J . CHEM. SOC., CHEM. COMMUN.,
1991
1019
Reactions of the Mixed-metal Clusters Prepared from Tungsten Acetylide Complexes;
X-Ray Structural Analyses of Two Novel Butterfly Clusters with
60
Valence Electrons
Yun Chi,*a Hsiu-Fu H s u , ~
Shie-Ming Pengt
band Gene-Hsiang Leeb
a Department of Chemistry, National Tsing Hua University, Hsinchu 30043, Taiwan b Department of Chemistry, National Taiwan University, Taipei 10764, Taiwan
Reaction of WL(C0)3C-CPh and O S ~ ( C O ) , ~ ( C ~ R ~ ) produced WOs3L(CO)&-CPh)(CCRCR), L = Cp,
R
= To1 (p-MeC6H4) l a ; L = C5Me5, R = Me, I b ; treatment of complexes 1 with ditolylacetylene yielded planar clustersWOs3L(CO)8(p3-CPh)[C(Tol)C(Tol)CCRCR], L = Cp, R = Tol, 2a; L = C5Me5,
R
= Me, 2b; complex 2b underwentreversible loss of CO to produce an unsaturated cluster W O S ~ C ~ M ~ ~ ( C O ) ~ ( ~ ~ - C P ~ ) [ C M ~ C M ~ C C ( T O I ) C ( T O I ) ] 3b; thermolysis of 3b in refluxing xylene induced the rearrangement of the cluster core, giving another unsaturated
complex 4b with identical molecular formula; complexes l a , 3b and 4b have been examined by X-ray diffraction studies.
We have recently devised syntheses of several polynuclear
complexes, such
as
Osg(
CO) lo(NCMe)2, Ru3(CO)
12,
cluster complexes from reactions
of group 6 mononuclear
R u ~ ( C O ) ~ ~ (
p3-NPh) and R u ~ ( C O ) ~ O (
p-H)(p-COMe), and
metal acetylide and hydride complexes with group
8cluster
examined the crystal structures and reactivities
of the new
heterometallic clusters prepared.1 We report here the pre-
paration
ofOs3W clusters W O S ~ L ( C O > ~ ( ~ ~ - C P ~ ) ( C C R C R )
,
L
=Cp, R
=Tol(p-MeC6H4),
la; L =CsMes, R
=Me, l b
via
1020
J . CHEM. SOC., CHEM. COMMUN.,r991
4b R = Me,
L
= C5Me5 1 a R = T o l , L = C p 2 a R = T o l , L = C p 3b R = Me,L
= C5Me5b
R
= Me,L
= C5Me5 b R = Me, L = C5Me5Scheme 1 i, excess C2T012; ii, -CO, 140"C, 20 min; iii, +CO, 11O"C, 5 min; iv, 140°C 65 min
cleavage of the acetylide C-C triple-bond and some results of
the subsequent reactivity studies. The reaction of l b with
ditolylacetylene coupled the alkyne to the coordinated C3
hydrocarbon, giving one saturated cluster compound with a
multi-site bound C5 ligand. On heating this compound loses a
CO ligand, yielding two novel, 6O-electron, coordinatively
unsaturated cluster compounds, sequentially? Our study
reported in this paper highlights the role and action of the
accessory ligands in responding to the creation of coordination
unsaturation.
Treatment of tungsten acetylide WCp(C0)3CzCPh3 with
the triosmium alkyne complex O S ~ ( C O ) ~ O ( C ~ T O ~ ~ ) ~
in reflux-
ing toluene (11O"C, 30 min) yielded a novel heterometallic
cluster (la, 20%), while the analogous derivative (lb, 90/)
was prepared from the reaction between WC5Me5( C0)3-
G C P h and Osj( CO) C2Me2)5 under similar conditions
(Scheme 1). Both complexes l a and b were initially charac-
terized by mass, IR and NMR spectroscopy;$ in addition,
complex l a was examined by X-ray diffraction in an attempt to
establish the exact molecular structure.
9
The ORTEP dia-
gram and some bond parameters are presented in Fig.
l .Consistent with its molecular structure, we deduce that the
formation of these Os3W clusters involves two unique
processes. One is the scission of the acetylide carbon-carbon
triple-bond and the other is the coupling oi the acetylide
a-carbon with the coordinated alkyne ligand, producing the
observed alkylidyne and C3 hydrocarbon ligands
, respec-
tively.
Reactions with disubstituted alkyne have also been exam-
ined. The reaction of l a with excess ditolylacetylene in toluene
$ Spectral data for la: MS (FAB, 1920s, lg4W) mlz 1384(M+); IR
(C6H12) v(CO)/cm-1 2 0 7 7 ~ ~ 2 0 4 8 ~ s ~ 2036m, 2018s, 1997vw, 1974m, 1969m and 1909w; 1H NMR (400 MHz, CDC13, 294 K) 6 7.24-6.75 (m, 13H), 5.49 (s, 5H), 2.33 (s, 3H) and 2.19 (s, 3H). For lb: MS 2040vs, 2032m, 2012s, 1991vw, 1970m, 1952s and 1912br, w; 1H NMR (FAB, 184W, 1 9 2 0 ~ ) , mlz 1302(M+). IR(C6H12) v(CO)/cm-l 2072~,
(400 MHz, CD2C12, 294 K) S 7.09 (t, 2H, JH-H 6.8 Hz), 6.70 (t, l H , J H - H 7.2 Hz), 6.91 (d, l H , J H - H 7.4 Hz), 6.84 (d, l H , J H - H 7.8 Hz),
CDZC12,294 K), 6 187.8,185.6,181.3,180.2,177.2,176.2 (3C), 172.7 3.21 (s, 3H), 2.07 (s, 3H) and 1.80 (s, 15H); 13C NMR (100 MHz, and 239.5 (Jw-c 111 Hz, p3-CPh). Satisfactory elemental analyses
were obtained for both l a and b.
Q Crystal data for la: C38H24090~3W1, M=1379.05, monoclinic, space group n 1 / n , a = 19.440(7), b = 9.655(2), c = 19.976(6)
A,
p
=105.85(3)", V = 3607(2) A3,Z = 4,
D,
= 2.540 gcm-3, F(000) = 2503, Nonius CAD-4 diffractometer with graphite-monochromated Mo-Ka radiation, h = 0.70930A,
p(Mo-Ka) = 13.85 mm-l. The min. and max. transmission factors are 0.416 and 0.997,6342 unique reflections were measured, and 4603 reflections with I>
2.0 a(Z) were used in refinement. Refinement of 75 atoms and 461 parameters converged to Rf = 0.037 and R, = 0.032, goodness of fit (GOF) = 1.89. Atomic coordinates, bond lengths and angles, and thermal parameters have been deposited at the Cambridge Crystallographic Data Centre for l a ,3b and 4b. See Notice to Authors, Issue No. 1.
(110 "C, 50 h) led to the isolation of a dark green complex (2a,
53%)6
and unreacted starting material l a (35%). On the other
hand, reaction of the analogous compound l b with ditolyl-
acetylene in refluxing xylene solvent (140 "C, 30 min) pro-
duced three cluster compounds 2b (yellowish green), 3b
(red-brown) and 4b (brown) in 14, 41 and
22% yields,
respectively, in addition to about
8% starting material l b
(Scheme 1). These cluster compounds were separated by TLC
and purified by recrystallization, although we have observed
that the silica gel tends to accelerate the decomposition of 3b
and 4b. The structure of 2b
is closely related to 2a as indicated
by its spectral data, whereas the FAB mass and 13C
NMR data
suggest that both 3b and 4b possess one carbonyl ligand less
than that of complex 2b.q
Red-brown, air-stable, plate-shaped crystals of 3b were
obtained from a solution of CH2C12-heptane at room tem-
perature and an X-ray diffraction study was carried out.
**
An
ORTEP diagram is shown in Fig. 2, which also provides
selected bond distances. The molecule contains a planar
triangulated rhomboidal arrangement with W and
Os(2)
atoms at the bridgehead position, the dihedral angle between
the W-Os(2)-Os( 1) and W-Os(2)-Os(3)
planes being
171.8(1)". There is an alkylidyne ligand (p3-CPh) which is
associated with the face defined by atoms Os(2), Os(3) and W
and, on the opposite side of the alkylidyne ligand, is a C5
hydrocarbon ligand which is coordinated to all four metal
atoms. The central carbon atom C( 12) is linked to three metal
fi
Spectral data for 2b: MS (FAB, 1*4W, I92Os), mlz 1480(M+).IR(C6H12) v(CO)/cm-l 2066s, 2 0 2 9 ~ s ~ 2008s, 1992m, 1964m, 1959s, 1950m and 1943w; l H NMR (400 MHz, CD2C12, 250 K) 6 7.74 (d, 1H,
JH-H 8.2 Hz), 7.20-7.09 (m, 5H), 6.99 (t, l H , JH-H 7 Hz), 6.86-6.77
(m,6H),3.40(s,3H),2.32(s,3H),2.19(s,3H),1.80(s,15H)and1.37
(s, 3H); '3C NMR (100 MHz, CD2C12, 296 K): 6 189.8, 187.7, 182.3 (3C, br), 180.9,178.6,173.9 and 247.2 (Jw-c 318 Hz, y3-CPh). For3b:
MS (FAB, 184W, 1920~), mlz 1452(M+). IR(C6H12) v(CO)/cm-l
2064s, 2 0 0 4 ~ 1959s, 1940s and 1878m; l H NMR (400 MHz, CD2C12, 294 K) 6 7.22 (d, 2H, 1H-H 8 Hz), 7.12 (m, 2H), 7.03 (d, 2H, JH-H 8 Hz), 7.01-6.95 (m, 5H), 6.67 (d, 2H, JH-H 8 Hz), 3.15 (s, 3H), 2.32 (s, 3H),2.29(s,3H), 1.87(s, 15H)and 1.23(s73H);13CNMR(100MHz, CD2C12,250 K) 6 209.2,189.3,189.2,186.1,181.3,178.4,171.9 (CO) and 257.0 ( J w - ~ 113 Hz, y3-CPh). For 4b: MS (FAB, 184W, 192Os),
mlz 1452(M+). IR(C6HI2) v(CO)/cm-1 2066s, 2 0 2 9 ~ s ~ 2008s, 1992m, 1964m, 1959s, 1950m and 1943w; lH NMR (400 MHz, CD2C12, 294 K) JH-H 8.0 Hz), 7.02-6.91 (m, 7H), 3.27 (s, 3H), 2.28 (s, 3H), 2.27
( s , 3H), 2.09 (s, 3H) and 1.61 (s, 15H); satisfactory elemental analyses were obtained for compounds 2b, 3b and 4b.
6 7.37 (d, 2H, JH-H 7.7 Hz), 7.27 (t, 2H, J H - H 7.7 Hz), 7.14 (d, 2H,
**
Crystal data for 3b: C45H40070s3WI, M = 1447.26, monoclinic,space group E l l n , a = 14.532(2), b = 18.485(2), c = 15.654(2)
A, 6
=91.52(1)", V = 4203(1)
A3,
Z = 4,D,
= 2.287 g ~ r n - ~ , F(000) = 2671, p(Mo-Ka) = 11.88 mm-l. The min. and max. transmission factors are 0.396 and 0.999, 5482 unique reflections were measured, and 3523 reflections with I>
2.0 o(l) were used in refinement. Refinement of 96 atoms and 501 parameters converged to Rf = 0.040 and R , = 0.036, G O F = 1.81.J. CHEM. S O C . , CHEM. COMMUN.,
1991
1021
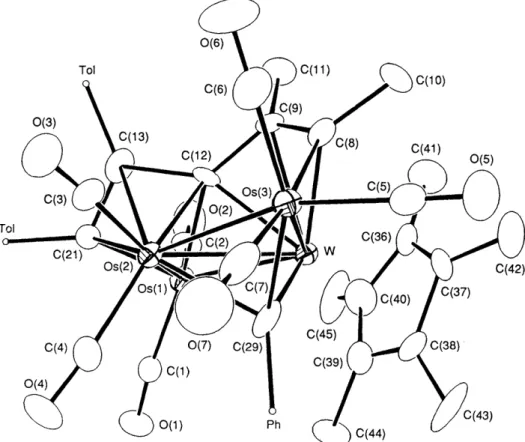
Fig. 1 The molecular drawing of l a . Bond lengths
(A):
Os(l)-Os(2) 2.769(1), Os(1)-W 2.830(1), Os(2)-W 2.849(1), Os(3)-W 2.857(1), 2.15(1), W-C(23) 2.29(1), Os(1)-C(24) 2.18(1), W-C(24) 2.21(1), C(22)-C(23) 1.44(2) and C(23)-C(24) 1.48(2). Dihedral angle between the planes Os( 1)-0s(2)-W and Os(2)-Os(3)-W 162.1(1)'.0 ~ ( 2 ) - 0 ~ ( 3 ) 2.818( 1) , OS( 1)-C(15) 2.16( l ) , 0~(2)-C( 15) 2.23( l ) , W-C( 15) 1.96( 1), 0~(2)-C(22) 2.11( 1), 0~(3)-C(22) 1.97( l ) , W-C(22)
Fig. 2 The molecular drawing of 3b. Bond lengths
(A):
Os(l)-Os(2) 2.803(1), Os(1)-W 2.681(1), Os(2)-W 2.887(1), Os(3)-W 2.873(1), 0 ~ ( 2 ) - 0 ~ ( 3 ) 2.770(1), 0~(2)-C(29) 2.19(2), 0~(3)-C(29) 2.15(2), W-C(29) 2.04(2), 0~(3)-C(8) 2.24(2), W-C(8) 2.27(2), W-C(9) 2.36(2), W-C(12) 2.19(2), Os(l)-C(12) 2.28(2), 0~(2)-C(12) 2.17(2), 0~(2)-C(13) 2.30(2), Os(l)-C(13) 2.56(2), Os(l)-C(21) 2.03(2), 0~(2)-C(21) 2.14(2), C(8)-C(9) 1.32(3), C(9)-C(12) 1.44(2), C(12)-C(13) 1.54(3) and C(13)-C(21) 1.39(3). Dihedral angle between the planes Os( 1)-0s(2)-W and Os(2)-Os(3)-W 171.8( 1)".1022
J. CHEM. SOC., CHEM. COMMUN.,1991
C(41 A)
C (43A)
C(11 A)
C(44A) W(1A) r / i Q A \
n
P
To'O(7A) C(7A)
n
c
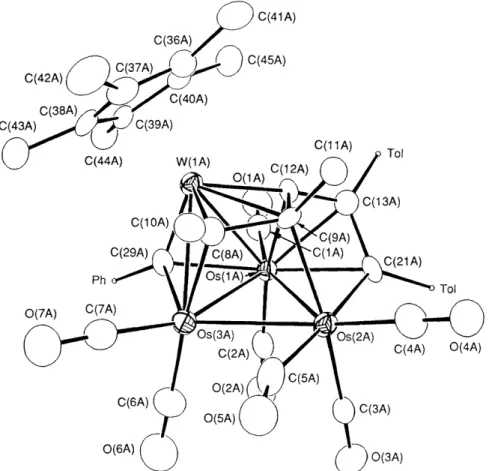
Fig. 3 The molecular drawing of 4b. Bond lengths
(A):
Os(lA)-Os(2A) 2.771(2), Os(lA)-Os(3A) 2.803(2), Os(1A)-W(1A) 2.791(2), 0 ~ ( 2 A ) - 0 ~ ( 3 A ) 2.723(2), Os(3A)-W(lA) 2.794(2), Os(lA)-C(29A) 2.18(2), 0~(3A)-C(29A) 2.04(2), W(lA)-C(29A) 2.03(2), Os(3A)-C(8A) 2.19( 2), W( 1 A)-C( 8A) 2.02(2), OS( 2A)-C(9A) 2.24(2), W( 1A)-C(9A) 2.40(2), OS( 1 A)-C( 12A) 2.17( 2), W( 1 A)-C( 12A) 1.94( 2), OS( 1A)- C( 13A) 2.22(2), 0~(2A)-C(21A) 2.14(2), OS( 1 A)-C(2 1A) 2.25 (2), C(8A)-C( 9A) 1.54( 3), C( 9A)-C( 12A) 1.57 (3), C( 12A)-C( 13A) 1.44( 3),OS( 1 A)-Os( 3A)-W (1 A) 108.9( 1)".
C(13A)-C(21A) 1.49(3), Os(2A)-C(5A) 1.92(3) and Os(3A)-C(5A) 2.48(3). Dihedral angle between the planes Os(lA)-Os(2A)-Os(3A) and
atoms W,
Os(1)
and Os(2), and two C2 alkyne fragments. The C(9)-C(8) fragment is linked to the Os(3) atomvia
a o-bonding and to the W atomvia
a n-bonding, whereas the C(13)-C(21) fragment is coordinated toOs(1)
atom and theOs(2)
atomvia
a o-bonding and a n-bonding.Basically, the core arrangement of 3b is similar to that
of
the structurally characterized 2a, except that the 'wing-tip' metal atom Os(1) in this case carries two C O ligands and that the W-Os(1) bond [2.681(1)A] is much shorter than the respec- tive W-0s distance of 2a [3.004(1)A1.6 Moreover, the conformation of the C(13)-C(21) fragment, with respect to theOs(
l)-Os(2)-C( 12) triangle, is reminiscent of the unusual p3( ~ 2 - 1 ) alkyne arrangement observed in the unsaturated,
46-electron trinuclear alkyne complexes,7 such as Fe3(CO)9(C2Ph2)g and Cp2W2Fe(C0)6(C2To12) .9 It is possible that the electron-donating ability
of
the C5Me5 ligand, the perpendicular arrangement of the alkyne fragment and the potential multiple-bonding character of the W-Os(1)
bond contribute substantially to the remarkable stability of 3b.Crystals of
4b
suitable for X-ray diffraction study were obtained from a solution of CC14-heptane. According to the X-ray analysis,?? the unit cell contains two crystallographic-il. Crystal data-for 4b: C 4 5 H 4 0 0 7 0 ~ 3 W l r M = 1447.26, monoclinic, space group P 1, a = 11.179(5), b = 17.689(4), c = 24.41(2)
A,
a =99.36(5), (j = 96.92(5), y = 88.75(3)", V = 4728(5) A3, 2 = 4,
D,
=2.033 g ~ m - ~ , F(000) = 2671, ~ ( M o - K a ) = 10.57 mm-l. The min. and max. transmission factors are 0.478 and 1.000, 12326 unique reflec- tions were measured, and 8668 reflections with I > 2.0 o(1) were used in refinement. Refinement of 192 atoms and 940 parameters converged to Rf = 0.048 and R, = 0.057, GOF = 3.68.
ally distinct, but structurally similar molecules. An
ORTEP
diagram of one of these molecules is shown in Fig. 3. This molecule displays a 'butterfly' arrangement with Os(2A) and W(1A) defining the 'wing-tip' positions and withOs(
1A) and Os(3A) atoms occupying the 'hinge' positions. The alkylidyne ligand is located at the exteriorof
the Os(lA)-Os(3A)-W(
1A) surface, and the C5 hydrocarbon ligand now adopts an S-shaped arrangement and is encapsulated in the interior of the butterfly core arrangement. Again, the C(8A)-C(9A) alkyne fragmentof
the coordinated C5 ligand may also exhibit the uncommon p3 ( ~ 2 - 1 ) bonding character.After completion
of
the structural assignment, the relation- ship between complexes2, 3b
and4b
can be readily understood and established. Thermolysis of 2a in xylene solution (140"C,
60 min) is fraught with much decomposition. However, thermolysisof
its C5Me5 analogue2b
under similar conditions (140"C, 20 min) induced elimination of C O to produce 3b in85%
yield; as expected, exposure of the toluene solution of3b
toCO
(1 atm, 110 "C,5
min) regenerated2b
in nearly quantitative yield. Further heating of3b
in xylenes (65 min) led to4b
in 25% yield, in addition to 48% unreacted 3b. In contrast, carbonylationof 4b
in refluxing toluene (1 atm, llO°C, 45 min) failed to produce its saturated, 62-electron precursor 2b, but gave instead two additional cluster com- plexes. This result is clearly due to the fact that the metal skeleton has encountered extensive, irreversible rearrange- ment during the formation of4b.
We thank the National Science Council of the Republic of China for financial support of this research (Grant No. NSC80-0208-M007-60).
J . CHEM. SOC., CHEM. COMMUN.,
1991
1023
References
1 Y. Chi, F.-J. Wu, B.-J. Liu, C.-C. Wang and S.-L. Wang, J. Chem.
SOC., Chem. Commun., 1989,873; Y. Chi, G.-H. Lee, S.-M. Peng and B.-J. Liu, Polyhedron, 1989, 8, 2003; Y. Chi, D.-K. Hwang, S.-F. Chen and L.-K. Liu, J. Chem. SOC., Chem. Commun., 1989, 1540; Y. Chi, G.-H. Lee, S.-M. Peng and C.-H. Wu, Organometal-
lics, 1989,8, 1574; C.-H. Wu, Y. Chi, S.-M. Peng and G.-H. Lee, J. Chem. SOC., Dalton Trans., 1990, 3025; Y. Chi, G. Huttner and
W. Imhof, J . Organomet. Chem., 1990,384, 93; D.-K. Hwang, Y. Chi, S.-M. Peng and G.-H. Lee, J. Organomet. Chem., 1990, 389,
c7.
2 D. M. P. Mingos and A . May, in The Chemistry of Metal Clusters,
eds. D. F. Shriver, H. D . Kaesz and R. D. Adams, VCH; New York, 1990; ch. 2; R. D. Adams and I. T. Horvath, Prog. Znorg.
Chem., 1985,33, 127.
3 M. I. Bruce, M. G. Humphrey, J. G. Matisons, S. K. Roy and
A . G. Swincer, Aust. J. Chem. 1984, 37, 1955.
4 M. Tachikawa, J. R . Shapley and C. G . Pierpont, J. Am. Chem.
Soc., 1975, 97, 7172.
5 B. F. G. Johnson, R. Khattar, J. Lewis, P. R. Raithby and D . N.
Smit, J . Chem. SOC., Dalton Trans., 1988, 1421.
6 Complex 2a was first prepared from the reaction between an acetylide cluster complex WOs3Cp(CO)11(GCPh) and ditolyl- acetylene, see: Y. Chi, C.-H. Wu, S.-M. Peng and G.-H. Lee,
Organometallics, 1990, 9, 2305.
7 The cluster core of these 46-electron complexes can be rationalized to adapt a closo-trigonal bipyramidal arrangement in terms of Wade’s rules, see: K. Wade, Adv. Znorg. Radiochem., 1976, 18, 1.
8 J . F. Bount, L. F. Dahl, C. Hoogzand and W. Hiibel, J. Am. Chem.
SOC., 1966, 88, 292.
9 L. Busetto, J. C. Jeffery, R. M. Mills, F. G. A . Stone, M. J. Went and P. Woodward. J. Chem. SOC., Dalton Trans., 1983, 101.