Beckmann rearrangement of ketoximes induced by phenyl dichlorophosphate at
ambient temperature
Chun-Wei Kuo
a,b, Min-Tsang Hsieh
c, Shijay Gao
b, Yi-Ming Shao
a, Ching-Fa Yao
band Kak-Shan Shia
a, a Institute of Biotechnology and Pharmaceutical Research, National Health Research Institutes, Miaoli County 35053, Taiwan, R.O.C.b Department of Chemistry, National Taiwan Normal University, Taipei 116, Taiwan R.O.C.
c Chinese Medicinal Research and Development Center, China Medical University Hospital, 2 Yude Road, Taichung 40447, Taiwan, R.O.C.
A R T I C L E I N F O A B S T R A C T
Article history:
Received
Received in revised form Accepted
Available online
Upon treatment with phenyl dichlorophosphate (PhOP=OCl2) in acetonitrile at ambient
temperature, a variety of ketoximes underwent Beckmann rearrangement in an effective manner to afford the corresponding amides in moderate to high yields.
. Keywords: Beckmann rearrangement ketoximes N-substituted amide Phenyl dichlorophosphate Ambient temperature 1. Introduction
The Beckmann rearrangement is a well-documented reaction for converting ketoximes into N-substituted amides, and serves constantly as a topic of great interest in organic synthesis.1-4
However, many conventional protocols of Beckmann rearrangement were found to be drastic and usually accompanied with environmentally harmful byproducts.5,6 Thus, enormous
efforts have been devoted to modification of the reaction in a more effective and milder fashion. Along this line, a wealth of useful reagents, including cyanuric chloride (TCT),7 TAPC,9
PTSA/ZnCl2,10 BOP-Cl,11 H2NSO3H,12 (EtO)2P=OCl,13 TISC,14
HgCl2,15 Ru(PPh3)3(CO)H2/dppe/TsOH,16 BDMS/ZnCl2,17 TsCl,18
PMSCl,19 I
2,20 and cyclopropenium ion,21 have been developed to
fulfill Beckmann rearrangement. Though these processes appeared effective, elevated temperatures, however, are usually required in order to obtain satisfactory yields. Recently, supercritical water (scH2O)22 was reported to be able to replace
both common organic solvents and conventional acid catalysts to facilitate the rearrangement; however, this approach was found synthetically useless in that high temperature and pressure are essential for the reaction to occur. Herein, we wish to report that a convenient new procedure, making use of phenyl dichlorophosphate (3) as a key activating agent, has been developed to facilitate the titled reaction in our laboratories.
2. Results and discussion
Phosphorous chemicals are known as versatile activating/dehydrating agents in organic synthesis as demonstrated by many historical cases.23-25 To our knowledge,
employing phosphorous reagents, such as phenyl dichlorophosphate 3, ethyl dichlorophosphate 4, diphenyl chlorophosphate 5, diethyl chlorophosphate 6,26 and
N,N-dimethylphosphoramidic dichloride 7, to induce Beckmann rearrangement at ambient temperature appears unprecedented.
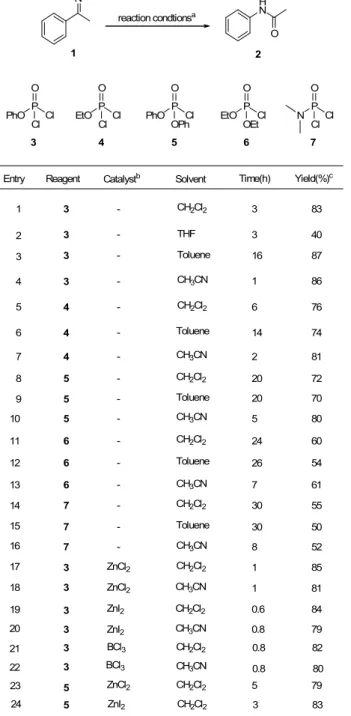
Compiled in Table 1 was the screening study using acetophenone oxime 1 as a model substrate to afford the desired product N-phenylacetamide 2 under various reaction conditions, wherein aforementioned phosphorous agents (e.g., 37), solvents, catalysts, temperature and time were systematically examined. According to experimental results, when oxime 1 was treated with phosphate 3 in low polar solvents, such as CH2Cl2, toluene,
or CH3CN, at room temperature, product 2 was obtained in high
yields (Entries 1, 3, and 4); among them, CH3CN appeared to be
the solvent of choice in terms of reaction rate and isolated yields (Entry 4). Interestingly, it was observed that a highly polar solvent (i.e., THF) seems incompatible with 3 to effect the above 1,2-migration (Entry 2).
Table 1 Beckmann rearrangement of acetophenone oxime promoted by phosphorous reagents under various reaction conditions
NOH H N O 1 2 reaction condtionsa EtO P Cl O Cl PhO P Cl O Cl EtO P Cl O OEt PhO P Cl O OPh N P Cl O Cl 3 4 5 6 7
Entry Reagent Catalystb Solvent Time(h) Yield(%)c
1 3 2 4 7 8 11 14 3 - CH2Cl2 3 83 4 - CH3CN 2 81 3 - Toluene 16 87 3 - THF 3 40 3 - CH3CN 5 - CH2Cl2 6 - CH2Cl2 24 60 7 - CH2Cl2 17 5 19 5 ZnCl2 CH2Cl2 9 5 - Toluene 30 55 30 50 7 - Toluene 15 20 72 20 70 1 86 5 79 3 83 5 4 - CH2Cl2 6 76 6 4 - Toluene 14 74 10 5 - CH3CN 5 80 12 6 - Toluene 26 54 13 6 - CH3CN 7 61 8 52 7 -16 CH3CN 20 3 ZnI2 CH2Cl2 0.6 84 22 3 ZnI2 CH3CN 0.8 79 18 3 ZnCl2 CH2Cl2 1 85 ZnI2 CH2Cl2 21 3 ZnCl2 CH3CN 1 81 23 3 BCl3 CH2Cl2 0.8 82 24 3 BCl3 CH3CN 0.8 80
aAll reactions were performed using oxime 1 (2 mmol) and phosphorous
reagent (3 mmol) in 4 mL of solvent as indicated above at room temperature.
b10 mol% of catalyst was added. cYields are for isolated, chromatographically
pure products.
Reagent 4 was almost as effective as 3 under similar reaction conditions (Entries 5
–
7) for the attempted Beckmann rearrangement, but in general, an extended reaction time was required as compared to 3. Along this line, an array of reactions, individually examined with the structurally closely related phosphorus reagents 5, 6 and 7 in low polar solvent systems, were also investigated. As a result, all these phosphorus reagents were found to be inferior to phosphate 3 in effecting the desired migration (Entries 8–
16), presumably due to less active phosphorous centers in comparison with 3. As well, inspired with the fact that Lewis acids might play a critical role in inducing Beckmann rearrangement in a catalytic fashion.7 Attempts toutilize Lewis acid as the additive to accelerate the reaction rate
were then made. As suggested by above studies, reagent 3 in low polar solvent was considered the most appropriate reaction
Table 2 Conversion of ketoximes into amides under treatment with phenyl dichlorophosphate
a No reaction occurred at room temperature or under refluxing conditions.
to test the concept. Unfortunately, as reflected in Entries 17
–
22 (Table 1),no significant synergistic effects were observed for the reactions in the presence of an additional catalyst. However, it is noteworthy that when the less reactive reagent 5 was used in conjunction with either ZnCl2 or ZnI2 as catalyst (Entries 23 and24), reaction rates are significantly enhanced with improvement in yields by ca. 10% as compared to the corresponding reaction without catalyst (Entry 8). Taken together, the reaction system (PhOP=OCl2/CH3CN/rt) exhibited in Entry 4 was tentatively
considered optimum and adopted as a typical procedure as delineated as follows. To a solution of acetophenone oxime 1 (0.270 g, 2 mmol) in anhydrous acetonitrile (4 mL) was added phenyl dichlorophosphate 3(0.633 g, 3 mmol) dropwise at room temperature. The resulting mixture was stirred at the same temperature for ca. 1 h, and then saturated sodium bicarbonate
R1 R2 NOH R1 H N R2 O 1 2 3 4 12 13
Entry Substrate Time (h) Product Yield (%) NOH 1 86 NOH MeO H N MeO O NOH H N O O O O O 0.5 90 0.5 88 C5H11 C5H11 NOH C5H11 H N C5H11 O 7 77 9 N H O 72 C7H15 NOH 3.5 H N 83 O N OH NH O NOH H N O MeO MeO NOH NH O NOH HN O Br Br NOH O2N H N O2N O NOH Cl Cl H N O S N OH S H N O N NOH 5 9 67 6 13 75 7 13 76 8 13 73 9 1.5 84 10 12 33 11 4.5 79 14 6 PhOP(O)Cl2 (1.5 eq) CH3CN, rt S.M. C7H15 N OH H N O NRa
solution (5 mL) was added to quench the reaction. The aqueous layer was separated and extracted with ethyl acetate (3 x 10 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated to give the crude product,
which was purified by flash chromatography on silical gel (30% EtOAc in n-hexane) to afford amide 2 (0.232 g, 86%) as a white solid (mp 114-115 0C).In fact, the gram-scale synthesis of 2 has
also been explored and was found to be equally operational simple and efficient.
To examine the generality of the newly developed protocol, structurally diverse ketoximes were further selected. As listed in Table 2, substrates bearing electron-donating groups at para position on the phenyl ring (Entries 1–3) were found to undergo 1,2-migration much faster than those with electron-withdrawing functionalities (Entries 6
–
8), and gave rise to higher yields of products. Ketoximes possessing a constrained six-membered ring, such as Entries 5 and 10, required a prolonged reaction time to complete the conversion with relatively lower yields (3367%), but it appeared not to be problematic with the case containing a large-ring system (Entry 11), suggesting that 1,2-migration process might be susceptibleto the ring strain imposed. More interestingly, when the migratory groups are heterocyclic rings, a significant difference in reactivity was observed. Thiophene-containing ketoxime (Entry 9) afforded the desired product within 1.5 h with a high yield of 84%; however, when the corresponding pyridine-containing substrate (Entry 14) was employed, the starting material was recovered intact even if the reaction time was prolonged under refluxing conditions for an additional 6 h. The latter was assumed to be retarded by the formation of hydrochloride salt via protonation of the pyridine nitrogen, wherein resulting poor electron density at the C-2 center in the pyridine ring dramatically stopped the migration process. Aforementioned results also indicate that the migratory aptitude of the alkyl linker appears to be poorer than that of the phenyl ring bearing either an donating or electron-withdrawing substituent (Entries 1–4 and Entries 6–8). Moreover, as different alkyl linkers are present in the ketoxime, the longer one seems to have a greater migratory aptitude (Entry 12).Scheme 1 Proposed mechanism of phenyl
dichlorophosphate-induced Beckmann rearrangement of oximes
The proposed mechanism of phenyl dichlorophosphate-induced Beckmann rearrangement is depicted in Scheme 1. Taking acetophenone oxime 1 as a typical example, it is widely accepted that product 2 was formed following pathway a; however, a shunt pathway b might not be ruled out in that as monitored by the TLC, product 2 appeared to grow at the expense of a potential intermediate 8 during the progress of reaction,27 the structure of which was unambiguously identified
by an X-ray analysis.28 The similar TLC behavior, however, was
not observed when the same reaction was carried out under refluxing conditions. An explanation for this could be that an extremely rapid conversion of 8 into product 2 (ca. 5 min, 91% yield) might take place at the elevated temperature; thus, existence of the intermediate was no longer detected. Dimer 8 thus obtained could also demonstrate the existence of chloroimine and its delocalizing nitrilium ion as proposed in the long-standing acceptable pathway a.
3. Conclusion
In summary, we have developed a mild and effective reaction system to induce Beckmann rearrangement of ketoximes at ambient temperature, leading to the corresponding amides in moderate to high yields. It is believed that the title system will be a valuable addition to synthetic organic chemistry in terms of its operational simplicity, up-scaled practicability and easy access to phosphorous reagents.
4. Experimental section 4.1. General
Unless otherwise stated, all materials used were commercially available and used as supplied. Reactions requiring anhydrous conditions were performed in flame-dried glassware, and cooled under an argon or nitrogen atmosphere. Unless otherwise stated, reactions were carried out under argon or nitrogen and monitored by analytical thin layer chromatography performed on glass-backed plates (5 × 10 cm) precoated with silica silica gel 60 F254
as supplied by Merck. Visualization of the resulting chromatograms was done by looking under an ultraviolet lamp (λ = 254 nm) followed by dipping in an ethanol solution of vanillin (5% w/v) containing sulfuric acid (3% v/v) or phosphomolybdic acid (2.5% w/v), and charring by heat gun. Flash chromatography was used routinely for purification and separation of product mixtures using silica gel 60 of 230−400 mesh size as supplied by Merck. Eluent systems are given in volume/volume concentrations. 1H-NMR and 13C-NMR spectra were recorded at
400 and 100 MHz, respectively, on a Bruker Avance 400 FT-NMR instrument. Chloroform-d or dimethyl sulfoxide-d6 was used as the solvent and TMS (δ 0.00 ppm) as an internal standard. Chemical shift values are reported in ppm relative to the TMS in delta (δ) units. Coupling constants (J) are expressed in Hz. High resolution electron impact mass spectra (HRMS (EI)) were recorded using a FINNIGAN MAT-95XL mass spectrometer. Spectral data were recorded as m/z values. Combustion elemental analyses using Heraruc CHN-O Rapid Elemental Analyzer were performed by the microanalytical laboratory at National Chung Hsing University, Taiwan.
4.2. General procedure for the synthesis of compounds in Table 2
To a solution of acetophenone oxime (1) (0.270 g, 2 mmol) in anhydrous acetonitrile (4 mL) was added phenyl dichlorophosphate (3) (0.633 g, 3 mmol) dropwise at room temperature. The resulting mixture was stirred at the same temperature for ca. 1 h, and then saturated sodium bicarbonate solution (5 mL) was added to quench the reaction. The aqueous layer was separated and extracted with ethyl acetate (3 x 10 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated to give the crude product,
NO P O OPh Cl Cl N Cl HN O N Cl H N Cl N H2O 8 N Cl a b N Cl HCl 1 2
which was purified by flash chromatography on silical gel (30% EtOAc in n-hexane) to afford amide 2 (0.232 g, 86%) as a white solid.
4.2.1 N-phenylacetamide (2) as a white solid: mp 114-115
oC; 1H NMR (400 MHz, CDCl
3) δ 8.27 (brs, 1H, NH), 7.50 (d, J
= 7.9 Hz, 2H), 7.27 (d, J = 7.6 Hz, 2H), 7.07 (t, J = 7.4 Hz, 1H), 2.12(s, 3H); 13C NMR (100 MHz, CDCl
3) δ 169.2, 138.2, 129.0,
124.4, 120.3, 24.5; Anal. Calcd. for C8H9NO: C 71.29, H 6.71, N
10.36; Found: C 71.09, H 6.71, N 10.26.
4.2.2 N-(4-methylphenyl)acetamide(Table 2, Entry 1) as a white solid: mp 149-150 oC; 1H NMR (400 MHz, CDCl
3) δ 7.74
(brs, 1H, NH), 7.37 (d, J = 7.8 Hz, 2H), 7.08 (d, J = 7.8 Hz, 2H), 2.29 (s, 3H), 2.12 (s, 3H); 13C NMR (100 MHz, CDCl
3) δ 168.8,
135.6, 134.0, 129.6, 120.4, 24.6, 21.0. Anal. Calcd. for C9H11NO:
C 72.46, H 7.43, N 9.39; Found: C 72.73, H 7.47, N, 9.08.
4.2.3 N-(4-methoxyphenyl)acetamide (Table 2, Entry 2) as a
brown solid: mp 127-129 oC; 1H NMR (400 MHz, CDCl 3) δ 7.67
(brs, 1H, NH), 7.38 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.8 Hz, 2H), 3.77 (s, 3H), 2.12 (s, 3H); 13C NMR (100 MHz, CDCl
3) δ 168.7,
156.6, 131.3, 122.2, 114.3, 55.6, 24.4; Anal. Calcd. for C9H11NO2: C 65.44, H 6.71, N 8.48; Found: C 65.31, H 6.86, N
8.13.
4.2.4 N-(2,3-dihydro-1,4-benzodioxin-6-yl)acetamide
(Table 2, Entry 3) as a colorless oil: 1H NMR (400 MHz, CDCl 3)
δ 7.08 (brs, 1H, NH), 7.09 (s, 1H), 6.85-6.76 (m, 2H), 4.20 (m, 4H), 2.11 (s, 3H); 13C NMR (125 MHz, CDCl
3) δ 168.2, 143.4,
140.5, 131.5, 117.1, 113.7, 109.9, 64.4, 64.2, 24.4; HRMS (FAB) calcd for C10H12NO3 (M+H)+: 194.0817; found: 194.0829.
4.2.5 N-(2-naphthyl)acetamide (Table 2, Entry 4) as a
colorless oil: 1H NMR (400 MHz, CDCl
3) δ 8.16 (s, 1H), 7.94
(brs, 1H, NH), 7.75-7.70 (m, 3H), 7.46-7.24 (m, 3H), 2.19 (s, 3H); 13C NMR (100 MHz, CDCl
3) δ 169.1, 135.6, 134.0, 130.8,
128.9, 127.7, 126.6, 125.1, 120.2, 117.0, 24.8; Anal. Calcd. for C12H11NO: C 77.81, H 5.99, N 7.56; Found: C 77.80, H 5.89, N
7.68.
4.2.6 1,2,3,4-teteahydro-7-methoxy-5H-benzazepin-2-one
(Table 2, Entry 5) as a colorless oil: 1H NMR (400 MHz, CDCl 3)
δ 7.63 (m, 1H), 7.12 (brs, 1H, NH), 6.80 (m, 1H), 6.68 (s, 3H), 3.80 (s, 3H), 3.11-1.95 (m, 6H); 13C NMR (125 MHz, CDCl
3) δ
174.1, 161.6, 140.6, 130.7, 127.4, 114.2, 111.8, 55.2, 39.7, 30.7, 30.4; HRMS (FAB) calcd for C11H14NO2 (M+H)+: 192.1025;
found: 192.1021.
4.2.7 N-(4-chlorophenyl)acetamide (Table 2, Entry 6) as a
colorless oil: 1H NMR (400 MHz, DMSO-d
6) δ 10.1 (brs, 1H,
NH), 7.60 (d, J = 7.8 Hz, 2H), 7.32 (d, J = 7.8 Hz, 2H), 2.04 (s, 3H); 13C NMR (100 MHz, DMSO-d
6) δ 168.4, 138.3, 128.5,
126.5, 120.5, 24.0; Anal. Calcd. for C8H8ClNO: C 56.65, H 4.75,
N 8.26; Found: C 56.70, H 4.92, N 8.12.
4.2.8 N-(4-nitrophenyl)acetamide (Table 2, Entry 7) as a
colorless oil: 1H NMR (400 MHz, DMSO-d6) δ 10.5 (brs, 1H, NH), 8.20 (d, J = 9.2 Hz, 2H), 7.81 (d, J = 9.2 Hz, 2H), 2.11 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.3, 145.4, 142.0, 125.0, 118.5, 24.2; Anal. Calcd for C8H8N2O2: C 53.33, H 4.48, N 15.55; Found: C 53.08, H 4.55, N 15.24.
4.2.9 N-(2-bromophenyl)acetamide (Table 2, Entry 8) as a
yellow solid: mp 98-99 oC; 1H NMR (400 MHz, DMSO-d
6) δ
9.45 (brs, 1H, NH), 7.65-7.10 (m, 4H), 2.07 (s, 3H); 13C NMR
(100 MHz, DMSO-d6) δ 168.5, 136.5, 132.6, 127.9, 127.2, 126.8,
117.8, 23.3; Anal. Calcd for C8H8BrNO: C 44.89, H 3.77, N 6.54;
Found: C 44.96, H 3.75, N 6.33.
4.2.10 N-(2-thienyl)acetamide (Table 2, Entry 9) as a
colorless oil: 1H NMR (400 MHz, DMSO-d
6) δ 11.1 (brs, 1H,
NH), 6.90-6.61 (m, 3H), 2.05 (s, 3H); 13C NMR (100 MHz,
DMSO-d6) δ 166.2, 139.9, 123.5, 116.6, 110.3, 22.5; Anal. Calcd
for C6H7NOS: C, 51.04; H, 5.00; N, 9.92; S, 22.71; Found: C,
50.92; H, 5.29; N, 9.66; S, 22.50.
4.2.11 -Caprolactam (Table 2, Entry 10) as a colorless oil:
1H NMR (400 MHz, CDCl
3) δ 7.11 (brs, 1H, NH), 3.14-3.11 (m,
2H), 2.39-2.36 (m, 2H), 1.68-1.57 (m, 6H); 13C NMR (100 MHz,
CDCl3) δ 179.5, 42.8, 36.8, 30.7, 29.8, 23.3; Anal. Calcd for
C6H11NO: C 63.68, H 9.80, N 12.38; Found: C 63.60, H 9.72, N
12.43.
4.2.12 Azacyclotridecan-2-one (Table 2, Entry 11) as a
colorless oil: 1H NMR (400 MHz, DMSO-d
6) δ 7.86 (brs, 1H,
NH), 3.07-3.03 (m, 2H), 2.06-2.03 (m, 2H), 1.55-1.24 (m, 18H);
13C NMR (100 MHz, DMSO-d
6) δ 172.7, 37.8, 35.4, 27.7, 26.3,
25.8, 25.7, 25.5, 24.8, 24.5, 24.2, 23.5; HRMS (FAB) calcd for C12H24NO (M+H)+: 198.1858; found: 198.1858.
4.2.13 N-heptyl-acetamide (Table 2, Entry 12) as a colorless
oil: 1H NMR (500 MHz, CDCl
3) δ 6.20 (brs, 1H, NH), 3.20 (s,
3H), 1.35-1.21 (m, 12H), 0.91-0.79 (m, 3H); 13C NMR (125
MHz, CDCl3) δ 170.7, 39.9, 31.7, 29.7, 29.4, 28.9, 26.8, 22.5,
14.0; HRMS (EI) calcd for C9H20NO (M+H)+: 158.1545; found:
158.1547.
4.2.14 N-pentyl-hexanamide (Table 2, Entry 13) as a
colorless oil: 1H NMR (400 MHz, DMSO-d
6) δ 7.76 (brs, 1H,
NH), 3.03-2.98 (m, 2H), 2.04-2.01 (m, 2H), 1.50-1.20 (m, 12H), 0.86-0.82 (m, 6H); 13C NMR (100 MHz, DMSO-d
6) δ 170.0,
38.4, 35.4, 31.0, 28.9, 28.7, 25.1, 22.1, 22.0, 13.7; HRMS (FAB) calcd for C11H24NO (M+H)+: 186.1858; found: 186.1857.
4.2.15 N-(1-Chloro-vinyl)-N,N'-diphenyl-acetamidine (8) as a white solid: mp 90-92 oC; 1H NMR (400 MHz, CDCl 3) δ 7.44-7.39 (m, 4H), 7.30-7.26 (m, 3H), 7.04-7.00 (m, 1H), 6.81-6.79 (m, 2H), 5.45 (d, J = 1.2 Hz, 1H), 5.38 (d, J = 1.2 Hz, 1H), 1.93 (s, 3H) ; 13C NMR (100 MHz, CDCl 3) δ 155.4, 150.3, 142.3, 140.0, 129.3, 128.8, 126.9, 126.8, 122.6, 130.0, 114.6, 16.5; HRMS (FAB): calcd for C16H16N2Cl (M+H)+: 271.1002; found:
271.1003.
Acknowledgement
We are grateful to the National Health Research Institutes and National Science Council of the Republic of China (NSC-100-2113-M-400-002) for financial support.
References
1. Xiao, L.; Xia, C.; Chen, J. Tetrahedron Lett. 2007, 48, 7218.
2. Li, D.; Shi, F.; Guo, S.; Deng, Y. Tetrahedron Lett.
2005, 46, 671.
3. Chandrasekhar, S.; Gopalaiah, K. Tetrahedron Lett.
2003, 44, 755.
4. Chandrasekhar, S.; Gopalaiah, K. Tetrahedron Lett.
5. Gawly, R. E. Org. React. 1988, 35, 1.
6. Smith, M. B.; March, J. in Advanced Organic
Chemistry, John Wiley & Sons, New York, 5th edn.,
2001, pp. 1415.
7. Furuya, Y.; Ishihara, K.; Yamamoto, H. J. Am. Chem.
Soc. 2005, 127, 11240. TCT along with DMF serving as
a solvent and reagent has been reported to undergo Beckmann rearrangement at room temperature.8
8. Luca, L. D.; Giacomelli, G.; Porcheddu, A. J. Org.
Chem. 2002, 67, 6272.
9. Hashimoto, M.; Obora, Y.; Sakaguchi, S.; Ishii, Y. J.
Org. Chem. 2008, 73, 2894.
10. Xiao, L. F.; Xia, C. G.; Chen, J. Tetrahedron Lett.
2007, 48, 7218.
11. Zhu, M.; Cha, C.; Deng, W. P.; Shi, X. X. Tetrahedron
Lett. 2006, 47, 4861.
12. Wang, B.; Gu, Y.; Luo, C.; Yang, T.; Yang, L.; Suo, J.
Tetrahedron Lett. 2004, 45, 3369.
13. Sardarian, A. R.; Shahsavari-Fard, Z.; Shahsavari, H. R.; Ebrahimi, Z. Tetrahedron Lett. 2007, 48, 2639. 14. Gui, J.; Deng, Y.; Hu, Z.; Sun, Z. Tetrahedron Lett.
2004, 45, 2681.
15. Ramalingan, C.; Park, Y. T. J. Org. Chem. 2007, 72, 4536.
16. Owston, N. A.; Parker, A. J; Williams, J. M. J. Org.
Lett. 2007, 9, 3599.
17. Yadav, L. D. S.; Patel, R.; Srivastava, V. P. Synthesis,
2010, 1771.
18. Pi, H. J.; Dong, J. D.; An, N.; Du, W.; Deng, W. P.
Tetrahedron, 2009, 65, 7790.
19. Karimi, B.; Behzadnia, H. Synlett, 2010, 2019. 20. Ganguly, N. C.; Mondal, P. Synthesis, 2010, 3705. 21. Srivastava,V. P.; Patel, R.; Yadav, L. D. S. Chem.
Commun., 2012, 5808.
22. (a) Ikushima, Y.; Hatakeda, K.; Sato, O.; Yokoyama, T.; Arai, M. J. Am. Chem. Soc., 2000, 122, 1908; (b) Boero, M.; Ikeshoji, T.; Liew, C. C.; Terakura, K.; Parrinello, M. J. Am. Chem. Soc., 2004, 126, 6280. 23. (a) Liu, H. J.; Chan, W. H.; Lee, S. P. Tetrahedron Lett.
1978, 19, 4461. (b) Liu, H. J.; Sabesan, S. I. Can. J.
Chem. 1980, 58, 2645. (c) Liu, H. J.; Lamoureux, G. V.;
Brunet, M. L. Can. J. Chem. 1986, 64, 520. (d) Liu, H. J.; Nyangulu, J. M. Tetrahedron Lett. 1989, 30, 5097. (e) Liu, H. J.; Nyangulu, J. M. Tetrahedron Lett. 1988,
29, 3267. (f) Liu, H. J.; Nyangulu, J. M. Synth. Commun. 1989, 19, 3407. (g) Liu, H. J.; Yu, S. Y. Synth. Commun. 1986, 16, 1357. (h) Liu, H. J.;
Wiszniewski, V. Tetrahedron Lett. 1988, 29, 5471. 24. Kuo, C. W.; Zhu, J. L.; Wu, J. D.; Chu, C. M.; Yao, C.
F.; Shia, K. S. Chem. Commun. 2007, 301.
25. Zhu, J. L.; Lee, F. Y.; Wu, J. D.; Kuo, C. W.; Shia, K. S. Synlett, 2007, 1317.
26. For an isolated case of Beckmann rearrangement of ketoximes with diethyl chlorophosphate 6, see reference 13.
27. As indicated by TLC, a potential intermediate 8 was detected under the titled system (PhOP=OCl2/CH3CN/rt); however, compound 8 is so
unstable that it is extremely difficult to be isolated.
28. CCDC 690116 (8) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via
www.ccdc.cam.ac.uk/conts/retrieving.html or deposite@ccdc.cam.ac.uk.