Triggering Apoptotic Death of HumanMalignant Melanoma
A375.S2 Cells by Bufalin: Involvement of Caspase
Cascade-Dependent and Independent Mitochondrial
Signaling
Pathways
Yu-Ping Hsiao,
1, 2, 3Chun-Shu Yu,
4Chien-Chih Yu,
4Jai-Sing Yang,
5Jo-Hua Chiang,
6Chi-Cheng Lu,
6Hui-Ying Huang,
7Nou-Ying Tang,
8Jen-Hung Yang,
9, 10An-Cheng Huang,
11and Jing-Gung Chung
12, 131 Institute of Medicine, Chung Shan Medical University, Taichung 402, Taiwan 2 Department of Dermatology, Chung Shan Medical University Hospital, Taichung 402, Taiwan
3 Department of Dermatology, Jen-Ai Hospital, Taichung 412, Taiwan 4 School of Pharmacy, China Medical University, Taichung 404, Taiwan
5 Department of Pharmacology, China Medical University, Taichung 404, Taiwan 6 Department of Life Sciences, National Chung Hsing University, Taichung 402, Taiwan
7 Department of Nutrition, China Medical University, Taichung 404, Taiwan 8 School of Chinese Medicine, China Medical University, Taichung 404, Taiwan 9 School of Medicine, Tzu Chi University, Hualien 970, Taiwan
10Department of Dermatology, Buddhist Tzu Chi General Hospital, Hualien 970, Taiwan
11Department of Nursing, St. Mary’s Medicine Nursing and Management College, Yilan 266, Taiwan
12Department of Biological Science and Technology, China Medical University, Taichung 404, Taiwan
13Department of Biotechnology, Asia University, Taichung 413, Taiwan
Bufalin was obtained from the skin and parotid venom glands of toad and has been shown to induce cytotoxic effects in various types of cancer cell lines, but there is no report to show that whether bufalin affects human skin cancer cells. The aim of this investigation was to study the effects of bufalin on human malignantmelanoma A375.S2 cells and to elucidate possiblemechanisms involved in induction of apoptosis. A375.S2 cells were treated with different concentrations of bufalin for a specific time period and investigated for effects on apoptotic analyses. Our results indicated that cells after exposure to bufalin significantly decreased cell viability, and induced cell morphological changes and chromatin condensation in a concentration-dependent manner. Flow cytometric assays indicated that bufalin promoted ROS productions, loss of mitochondrial membrane potential (ΔΨm), intracellular Ca2+ release, and nitric oxide (NO) formations in A375.S2 cells. Additionally, the
apoptotic induction of bufalin on A375.S2 cells resulted from mitochondrial
dysfunction-related responses (disruption of the ΔΨm and releases of cytochrome c, AIF, and Endo G), and activations of caspase-3, caspase-8 and caspase-9 expressions. Based on those observations, we suggest that bufalin-triggered apoptosis in A375.S2 cells is correlated with extrinsic- and mitochondria-mediated multiple signal
pathways.
1. Introduction
Skin cancer is one of the major causes of cancer death worldwide [1, 2]. Human cutaneous malignant melanoma is an aggressive skin cancer, and its incidence still continues to rise in individuals of European origin worldwide [3]. About 4%of all dermatologic cancers are melanoma, but it is responsible for more than 80% of deaths from skin cancers, and patients with metastatic melanoma have a 10-year survival rate that is less than 10% [4, 5]. It is recognized that melanoma is highly resistant to conventional chemotherapy, which has preferential metastasis to the brain, lung, liver, and skin [6, 7]. Currently, the effective cure rate of melanoma has not been achieved with surgery, radiation, or chemotherapy.
For inhibiting the development of cancer, chemoprevention and chemotherapy are commonly used, and those agents belong to pharmacological or natural agents [8– 10]. Chemoprevention has been shown to prevent a wide variety of cancers in
multiple animal models [11]. Natural agents include antioxidants and cancer preventative agents or even act as cancer therapy drugs [12, 13].
Bufalin, one of the major digoxin-like components, is a bufadienolide purified from Chan-Su extracts from the venom of Bufo bufo gargarizan [14, 15], and it has long been used as a treatment for heart failure in Chinese medicine in Asian countries [16, 17]. Bufalin acts as a Na+-K+-ATPase inhibitor for increasing the intracellular Ca2+ concentration [14, 18] and as a topoisomerase II inhibitor [19, 20]. Bufalin induced leukemia cell differentiation and apoptotic death in prostate cancer cells [21– 23], and it inhibited hepatocellular carcinoma solid tumor growth in vivo [24]. Recently, the reports have shown that bufalin inhibited cell proliferation of human lung cancer cells [25] and induced apoptosis of hepatomaHep G2 cells [26]. In our laboratory, we also found that bufalin suppressed the migration and invasion of human osteosarcoma U-2 OS cells by suppression of the matrix metalloproteinase-2 (MMP-2), extracellular signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK) signaling pathways [27].However, there is no report regarding bufalininduced apoptotic death in human malignant skin cancer cells in vitro. The present study investigated bufalin-triggered cell death in human malignant melanoma A375.S2 cells
in vitro. Due to our observations, the induction of apoptotic death in A375.S2 cells by
mitochondrial signaling pathways.
2.Materials andMethods
2.1. Chemicals and Reagents. Bufalin, dimethyl sulfoxide (DMSO), propidium iodide
(PI), RNase A, and Triton X-100 were obtained from the Sigma-Aldrich Corp. (St. Louis, MO, USA). Minimum essential media (MEM), penicillinstreptomycin, trypsin-EDTA, fetal bovine serum (FBS) and the L-glutamine were obtained fromGibco/Life Technologies (Carlsbad, CA, USA). 4,6-diamidino-2-phenylindole dihydrochloride (DAPI), 2_,7_-dichlorfluorescein-diacetate (DCFH-DA),
3_,3-dihexyloxacarbocyanine iodide (DiOC6), Fluo-3/AM, and 4-amino-5-methylamino-2_,7_-difluorofluorescein diacetate (DAF-FM) were obtained from theMolecular Probes/Life Technologies (Eugene, OR, USA). Sources of antibodies used in this study were as follows. Polyclonal antibodies specific for caspase-3, caspase-8, and caspase-9 were obtained from the Cell Signaling Technology Inc. (Danvers, MA, USA). Monoclonal antibodies specific for cytochrome c, apoptosis-inducing factors (AIF), Bcl-X, Fas, Fas ligand (FasL), glucose-regulated protein 78 (GRP78), β-actin, and horseradish peroxidase- (HRP-) conjugated secondary antibodies were obtained from the Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). Anti-endonuclease G (Endo G) (Cat. AB3639) and anti-Bax (Cat. 04-434) were bought from Merck Millipore (Billerica,MA,USA).
2.2. Cell Culture. The human malignant melanoma cell line (A375.S2) was purchased
from the Food Industry Research and Development Institute (Hsinchu, Taiwan). About 1 × 106 cells/mL were maintained in 75 cm2 tissue culture flasks with 90% MEM supplemented with 10% FBS, penicillinstreptomycin (100 U/mL penicillin and 100 μg/mL streptomycin), and 2mM L-glutamine and grown at 37◦C in 100%
humidity, 5% CO2, and 95% air [7, 28].
2.3. Assessment of Cell Morphological Changes and Viability. A375.S2 cells at the
density of 2 × 105 cells/well were placed onto 12-well plates and incubated at 37◦C for 24 h before being treated with or without various concentrations (18.75, 37.5, 75, 150, 250, 350, and 450 nM) of bufalin for 24 or 48 h. About 0.5% DMSO (solvent) was used for the vehicle control regimen. At the end of incubation, cells were examined and photographed under contrast phase microscope for morphological changes determination as described elsewhere [29]. Then cells (1 × 105 cells per sample) were centrifuged at 1000×g for 5min, and cell pellets were resuspended with 0.5mL of PBS containing 5 μg/mL PI. The viable cells were determined by a PI-exclusion method and using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA) for determination of viable cells as previously described [27, 30].
2.4. DAPI Staining for Apoptotic Cells. A375.S2 cells at a density of 2 × 105
cells/well were placed onto 6-well plates and treated with bufalin (0, 150, 250, 350, 450, and 550 nM) for 24 and 48 h before cells from each treatment were isolated for DAPI staining as described previously [31, 32]. After staining, the cells were
examined and photographed using a fluorescence microscope at 200x magnification [33, 34].
2.5. Determinations of the Reactive Oxygen Species (ROS) Production, the Levels of Mitochondrial Membrane Potential (ΔΨm), Intracellular Ca2+ Release, and Nitric Oxide (NO) Generation. A375.S2 cells at a density of 2 × 105 cells/well onto 12-well
plates were exposed to 450nM bufalin for 0, 1, 3, 6, 12, or 24 h to determine the changes in the levels of ROS, ΔΨm, intracellular Ca2+ release, and NO formation, respectively. Cells were harvested from each treatment then resuspended in 500 μL of DCFH-DA (10 μM) for ROS (H2O2) determination, DiOC6 (1 μmol/L) for ΔΨm, Fluo- 3/AM (2.5 μg/mL) for intracellular Ca2+ concentrations, and DAF-FM (a nitric oxide indicator) for NO assessment and incubated at 37◦C for 30min and then were analyzed by flow cytometry [29, 34, 35].
2.6. Apoptotic Death-Associated Protein LevelsWere Examined by Western Blotting.
A375.S2 cells at a density of 1 × 106 A375.S2 cells in 6-well plates were treated with 450nM bufalin for 0, 6, 12, 24, and 48 h. Cells were harvested from each treatment by centrifugation for the total protein determination as PRO-PREP protein extraction solution (iNtRON Biotechnology, Seongnam-si, Gyeonggi-do, Korea) forWestern blotting. The protein levels of caspase-3, caspase- 8, caspase-9, cytochrome c, AIF, Endo G, Bcl-X, Bax, Fas, FasL, and GRP78 were examined by using sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) as described previously [34–36]. After electrophoresis, the proteins were transferred to the Immobilon-P transfer membrane (Cat. IPVH00010, Merck Millipore) as described elsewhere [30, 33]. The appropriate horseradish peroxidase- (HRP-) conjugated secondary antibodies was used to visualize by ImmobilonWestern
ChemiluminescentHRP substrate (Millipore) and X-ray film (GE Healthcare, Piscataway, NJ, USA). The density of bands was quantified using ImageJ 1.45 program [30].
2.7. Statistical Analysis. The statistical differences between
∗P < 0.05 was considered significant. The quantitative data are shown as mean ± SD.
The results from the in vitro studies are representative of at least two or three independent experiments.
3. Results
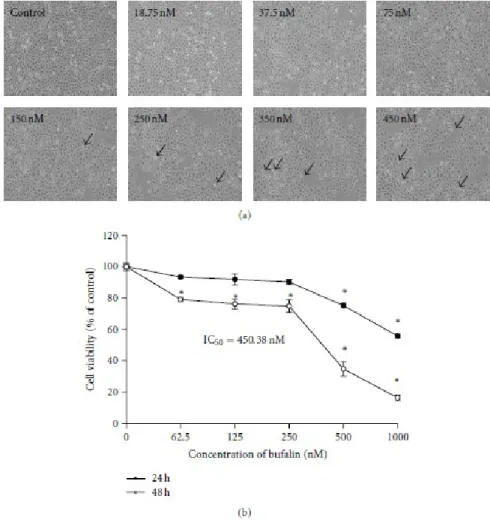
3.1. Bufalin Affects Cell Morphological Changes and Reduces Percentage of Cell Death in A375.S2 Cells. In order to investigate the biological effects of bufalin,
A375.S2 cells were treated with various concentrations of bufalin for 24 and 48 h, and cell morphological changes and cell death were determined. The results are shown in Figures 1(a) and 1(b), which indicated that bufalin induced morphological changes (Figure 1(a)) and caused cell death in a concentrationdependent manner (Figure 1(b)). We found that the half maximal inhibitory concentration (IC50) is 450.38nM in bufalin-treated A375.S2 cells at a 48 h incubation. Based on these observations, we selected the concentration of 450nM bufalin, which is close to IC50, for further assessing whether the growth-inhibitory and cell death effects of bufalin are accompanied by its effect on apoptotic cell death.
viable A375.S2 cells. Cells were plated onto MEM + 10% FBS with various concentrations of bufalin for 24 or 48 h. (a) The morphological changes were examined and photographed under phase contrast microscope, and (b) the total percentages of viable cells were determined utilizing a PI-exclusion method and by flow cytometry as described in Section 2. Each point is mean ± SD of three
experiments, ∗P < 0.05 was significantly different from the untreated control
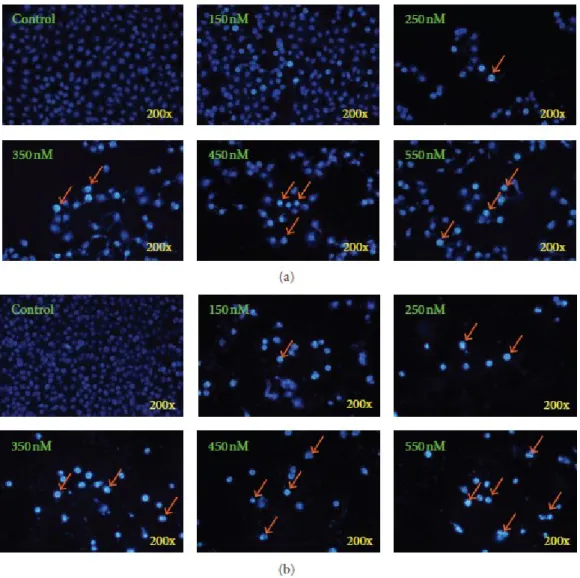
3.2. Bufalin Triggers Apoptotic Cell Death in A375.S2 Cells. In order to confirm if
bufalin-induced cell death in A375.S2 cells was through the induction of apoptosis, we used DAPI staining assay. Results shown in Figures 2(a) and 2(b) revealed that chromatin condensation and apoptotic cells were present in bufalin-treated A375.S2 cells for 24 and 48-h treatments. Moreover, the percentage of apoptotic cells is calculated (Figure 2) compared with intact control cells and this effect was concentration dependent.
Figure 2: Bufalin induces chromatin condensation and apoptosis in A375.S2 cells. Cells were incubated with various concentrations of bufalin for 24 (a) and 48 h (b). Apoptotic cells were determined by DAPI staining and were photographed at a 200x magnification under fluorescence microscopy as described in Section 2. Each
experiment was done with triple sets with similar results.
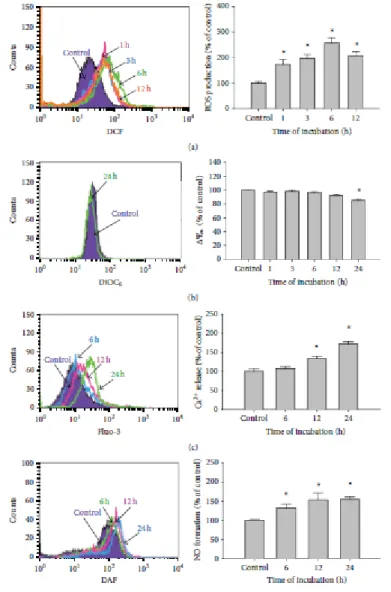
3.3. Bufalin Affects the Levels of ROS, ΔΨm, and Intracellular Ca2+ and NO in A375.S2 Cells. In order to further examine whether bufalin-induced apoptotic death is
due to affecting the levels of ROS, ΔΨm, intracellular Ca2+ andNOin A375.S2
cells. Cells were incubated with 450nM bufalin for various time periods. The levels of ROS production, ΔΨm, and Ca2+ and NO generation were measured by flow
demonstrated that bufalin promoted ROS (Figure 3(a)), intracellular Ca2+ production (Figure 3(c)), and NO formation (Figure 3(d)) in a time-dependent manner. We also found that bufalin decreased the levels of ΔΨm in A375.S2 cells at a 24 h exposure (Figure 3(b)).
Figure 3: Bufalin influences the levels of reactive oxygen species (ROS),
mitochondria membrane potential (ΔΨm), intracellular Ca2+, and nitric oxide (NO) in A375.S2 cells. Cells were incubated with 450nM bufalin for various time periods, before being stained by DCFH-DA for ROS level (a), by Fluo-3/AM for the
intracellular Ca2+ level determined (b), and stained with DiOC6 for the ΔΨm levels (c), or cells were analyzed for NO production after being stained with DAF-FM (d) by flow cytometric analysis as described in Section 2. Each point is performed mean
±S.D. and was done in triplicate; ∗P < 0.05 was significantly different from the
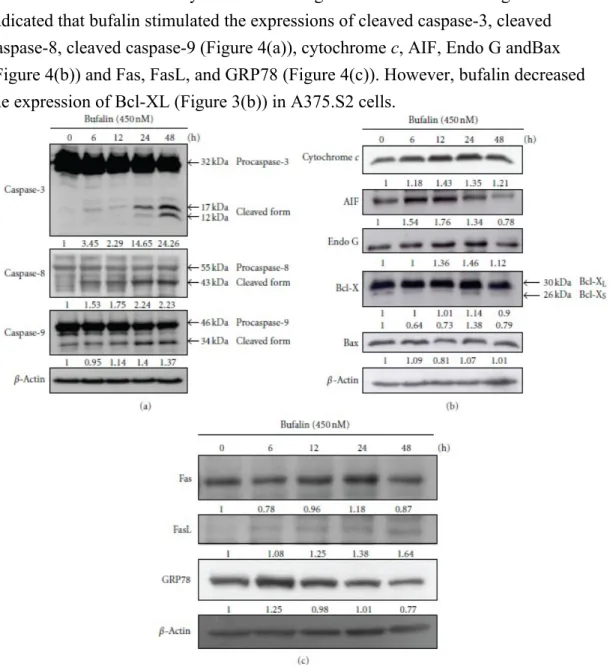
3.4. Bufalin Alters in Apoptotic-Associated Proteins in A375.S2 Cells. To further
examine whether or not bufalin induces apoptosis via alterations of associated protein levels in A375.S2 cells, cells were treated with 450nM bufalin for 0, 6, 12, 24, and 48 h and then were examined by Western blotting. The results shown in Figure 4
indicated that bufalin stimulated the expressions of cleaved caspase-3, cleaved caspase-8, cleaved caspase-9 (Figure 4(a)), cytochrome c, AIF, Endo G andBax (Figure 4(b)) and Fas, FasL, and GRP78 (Figure 4(c)). However, bufalin decreased the expression of Bcl-XL (Figure 3(b)) in A375.S2 cells.
Figure 4: Bufalin alters the apoptosis-related protein levels in
A375.S2 cells. A total of 1 × 106 A375.S2 cells in 6-well plates were treated with 450nM bufalin for 0, 6, 12, 24, and 48 h. Cells were harvested from each sample and associated proteins were
Section 2. The protein levels of caspase-3, caspase-8, and caspase-9 (a), cytochrome c, AIF, Endo
G, Bcl-X, and Bax (b), and Fas, FasL, and GRP78 (c) expressions were examined. β-Actin was used as an internal control to ensure equal loading.
4. Discussion
Several reports have shown that bufalin inhibited cell growth via cell cycle arrest and induction of apoptosis in many types of cancer cell lines [21–26]. However, there is no report to show that bufalin induced cell death in human melanoma A375.S2 cells. In the present study, we found that bufalin significantly induced cell morphological changes and decreased the percentage of viable A375.S2 cells (P < 0.05) at 24 and 48 h (Figures 1(a) and 1(b)). Furthermore, results from DAPI staining showed that bufalin induced apoptosis in A375.S2 cells in a concentration-dependent manner (Figures 2(a) and 2(b)). Although many experiments have showed that bufalin induced apoptosis in human cancer cell lines, the molecular mechanism of apoptotic induction in A375.S2 melanoma cells remains unclear. Thus, the present study further investigated the effect of bufalin on the intrinsic pathway of apoptosis in A375.S2 cells.
Apoptosis (programmed cell death type I) has been shown to perform via the death receptor-dependent pathway (extrinsic pathway) and mitochondria-dependent pathway (intrinsic pathway) [37–39]. In death receptor-dependent pathway, Fas and its receptor Fas ligand (FasL) and caspase- 8 play an important role in regulating the induction of apoptosis in diverse cell types and tissues [40]. In the present study, the results fromWestern blotting (Figure 4(c)) indicated that bufalin promoted the expression of Fas and FasL and active form of caspase-8 (Figure 4(a)); these data demonstrated that bufalin induced apoptosis in A375.S2 cells through the death receptor-dependent pathway (extrinsic pathway). Figure 4(a) also shows that bufalin promoted the active form of caspase-3 in a time-dependent manner, which
also indicated that bufalin induced apoptosis of A375.S2 cells via caspase-dependent pathway. It is well documented that caspase is normally present as an inactive procaspase but it can exist as a cleaved form when triggered [41, 42].
Those observations are in agreement with other reports which showed that bufalin induced apoptosis in human hepatocellular carcinoma cells and human prostate cancer cells via Fas and FasL pathway and caspase pathways [17, 26]. Results from flow cytometric assay showed that bufalin decreased the levels of ΔΨm (Figure 3(b)).
Furthermore, Figure 4(b) indicates that bufalin promoted the levels of cytochrome c, AIF, Endo G, and Bax but inhibited the Bcl-XL expression in A375.S2 cells. Bcl-2 family proteins have been shown to regulate the mitochondria-dependent pathway and death receptor-dependent pathway [43, 44]. Bcl-2 family proteins include the
proapoptotic proteins such as Bax, Bak, Bad, and Bcl-Xs and the antiapoptotic proteins such as Bcl- 2, Bcl-XL, and Mcl-1 [45]. The ratio of Bax/Bcl-2 affects the levels of ΔΨm in cells after exposure to inducer of apoptosis [45], and then
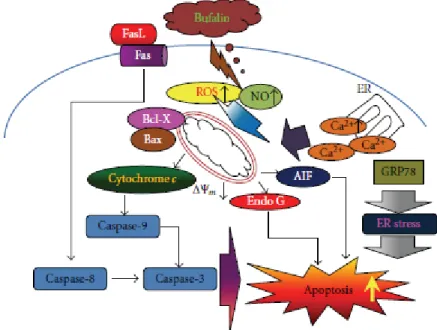
mitochondrial release of cytochrome c can be controlled by the ratio of Bax/Bcl-2 proteins and activated by proteolytic cleavage and heterodimerization [46]. Based on those observations, we suggest that bufalin-induced apoptosis in A375.S2 cells was partly through the mitochondriaas well as caspase-dependent and independent pathways. Overall, bufalin induced apoptosis in A375.S2 cells through cross-talk between the extrinsic and the intrinsic pathways. In conclusion, we proposed the possible signaling pathway of bufalin-induced apoptosis in A375.S2 cells that is shown in Figure 5. The possible flow chart indicated that bufalin triggers apoptosis
via Fas/FasL, caspase cascade (caspase-3, 8 and 9) or the loss of ΔΨm in
mitochondria and then led to AIF and Endo G release that is a novel finding for bufalininduced apoptosis in A375.S2 cells. These results provided a novel and more detailed molecular mechanism in bufalininduced apoptosis in A375.S2 cells in vitro.
Figure 5: A schematic diagram showing the proposed signaling pathways of bufalin-induced apoptosis in A375.S2 human malignant melanoma cells.
Authors’ Contribution
An-Cheng Huang and Jing-Gung Chung contributed equally to this work.
Acknowledgment
The authors would like to thank the support provided by the Grant CMU100-ASIA-4 from the China Medical University, Taichung, Taiwan.
References
[1] J. Girschik, L. Fritschi, T. Threlfall, and T. Slevin, “Deaths from non-melanoma skin cancer inWestern Australia,” Cancer Causes and Control, vol. 19, no. 8, pp. 879–885, 2008.
[2] C. Lo, T. Y. Lai, J. S. Yang et al., “Gallic acid inhibits the migration and invasion of A375.S2 human melanoma cells through the inhibition of matrix
metalloproteinase-2 and Ras,” Melanoma Research, vol. 21, no. 4, pp. 267–273, 2011.
[3] J. F. Thompson, R. A. Scolyer, and R. F. Kefford, “Cutaneous melanoma in the era of molecular profiling,” The Lancet, vol. 374, no. 9687, pp. 362–365, 2009. [4] A. J. Miller and M. C. Mihm, “Jr. melanoma,” The New England Journal of
Medicine, vol. 355, no. 1, pp. 51–65, 2006.
[5] S. Bhatia, S. S. Tykodi, and J. A. Thompson, “Treatment of metastatic melanoma: an overview,” Oncology, vol. 23, no. 6, pp. 488–496, 2009.
[6] B. Gava, S. Zorzet, P. Spessotto, M. Cocchietto, and G. Sava, “Inhibition of B16 melanoma metastases with the ruthenium complex imidazolium
trans-imidazoledimethylsulfoxide-tetrachlororuthenate and down-regulation of tumor cell invasion,” Journal of Pharmacology and Experimental Therapeutics, vol. 317, no. 1, pp. 284–291, 2006.
[7] C. Lo, T. Y. Lai, J. H. Yang et al., “Gallic acid induces apoptosis in A375.S2 human melanoma cells through caspase-dependent and -independent pathways,”
International Journal of Oncology, vol. 37, no. 2, pp. 377–385, 2010.
[8] J. H. Kuo, Y. L. Chu, J. S. Yang et al., “Cantharidin induces apoptosis in human bladder cancer TSGH 8301 cells through mitochondria-dependent signal
pathways,” International Journal of Oncology, vol. 37, no. 5, pp. 1243–1250, 2010.
[9] H. F. Lu, K. C. Lai, S. C. Hsu et al., “Curcumin induces apoptosis through FAS and FADD, in caspase-3-dependent and -independent pathways in the N18 mouse-rat hybrid retina ganglion cells,” Oncology Reports, vol. 22, no. 1, pp. 97–104, 2009.
[10] J. I. Bin-Chuan,W. H. Hsu, J. S. Yang et al., “Gallic acid induces apoptosis via caspase-3 and mitochondrion-dependent pathways in vitro and suppresses lung
xenograft tumor growth in vivo,” Journal of Agricultural and Food Chemistry, vol. 57, no. 16, pp. 7596–7604, 2009.
[11] W. K. Hong and M. B. Sporn, “Recent advances in chemoprevention of cancer,”
Science, vol. 278, no. 5340, pp. 1073–1077, 1997.
[12] M. A. I. AbouEl Hassan, S. R. Braam, and F. A. E. Kruyt, “Paclitaxel and vincristine potentiate adenoviral oncolysis that is associated with cell cycle and apoptosismodulation, whereas they differentially affect the viral life cycle in non-small-cell lung cancer cells,” Cancer Gene Therapy, vol. 13, no. 12, pp. 1105– 1114, 2006.
[13] F. D. T. Lung and J. Y. Tsai, “Grb2 SH2 domain-binding peptide analogs as potential anticancer agents,” Biopolymers, vol. 71, no. 2, pp. 132–140, 2003. [14] A. Y. Bagrov, N. I. Roukoyatkina, O. V. Fedorova, A. G. Pinaev, and M. V.
Ukhanova, “Digitalis-like and vasoconstrictor effects of endogenous digoxin-like factor(s) from the venom of Bufo marinus toad,” European Journal of
Pharmacology, vol. 234, no. 2-3, pp. 165–172, 1993.
[15] Z. Hong, K. Chan, and H. W. Yeung, “Simultaneous determination of
bufadienolides in the traditional Chinese medicine preparation, Liu-Shen-Wan, by liquid chromatography,” Journal of Pharmacy and Pharmacology, vol. 44, no. 12, pp. 1023– 1026, 1992.
[16] P. Datta and A. Dasgupta, “Interactions between drugs and asian medicine: displacement of digitoxin from protein binding site by bufalin, the constituent of Chinese medicines Chan Su and Lu-Shen-Wan,” Therapeutic Drug Monitoring, vol. 22, no. 2, pp. 155–159, 2000.
[17] C. H. Yu, S. F. Kan, H. F. Pu, E. Jea Chien, and P. S. Wang, “Apoptotic signaling in bufalin- and cinobufagin-treated androgen-dependent and
-independent human prostate cancer cells,” Cancer Science, vol. 99, no. 12, pp. 2467–2476, 2008.
[18] L. Krenn and B. Kopp, “Bufadienolides from animal and plant sources,”
Phytochemistry, vol. 48, no. 1, pp. 1–29, 1998.
[19] S. Hashimoto, Y. Jing, N. Kawazoe et al., “Bufalin reduces the level of topoisomerase II in human leukemia cells and affects the cytotoxicity of anticancer drugs,” Leukemia Research, vol. 21, no. 9, pp. 875–883, 1997. [20] N. Pastor, I. Dom´ınguez, S. Mateos, and F. Cort´es, “A comparative study of
genotoxic effects of anti-topoisomerase II drugs ICRF-193 and bufalin in Chinese hamster ovary cells,” Mutation Research, vol. 515, no. 1-2, pp. 171–180, 2002. [21] L. Zhang, K. Nakaya, T. Yoshida, and Y. Kuroiwa, “Induction by bufalin of
differentiation of human leukemia cells HL60, U937, and ML1 toward macrophage/monocyte-like cells and its potent synergistic effect on the
differentiation of human leukemia cells in combination with other inducers,”
Cancer Research, vol. 52, no. 17, pp. 4634–4641, 1992.
[22] M. Watabe, Y. Masuda, S. Nakajo, T. Yoshida, Y. Kuroiwa, and K. Nakaya, “The cooperative interaction of two different signaling pathways in response to bufalin induces apoptosis in human leukemia U937 cells,” Journal of Biological
Chemistry, vol. 271, no. 24, pp. 14067–14073, 1996.
[23] J. Y. Yeh, W. J. Huang, S. F. Kan, and P. S. Wang, “Effects of bufalin and cinobufagin on the proliferation of androgen dependent and independent prostate cancer cells,” Prostate, vol. 54, no. 2, pp. 112–124, 2003.
[24] K. Q. Han, G. Huang, W. Gu, Y. H. Su, X. Q. Huang, and C. Q. Ling, “Anti-tumor activities and apoptosis-regulated mechanisms of bufalin on the orthotopic transplantation tumormodel of human hepatocellular carcinoma in nude mice,”
World Journal of Gastroenterology, vol. 13, no. 24, pp. 3374– 3379, 2007.
[25] Y. Jiang, Y. Zhang, J. Luan et al., “Effects of bufalin on the proliferation of human lung cancer cells and its molecular mechanisms of action,”
Cytotechnology, vol. 62, no. 6, pp. 573– 583, 2010.
[26] F. Qi, Y. Inagaki, B. Gao et al., “Bufalin and cinobufagin induce apoptosis of human hepatocellular carcinoma cells via Fasand mitochondria-mediated pathways,” Cancer Science, vol. 102, no. 5, pp. 951–958, 2011.
[27] F. S. Chueh, Y. Y. Chen, A. C. Huang et al., “Bufalin-inhibited migration and invasion in human osteosarcoma U-2 OS cells is carried out by suppression of the matrix metalloproteinase- 2, ERK, and JNK signaling pathways,” Environmental
Toxicology. In press.
[28] S. H. Huang, L. W. Wu, A. C. Huang et al., “Benzyl isothiocyanate (BITC) induces G2/M phase arrest and apoptosis in human melanoma A375.S2 cells through reactive oxygen species (ROS) and both mitochondria-dependent and death receptor-mediated multiple signaling pathways,” Journal of Agricultural
and Food Chemistry, vol. 60, no. 2, pp. 665–675, 2012.
[29] C. L.Wu, A. C. Huang, J. S. Yang et al., “Benzyl isothiocyanate (BITC) and phenethyl isothiocyanate (PEITC)-mediated generation of reactive oxygen species causes cell cycle arrest and induces apoptosis via activation of caspase-3, mitochondria dysfunction and nitric oxide (NO) in human osteogenic sarcoma U-2 OS cells,” Journal of Orthopaedic Research, vol. U-29, no. 8, pp. 1199–1U-209, 2011.
[30] J. H. Chiang, J. S. Yang, C. Y. Ma et al., “Danthron, an anthraquinone derivative, induces DNA damage and caspase cascades-mediated apoptosis in SNU-1 human gastric cancer cells through mitochondrial permeability transition pores and Bax-triggered pathways,” Chemical Research in Toxicology, vol. 24, no. 1, pp. 20–29,
2011.
[31] K. C. Liu, A. C. Huang, P. P. Wu et al., “Gallic acid suppresses the migration and invasion of PC-3 human prostate cancer cells via inhibition of matrix metalloproteinase-2 and -9 signaling pathways,” Oncology Reports, vol. 26, no. 1, pp. 177– 184, 2011.
[32] C. C. Lin, C. L. Kuo, M. H. Lee et al., “Wogonin triggers apoptosis in human osteosarcoma U-2 OS cells through the endoplasmic reticulum stress,
mitochondrial dysfunction and caspase-3-dependent signaling pathways,”
International Journal of Oncology, vol. 39, no. 1, pp. 217–224, 2011.
[33] C. C. Lu, J. S. Yang, A. C. Huang et al., “Chrysophanol induces necrosis through the production of ROS and alteration of ATP levels in J5 human liver cancer cells,” Molecular Nutrition and Food Research, vol. 54, no. 7, pp. 967–976, 2010.
[34] S. H. Wu, L. W. Hang, J. S. Yang et al., “Curcumin induces apoptosis in human non-small cell lung cancer NCI-H460 cells through ER stress and caspase cascade- and mitochondria- dependent pathways,” Anticancer Research, vol. 30, no. 6, pp. 2125–2133, 2010.
[35] S. S. Lin, H. P. Huang, J. S. Yang et al., “DNA damage and endoplasmic reticulumstressmediated curcumin-induced cell cycle arrest and apoptosis in human lung carcinoma A-549 cells through the activation caspases cascade- and mitochondrial- dependent pathway,” Cancer Letters, vol. 272, no. 1, pp. 77–90, 2008.
[36] S. T. Kao, C. C. Yeh, C. C. Hsieh et al., “The Chinese medicine Bu-Zhong-Yi-Qi-Tang inhibited proliferation of hepatoma cell lines by inducing apoptosis via G0/G1 arrest,” Life Sciences, vol. 69, no. 13, pp. 1485–1496, 2001.
[37] N. A. Thornberry, T. A. Rano, E. P. Peterson et al., “A combinatorial approach defines specificities ofmembers of the caspase family and granzyme B: functional relationships established for key mediators of apoptosis,” Journal of Biological
Chemistry, vol. 272, no. 29, pp. 17907–17911, 1997.
[38] G. Kroemer, L. Galluzzi, and C. Brenner, “Mitochondrial membrane
permeabilization in cell death,” Physiological Reviews, vol. 87, no. 1, pp. 99– 163, 2007.
[39] E. Lai, T. Teodoro, and A. Volchuk, “Endoplasmic reticulum stress: signaling the unfolded protein response,” Physiology, vol. 22, no. 3, pp. 193–201, 2007.
[40] S. Nagata and P. Golstein, “The Fas death factor,” Science, vol. 267, no. 5203, pp. 1449–1456, 1995.
[41] J. G. Chung, J. S. Yang, L. J. Huang et al., “Proteomic approach to studying the cytotoxicity of YC-1 on U937 leukemia cells and antileukemia activity in
orthotopic model of leukemia mice,” Proteomics, vol. 7, no. 18, pp. 3305–3317, 2007.
[42] K. Sanjiv, T. L. Su, S. Suman et al., “The novel DNA alkylating agent BO-1090 suppresses the growth of human oral cavity cancer in xenografted and orthotopic mousemodels,” International Journal of Cancer, vol. 130, no. 6, pp. 1440–1450, 2011.
[43] I. N. Lavrik, A. Golks, and P. H. Krammer, “Caspase: pharmacological
manipulation of cell death,” Journal of Clinical Investigation, vol. 115, no. 10, pp. 2665–2672, 2005.
[44] S. Orrenius, “Reactive oxygen species in mitochondriamediated cell death,”
Drug Metabolism Reviews, vol. 39, no. 2-3, pp. 443–455, 2007.
[45] D. E.Merry and S. J. Korsmeyer, “Bcl-2 gene family in the nervous system,”
Annual Review of Neuroscience, vol. 20, pp. 245– 257, 1997.
[46] S. Cory and J. M. Adams, “The BCL2 family: regulators of the cellular life-or-death switch,” Nature Reviews Cancer, vol. 2, no. 9, pp. 647–656, 2002.