 9,10-diphenylan-thracene (ca. 5× 10-6M solution in cyclohexane, having a fluorescence quantum yield of 0.90),27with the results listed in Table 2. These PL efficiency values are con-siderably dependent not only on the size of the dendritic side chain but also on the concentration of solutions. The [G3] and [G2]-functionalized copolymers exhibit higher PL efficiencies than their [G1] analogue. Figure 3 illustrates the variation of the PL efficiency with the concentration of solutions. The ΦFL value of PF-G3 stays near 1.0 over a rather wide concentration range, with absorbance at λmax (380 nm) changing from 0.02 to 0.12. While the ΦFLvalue of PF-G2 is also very high under dilute concentration (∼0.99 at abs388nm) 0.02), it dramatically drops by 83% when the solution is concentrated (absorbance at 388 nm increases to 0.12). In the case of the lowest generation PF-G1, the ΦFL
Table 1. Molecular Weights and Thermal Properties of Polymers PF-GX (X ) 1-3)
polymer Mwa Mna Mw/Mn Tgb T5%c
PF-G1 65 000 35 000 1.9 90 351
PF-G2 40 000 18 000 2.2 59 371
PF-G3 38 000 15 000 2.5 56 360
aMolecular weight determined by GPC in THF, based on polystyrene standards.bGlass transition temperature (°C) deter-mined by DSC at a heating rate of 20 °C min-1.cTemperature (°C) at 5% weight loss measured by TGA at a heating rate 20 °C min-1under nitrogen.
Figure 2. UV-vis absorption and PL spectra (excited at 384 nm) in THF of (a) PF-G3, (b) PF-G2, (c) PF-G1, and (d) POF; also including (e) the absorption spectrum of the benzyl alcohol dendron G3-OH in THF.
Table 2. Optical Data and the Fluorescence Quantum Yields of PF-GX (X ) 1-3)
solution λmax(nm)a film λmax(nm)a PL efficiency
polymer abs emiss abs emiss solutionb filmc
PF-G1 278, 392 419 (443) 282, 394 424 (448) 0.92 0.26 PF-G2 278, 388 418 (442) 282, 392 424 (448) 0.99 0.58 PF-G3 278, 380 417 (439) 282, 385 423 (446) 1.0 0.75 aThe data in parentheses are the wavelengths of shoulders and subpeaks.bSolution fluorescence quantum efficiency measured in THF, relative to 9,10-diphenylanthracene (ΦFL ) 0.90).cPL quantum efficiency estimated relative to a sample of poly-2,7-(9,9-dioctylfluorene) (ΦFL) 0.55).
value is 0.92 under dilute concentration and further drops to 0.70 upon concentrating the solution. These results demonstrate that the polymer backbone of polyfluorene containing higher generation dendrons is efficiently shielded and prevented from collisional quench-ing (concentration quench) in the photoexcited state, attributed to the steric effect of the bulky dendrimer side chain.
To further investigate the effects of the dendritic side chains on the packing of polymer chains, the photo-physical properties of PF-GX (X ) 1-3) in the solid state were studied. Figure 4 shows the absorption and PL spectra of PF-GX (X ) 1-3) films spin-coated from toluene solutions onto quartz plates. In comparison to the corresponding dilute solutions, the absorption spec-trum of the PF-G1 film is broadened and has a 2 nm red shift. PF-G2 and PF-G3 films show a 4-5 nm red shift without broadening in absorption bands. The emission spectra of the three polymers, however, exhibit a 5-7 nm red shift and do not show any sign of aggregate or excimer formation. The small red shift of the emission observed in solid state is probably due to the different dielectric constant of the environment. To examine the effects of the size of side chains on the thermal stability of the dendronized copolymers, the polymer films were heated on a hot plate at 150 °C in
nitrogen atmosphere for 20 h. The results are illustrated in Figure 4. The absorption spectra of these polymers are nearly unchanged after annealing. The annealing of the PF-G1 film results in not only a significant reduction in emission intensity, but also the appearance of an additional emission band between 500 and 600 nm. In the case of PF-G2, the reduction in emission intensity and the appearance of the long wavelength tail is much less pronounced. The PL spectrum of the PF-G3 film, however, remains almost intact and no long wavelength emission is detected after thermal treat-ment. For comparison, the same experiments were conducted for the film of POF. Annealing of the POF film led to not only a spectral shift, but also the appearance of an additional emission band between 500 and 600 nm. It is apparent that the thermal stability of PF-G2 and PF-G3 is improved over that of POF,5 which is due to the incorporation of bulky dendrons into the C-9 position of the fluorene units. The large den-dritic framework is likely to encapsulate the conjugated backbone and prevent π-stacking interactions between polymer main chains, suppressing the formation of aggregates/excimers in the solid state. In the case of PF-G1, the [G1]-substituent is insufficiently bulky to completely shield the polymer backbone and, conse-quently, the long wavelength emission is observed due to the excimer formation. The incomplete suppression of long wavelength emission band with the first genera-tion dendrons has also been seen for the similar den-dronized polyfluorenes.16
The shielding effect of the dendritic side chains on the polymer backbone is also reflected in the emission efficiency of the dendronized polymers. The solid-state fluorescence yields (ΦFL) were estimated by comparison of the fluorescence intensity of the respective polymer films with that of a sample POF film (ΦFL ) 0.55, excited at 384 nm),28with the results listed in Table 2. The comparison data indicates the dependence of the polymer’s PL performance in the solid state on the size of the attached dendrons. PF-G1, which was incorpo-rated with the small [G1]-dendron, exhibits the highest excimer formation tendency and possesses the lowest PL quantum yield (ΦFL) 0.26). The PL efficiency of PF-G2 (ΦFL) 0.58) is comparable to that of POF. PF-G3 shows a remarkably high quantum efficiency (ΦFL ) 0.75), attributed to the three-dimensional shielding effect of the bulky dendrimer side chains on the polymer backbone, leading to the minimization of self-quenching and an enhancement in PL efficiency.
Summary
Using the macromonomer approach, we have synthe-sized dendronized polymers consisting of a conjugated polyfluorene backbone appended with different genera-tions of poly(benzyl ether) dendritic wedges.15 The dendritic monomers were prepared by coupling “Fre´-chet-type” benzyl bromide dendrons with 9,9-bis(4-hydroxyphenyl)-2,7-dibromofluorene (2) via the Will-iamson ether synthesis, and then copolymerized with the diboronate (4) by the Suzuki coupling reaction to afford the alternating copolymers. Photophysical studies revealed that the size of the dendrimer side chain has a significant influence on the luminescence activity of the dendronized polymers. The large framework of higher generation dendrons, such as [G3], is likely to sufficiently encapsulate the conjugated backbone as a “shell.” This not only prevents self-quenching, but also Figure 3. Fluorescence quantum yields (ΦFL) of (a) PF-G1,
(b) PF-G2, and (c) PF-G3 in THF solutions upon excitation of the polyfluorene backbone with absorbance at λmax ranging from 0.02 to 0.12.
Figure 4. UV-vis absorption and PL spectra of (a) PF-G1, (b) PF-G2, (c) PF-G3, and (d) POF films before annealing (solid lines) and after annealing at 150 °C for 20 h in a nitrogen atmosphere (dashed lines).
suppresses aggregate/excimer formation, leading to an enhancement in PL efficiency and luminescent stability. These results demonstrate that PF-G3 is a promising candidate for blue-emitting polymer LEDs. The applica-tion of this polymer as an emitting material in elec-troluminescent devices is currently under study.
Acknowledgment. We thank the National Science Council of the Republic of China for financial support. We also thank Professor Yu-Chie Chen and Mr. Ya-Shiuan Lin (National Chiao Tung University) for mea-suring the MALDI-TOF spectra.
Supporting Information Available: Text giving experi-mental details including materials, characterization, synthetic procedures, and polymerizations. This material is available free of charge via the Internet at http://pubs.acs.org. References and Notes
(1) Burroughes, J. H.; Bradley, D. D. C.; Brown, A. R.; Marks, R. N.; Mackay, K.; Friend, R. H.; Burns, P. L.; Holmes, A. B.
Nature (London) 1990, 347, 539.
(2) (a) Kraft, A.; Grimsdale, A. C.; Holmes, A. B. Angew. Chem.,
Int. Ed. 1998, 37, 402. (b) Friend, R. H.; Gymer, R. W.;
Holmes, A. B.; Burroughes, J. H.; Marks, R. N.; Taliani, C.; Bradley, D. D. C.; Dos Santos, D. A.; Bre´das, J. L.; Lo¨gdlund, M.; Salaneck, W. R. Nature (London) 1999, 397, 121. (c) Bernius, M. T.; Inbasekaran, M.; O’Brien, J.; Wu, W. Adv.
Mater. 2000, 12, 1737. (d) Mitschke, U.; Ba¨urele, P. J. Mater. Chem. 2000, 10, 1471.
(3) (a) Kido, J.; Hongawa, K.; Okuyama, K.; Nagai, K. Appl. Phys.
Lett. 1994, 64, 815. (b) Kido, J.; Shionoya, H.; Nagai, K. Appl. Phys. Lett. 1995, 67, 2281. (c) McGehee, M. D.; Bergstedt,
T.; Zhang, C.; Saab, A. P.; O’Regan, M. B.; Bazan, G. C.; Srdanov, V. I.; Heeger, A. J. Adv. Mater. 1999, 11, 1349. (d) Chen, F.-C.; Yang, Y.; Thompson, M. E.; Kido, J. Appl. Phys.
Lett. 2002, 80, 2308.
(4) (a) Pei, Q.; Yang, Y. J. Am. Chem. Soc. 1996, 118, 7416. (b) Leclerc, M. J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 2867. (c) Neher, D. Macromol. Rapid Commun. 2001, 22, 1365. (d) Becker, S.; Ego, C.; Grimsdale, A. C.; List, E. J. W.; Marsitzky, D.; Pogantsch, A.; Setayesh, S.; Leising, G.; Mu¨ llen, K. Synth. Met. 2002, 125, 73.
(5) (a) Lee, J. I.; Klaerner, G.; Miller, R. D. Synth. Met. 1999,
101, 126. (b) Teetsov, J.; Fox, M. A. J. Mater. Chem. 1999, 9,
2117. (c) Weinfurtner, K.-H.; Fujikawa, H.; Tokito, S.; Taga, Y. Appl. Phys. Lett. 2000, 76, 2502. (d) Yu, W.-L.; Pei, J.; Huang, W.; Heeger, A. J. Adv. Mater. 2000, 12, 828. (6) (a) Tomalia, D. A.; Naylor, A. M.; Goddard, W. A. III. Angew.
Chem., Int. Ed. Engl. 1990, 29, 138. (b) Mekelburger, H.-B.;
Jaworek, W.; Vo¨gtle, F. Angew. Chem., Int. Ed. Engl. 1992,
31, 1571. (c) Fre´chet, J. M. J. Science 1994, 263, 1710. (d)
Zeng, F.; Zimmerman, S. C. Chem. Rev. 1997, 97, 1681. (e)
Frey, H.; Lach, C.; Lorenz, K. Adv. Mater. 1998, 10, 279. (f) Smith, D. K.; Diederich, F. Chem.sEur. J. 1998, 4, 1353. (7) (a) Frey, H. Angew. Chem., Int. Ed. 1998, 37, 2193. (b)
Schlu¨ ter, A. D.; Rabe, J. P. Angew. Chem., Int. Ed. 2000, 39, 864.
(8) (a) Kaneko, T.; Horie, T.; Asano, M.; Aoki, T.; Oikawa, E.
Macromolecules 1997, 30, 3118. (b) Jahromi, S.; Coussens,
B. Meijerink, N.; Braam, A. W. M. J. Am. Chem. Soc. 1998,
120, 9753. (c) Jahromi, S.; Litvinov, V.; Coussens, B. Mac-romolecules 2001, 34, 1013.
(9) Yin, R.; Zhu, Y.; Tomalia, D. A. J. Am. Chem. Soc. 1998, 120, 2678. (b) Shu, L.; Scha¨fer, A.; Schlu¨ ter, A. D. Macromolecules 2000, 33, 4321. (c) Grayson, S. M.; Fre´chet, J. M. J.
Macro-molecules 2001, 34, 6542.
(10) (a) Karakaya, B.; Claussen, W.; Gessler, K.; Saenger, W.; Schlu¨ ter, A. D. J. Am. Chem. Soc. 1997, 119, 3296. (b) Stocker, W.; Karakaya, B.; Schu¨rmann, B. L.; Rabe, J. P.; Schlu¨ ter, A. D. J. Am. Chem. Soc. 1998, 120, 7691. (b) Bo, Z.; Zhang, C.; Severin, N.; Rabe, J. P.; Schlu¨ter, A. D.
Macromolecules 2000, 33, 2688.
(11) (a) Bao, Z.; Amundson, K. R.; Lovinger, A. J. Macromolecules 1998, 31, 8647. (b) Jakubiak, R.; Bao, Z.; Rothberg, L. Synth.
Met. 2000, 114, 61.
(12) Sato, T.; Jiang, D.-L.; Aida, T. J. Am. Chem. Soc. 1999, 121, 10658.
(13) Malenfant, P. R. L.; Fre´chet, J. M. Macromolecules 2000, 33, 3634.
(14) Setayesh, S.; Grimsdale, A. C.; Weil, T.; Enkelmann, V.; Mu¨ llen, K.; Meghdadi, F.; List, E. J. W.; Leising, G. J. Am.
Chem. Soc. 2001, 123, 946.
(15) Hawker, C. J.; Fre´chet, J. M. J. J. Am. Chem. Soc. 1990, 112, 7638.
(16) Marsitzky, D.; Vestberg, R.; Blainey, P.; Tang, B. T.; Hawker, C. J.; Carter, K. R. J. Am. Chem. Soc. 2001, 123, 6965. (17) Tang, H.-Z.; Fujiki, M.; Zhang, Z.-B.; Torimitsu, K.; Motonaga,
M. Chem. Commun. 2001, 2426.
(18) Kla¨rner, G.; Lee, J.-I.; Lee, V. Y.; Chan, E.; Chen, J.-P.; Nelson, A.; Markiewicz, D.; Siemens, R.; Scott, J. C.; Miller, R. D. Chem. Mater. 1999, 11, 1800.
(19) Ego, C.; Grimsdale, A. C.; Uckert, F.; Yu, G.; Srdanov, G.; Mu¨ llen, K. Adv. Mater. 2002, 14, 809.
(20) Wu, F.-I.; Reddy, S.; Shu, C.-F.; Liu, M. S.; Jen, A. K.-Y.
Chem. Mater. submitted.
(21) Miller, T. M.; Neenan, T. X.; Zayas, R.; Bair, H. E. J. Am.
Chem. Soc. 1992, 114, 1018.
(22) Ranger, M.; Leclerc, M. Macromolecules 1999, 32, 3306. (23) Ranger, M.; Rondeau, D.; Leclerc, M. Macromolecules 1997,
30, 7686.
(24) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457. (25) Kreyenschmidt, M.; Uckert, F.; Mu¨ llen, K. Macromolecules
1995, 28, 4577.
(26) Klaerner, G.; Miller, R. D. Macromolecules 1998, 31, 2007. (27) Eaton, D. Pure Appl. Chem. 1998, 60, 1107.
(28) Grice, A. W.; Bradley, D. D. C.; Bernius, M. T.; Inbasekaran, M.; Wu, W. W.; Woo, E. P. Appl. Phys. Lett. 1998, 75, 629.