A Remarkable Effect of C
-
O
-
C Bond Angle Strain on the
Regioselective Double Nucleophilic Substitution of the Acetal
Group of Tetraacetal Tetraoxa-Cages and a Novel Hydride
Rearrangement of Tetraoxa-Cages
Hsien-Jen Wu* and Jyh-Haur Chern
Department of Applied Chemistry, National Chiao Tung University, Hsinchu, Taiwan, China Received December 9, 1996X
A remarkable effect of C-O-C bond angle strain on the regioselective double nucleophilic substitution of the acetal group of tetraacetal tetraoxa-cages and a novel regioselective and stereoselective hydride rearrangement of tetraoxa-cages are reported. Reaction of the tetraacetal tetraoxa-cages 1 with 3 equiv of triethylsilane (at-78 °C), cyanotrimethylsilane (at 25 °C), and allyltrimethylsilane (at -78 °C) in dichloromethane in the presence of TiCl4 gave the double nucleophilic substitution products 2, 6, and 7 in 85-90% yields, respectively. No detectable amount of other regioisomers was obtained. Reaction of 1a with (methylthio)trimethylsilane and (phen-ylthio)trimethylsilane in dichloromethane in the presence of TiCl4at-78 °C gave the symmetric products 10a,b and the unsymmetric products 11a,b in ratios of 8-10:1. The stereochemistry of the symmetric substitution products was proven by X-ray analysis of the crystalline compound
10a. The mechanism of the double nucleophilic substitution of the tetraoxa-cages 1 are discussed.
Treatment of the tetraoxa-cages 1a,c and 22a-c with 2 equiv of TiCl4 or MeSO3H in dichlo-romethane at 25 °C for 3 h regioselectively and stereoselectively gave the novel hydride rearrangement products 16a,b and 23a-c respectively. No detectable amount of other regioisomers was observed. The stereochemistry of the hydride rearrangement was proven by DIBAL-H reduction of 16 and 23 and X-ray analysis of the reduction product 24a. We attribute the high regioselectivity of the double nucleophilic substitution and the hydride rearrangement of the tetraoxa-cages 1 to the bond angle strain of the unusually large bond angle of C(3)-O(4)-C(5) of the tetraoxa-cages.
Introduction
The reaction chemistry of acetals has been greatly expanded by the use of Lewis acidic promoters particu-larly in conjunction with silicon-containing nucleophiles.1
In recent times much interest has been shown in the mechanism and origin of stereoselectivity of substitution of chiral acetals,2a concept initiated by Johnson et al.3
Usually, acyclic and monocyclic acetals are the objects
for study. Recently, we accomplished the synthesis of novel oxa-cage compounds, such as tetraacetal tetraoxa-cages,4tetraacetal petaoxa-cages,5triacetal trioxa-cages,6
diacetal trioxa-cages,7and pentaacetal pentaoxa-cages
(the pentaoxa[5]-peristylanes).8 For instance, the
tet-raoxa-cages B were synthesized by ozonolysis of 2,3-endo-diacylnorbornenes A (Scheme 1).4a Afterward, we
devel-oped a new entry for the synthesis of the unsubstituted (parent) compound 1c and its 3-alkyl-substituted deriva-tives D via ozonolysis of 2-endo-7-anti-diacylnorbornenes
C.4h All these oxa-cages contain acetal and ketal groups
on the molecule, and they are new systems for the study of the reaction chemistry of acetals. As part of a program that involves the synthesis, chemistry, and applications of new heterocyclic cage compounds, we report here a remarkable effect of the C-O-C bond angle strain on the regioselective double nucleophilic substitution of the acetal group of tetraacetal tetraoxa-cages. We also wish to demonstrate a novel hydride rearrangement of the acetal group of tetraoxa-cages mediated by Lewis acids.
XAbstract published in Advance ACS Abstracts, April 15, 1997.
(1) Reviews: (a) Mukaiyama, T.; Murakami, M. Synthesis 1987, 1043. (b) Hosomi, A.; Endo, M.; Sakurai, H. Chem. Lett. 1976, 941. (c) Hosomi, A. Acc. Chem. Res. 1988, 21, 200.
(2) (a) Denmark, S. E.; Almstead, N. G. J. Am. Chem. Soc. 1991,
113, 8089. (b) Sammakia, T.; Smith, R. S. J. Org. Chem. 1992, 57,
2997. (c) Denmark, S. E.; Wilson, T.; Willson, T. M. J. Am. Chem.
Soc. 1988, 110, 984. (d) Denmark, S. E.; Willson, T. M.; Almstead, N.
G. J. Am. Chem. Soc. 1989, 111, 9258. (e) Yamamoto, Y.; Nishii, S.; Yamada, J. J. Am. Chem. Soc. 1986, 108, 7116. (f) Mori, I.; Ishihara, K.; Flippin, L. A.; Nozaki, K.; Yamamoto, H.; Bartlett, P. A.; Heathcock, C. H. J. Org. Chem. 1990, 55, 6107. (g) Denmark, S. E.; Willson, T. M. J. Am. Chem. Soc. 1989, 111, 3475. (h) Denmark, S. E.; Almstead, N. G. J. Org. Chem. 1991, 56, 6485. (i) Ishihara, K.; Mori, A.; Yamamoto, H. Tetrahedron 1990, 46, 4595.
(3) (a) Johnson, W. S.; Harbert, C. A.; Stipanovic, R. D. J. Am. Chem.
Soc. 1968, 90, 5279. (b) Johnson, W. S.; Harbert, C. A.; Ratcliffe, B.
E.; Stipanovic, R. D. J. Am. Chem. Soc. 1976, 98, 6188. (c) Bartlett, P. A.; Johnson, W. S.; Elliott, J. D. J. Am. Chem. Soc. 1983, 105, 2088. (d) Johnson, W. S.; Crackett, P. H.; Elliott, J. D.; Jagodzinski, J. J.; Lindell, S. D.; Natarajan, S. Tetrahedron Lett. 1984, 25, 3951. (e) Elliott, J. D.; Choi, V. M. F.; Johnson, W. S. J. Org. Chem. 1983, 48, 2294. (f) Johnson, W. S.; Edington, C.; Elliott, J. D.; Silverman, I. R.
J. Am. Chem. Soc. 1984, 106, 7588. (g) Lindell, S. D.; Elliott, J. D.;
Johnson, W. S. Tetrahedron Lett. 1984, 25, 3947. (h) Choi, V. M. F.; Elliott, J. D.; Johnson, W. S. Tetrahedron Lett. 1984, 25, 591. (i) Elliott, J. D.; Steele, J.; Johnson, W. S. Tetrahedron Lett. 1985, 26, 2535. (j) Silverman, R. I.; Edington, C.; Elliott, J. D.; Johnson, W. S. J. Org.
Chem. 1987, 52, 180. (k) McNamara, J. M.; Kishi, Y. J. Am. Chem. Soc. 1982, 104, 7371. (l) For copper nucleophiles: Ghribi, A.; Alexakis,
A.; Normant, J. F. Tetrahedron Lett. 1984, 25, 3075. (m) Ghribi, A.; Alexakis, A.; Normant, J. F. Tetrahedron Lett. 1984, 25, 3083. (n) Richter, W. J. J. Org. Chem. 1981, 46, 5119. (o) Alexakis, A.; Mangeney, P. Tetrahedron Asymm. 1990, 1, 477.
(4) (a) Wu, H. J.; Lin, C. C. J. Org. Chem. 1995, 60, 7558. (b) Wu, H. J.; Lin, C. C. J. Org. Chem. 1996, 61. 3820. (c) Lin, C. C.; Wu, H. J. Tetrahedron Lett. 1995, 36, 9353. (d) Wu, H. J.; Huang, F. J.; Lin, C. C. J. Chem. Soc., Chem. Commun. 1991, 770. (e) Lin, C. C.; Wu, H. J. J. Chin. Chem. Soc. 1995, 42, 815. (f) Lin, C. C.; Huang, F. J.; Lin, J. C.; Wu, H. J. J. Chin. Chem. Soc. 1996, 43, 177. (g) Lin, R. L.; Wu, C. Y.; Chern, J. H.; Wu, H. J. J. Chin. Chem. Soc. 1996, 43, 289. (h) Wu, H. J.; Chern, J. H.; Wu, C. Y. Tetrahedron 1997, 53, 2401.
(5) Lin, C. C.; Wu, H. J. Synthesis 1996, 715.
(6) Wu, C. Y.; Lin, C. C.; Lai, M. C.; Wu, H. J. J. Chin. Chem. Soc.
1996, 43, 187.
(7) (a) Wu, H. J.; Tsai, S. H.; Chung, W. S. Tetrahedron Lett. 1996,
37, 8209. (b) Tsai, S. H.; Wu, H. J.; Chung, W. S. J. Chin. Chem. Soc.
1996, 43, 445. (c) Wu, H. J.; Tsai, S. H.; Chern, J. H.; Lin, H. C. J.
Org. Chem., submitted for publication.
(8) Wu, H. J.; Wu, C. Y. Tetrahedron Lett. 1997, in press.
Results and Discussion
Reaction of the tetraacetal tetraoxa-cages 1a,b4aand 1c4hwith 3 equiv of triethylsilane9in dichloromethane
at-78 °C in the presence of a catalytic amount of TiCl4 for 0.5 h regioselectively gave the substitution products
2a,b and 2c in 85-90% yields, respectively (Scheme 2). No detectable amount of the other regioisomers 3, 4, or
5 was obtained. We attribute the highly regioselective
nucleophilic substitution by cleavage of the C(3)-O(4) or C(5)-O(4) bond of 1 mediated by TiCl4to the unusually large bond angle of C(3)-O(4)-C(5). While the other C-O-C bond angles of tetraoxa-cages 1 are in between 111-108°, the C(3)-O(4)-C(5) bond angle is 117.5°, remarkably larger than the ordinary bond angles with sp3-hybridized atoms.4a Steric factor for the regioselective
nucleophilic substitution of the acetal groups of 1 was excluded since no detectable amount of 3c was obtained in the case of 1c.
Reaction of 1a-c with 3 equiv of cyanotrimethylsi-lane10 in dichloromethane at 25 °C in the presence of
TiCl4for 1 h regioselectively and stereoselectively gave 6a-c in 85-90% yields (Scheme 3). No detectable
amount of other regioisomeric substitution products was obtained. The stereochemistry of the cyano groups of 6 was assigned on the basis of NOE experiments and other similar chemical transformations, such as reaction of 1 with Me3SiSMe on Scheme 4. Irradiating the proton on
C2and C8of 6a (δ 4.73) gives 6.8% enhancement for the syn proton on the apical carbon C12 and less than 1% enhancement for the bridgehead C1proton. Treatment
of 1a,b with 3 equiv of allyltrimethylsilane11,12in
dichlo-romethane at-78 °C for 0.5 h gave compounds 7a,b in 90% yields. The nucleophilic substitution reactions took place regioselectively on the C(3)-O(4)-C(5) bonds of
1a-c.
Reaction of 1a with 1 equiv of triethylsilane and allyltrimethylsilane in dichloromethane at-78 °C gave
2a and 7a in 45% yields and the unreacted compound 1a. No detectable amount of the monosubstitution products 8 or 9 was obtained. Similarly, reaction of 1a with 1 equiv of cyanotrimethylsilane in dichloromethane at 25 °C gave 6 in 45% yield and unreacted 1a. There are two sequential nucleophilic substitution reactions present in each case of the reaction of 1a-c with the silicon-containing nucleophiles. The above experimental results indicate that the second nucleophilic substitution reaction of the acetal group of 1 is faster than the first one in reaction with the silane nucleophiles in the presence of TiCl4.
Reaction of 1a with 3 equiv of (methylthio)trimethyl-silane and (phenylthio)trimethyl(methylthio)trimethyl-silane in dichloromethane in the presence of TiCl4 at -78 °C for 1 h gave the symmetric products 10a and 10b (80-85%) and the unsymmetric products 11a and 11b (10-8%) in ratios of 8-10:1 (Scheme 4). The stereochemistry of the
substit-(9) (a) Frainnet, E.; Esclamadon, C. C. R. 1962, 254, 1814. (b) Loim, N. M.; Parnes, Z. N.; Vasil’eva, S. P.; Kursanov, D. N. Zh. Org. Khim.
1972, 8, 896. (c) For a review on ionic hydrogenation with silanes,
see: Kursanov, D. N.; Parnes, Z. N.; Loim, N. M. Synthesis 1974, 633. (d) Tsunoda, T.; Suzuki, M.; Noyori, R. Tetrahedron Lett. 1979, 4679. (e) Olah, G. A.; Yamato, T.; Iyer, P. S.; Prakash, G. K. S. J. Org. Chem.
1986, 51, 2826. (f) Mori, A.; Ishihara, K.; Yamamoto, H. Tetrahedron
Lett. 1986, 27, 987. (g) Sato, T.; Otera, J.; Nozaki, H. J. Am. Chem. Soc. 1990, 112, 901.
(10) (a) Evans, D. A.; Truesdale, L. K.; Carroll, G. L. J. Chem. Soc.,
Chem. Commun. 1973, 55. (b) Evans, D. A.; Truesdale, L. K.
Tetrahedron Lett. 1973, 4929. (c) Utimoto, K.; Wakabayashi, Y.;
Shishiyama, Y.; Inoue, M.; Nozaki, H. Tetrahedron Lett. 1981, 22, 4279. (d) Utimoto, K.; Wakabayashi, Y.; Horiie, T.; Inoue, M.; Shishiyama, Y.; Obayashi, M.; Nozaki, H. Tetrahedron 1983, 39, 967. (e) Kirchm-eyer, S.; Mertens, A.; Arvanaghi, M.; Olah, G. A. Synthesis 1983, 498.
(11) (a) Hosomi, A.; Sakurai, H. Tetrahedron Lett. 1976, 1295. (b) Hosomi, A.; Sakurai, H. J. Am. Chem. Soc. 1977, 99, 1673. (c) Hosomi, A.; Endo, M.; Sakurai, H. Chem. Lett. 1976, 941. (d) Mukaiyama, T.
Angew. Chem., Int. Ed. Engl. 1977, 16, 817. (e) Nishiyama, H.; Itoh,
K. J. Org. Chem. 1982, 47, 2496.
(12) Reviews: (a) Marshall, J. A. Chem. Rev. 1996, 96, 31. (b) Yamamoto, Y.; Asao, N. Chem. Rev. 1993, 93, 2207. (c) Chan, T. H.; Wang, D. Chem. Rev. 1992, 92, 995. (d) Hoffman, R. W. Angew. Chem.
1982, 94, 569; Angew. Chem., Int. Ed. Engl. 1982, 21, 555.
Scheme 1
Scheme 2
uents on C2and C8of the symmetric compounds 10a,b
was proven by X-ray analysis of the crystalline compound
10a (Figure 1).13
Lewis acid-mediated nucleophilic substitution of ac-etals can occur by direct displacement (SN2) or
oxocar-benium ion (SN1) mechanisms.2 Each case of the above
reactions involves double nucleophilic substitution. Since the unsymmetric substitution products 11a and 11b were obtained in the reaction of 1a with TMSX (X ) SMe, SPh), a mechanism via double SN2 direct displacement
may be excluded. A mechanism via the intermediates
12 and 13 may be proposed for this double nucleophilic
substitution reaction (Scheme 5). Nevertheless, the detailed mechanism of the double nucleophilic substitu-tion is not clear at present. The slight difference in the double nucleophilic substitution reaction of 1 with Me3
-SiSR from that with triethylsilane, allyltrimethylsilane, and cyanotrimethylsilane may depend on the nature of the nucleophiles.2
Reaction of 1a-c with 4 equiv of methanol in dichlo-romethane at 25 °C in the presence of TiCl4 for 2 h
regioselectively and stereoselectively gave the substitu-tion products 14a-c in 80-85% yields. Treatment of
1a-c with 3 equiv of m-chloroperoxybenzoic acid (m-CPBA) in the presence of BF3‚OEt2in dichloromethane at 25 °C for 1 h gave the bislactones 15a-c in 85% yields
(Scheme 6). This oxidation reaction also takes place regioselectively on the acetal carbons C3and C5of 1 even
in the case of unsubstituted compound 1c.
A novel regioselective and stereoselective hydride rearrangement was also discovered. Treatment of the symmetric tetraoxa-cages 1a and 1c with 2 equiv of Lewis acids, such as TiCl4, BF3-OEt2, and MeSO3H in dichloromethane at 25 °C for 12 h regioselectively gave the novel hydride rearrangement products 16a and 16b in 90% yields (Scheme 7). No detectable amount of the other regioisomer 17 was obtained. In the case of the unsubstituted (parent) compound 1c, no detectable amount of the other regioisomers 18 or 19 was obtained. Treat-ment of 1a and 1c with a catalytic amount of TiCl4at 25
°C for 12 h gave 16a and 16b in 15-20% yields and the unreacted 1a and 1b. Similar to the previous nucleo-philic substitution reactions, we attribute the high regio-selectivity of the hydride rearrangement to the bond angle strain of the unusually large bond angle of C(3) -O(4)-C(5) of the tetraoxa-cages 1. In the case of 1c, with no alkyl substituents on C1 and C7, the hydride
rear-rangement still took place regioselectively between C3
and C5. Thus, steric hindrance factor for the hydride
rearrangement of 1 to 16 was excluded. Treatment of the tetraoxa-cage 204gwith TiCl
4under the same reaction
conditions gave 21 in 90% yield. A three-membered spiro ring on the apical carbon did not interfere with the hydride rearrangement.
The IR spectra of 16a,b and 21 showed strong absorp-tion at 1770 cm-1
for the five-membered lactone carbonyl group. The 1H NMR spectrum of 16b revealed two (13) The authors have deposited atomic coordinates for these
structures with the Cambridge Crystallographic Data Centre. The coordinates can be obtained, on request, from the Director, Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK.
Figure 1. ORTEP diagram of 10a. Scheme 4
Scheme 5
Scheme 6
doublets at δ 6.04 and 5.97 for the two acetal protons on C4and C6and two doublets of doublet at δ 4.13 and 3.50
for the methylene protons on C2. The13C NMR spectrum
of 16b displayed a singlet at δ 179.09 for the lactone carbonyl, two peaks at δ 112.31 and 106.38 for the acetal carbons C4 and C6, and one peak at δ 71.59 for the
methylene carbon C2.
In order to understand the stereochemistry of the hydride rearrangement, we prepared compounds 22a
-c4gfor the rearrangement study. Treatment of 22a
-c with 2 equiv of TiCl4in dichloromethane at 25 °C for 12
h stereoselectively gave compounds 23a-c in 85-90% yields (Scheme 8). The stereochemistry of the alkyl group on C2 of 23 was assigned on the basis of NOE
experi-ments of 23a and proven by chemical transformation of
23. Irradiating the methyl group on C2of 23a gives 8.6%
enhancement of the intensity of the C1proton. Reduction
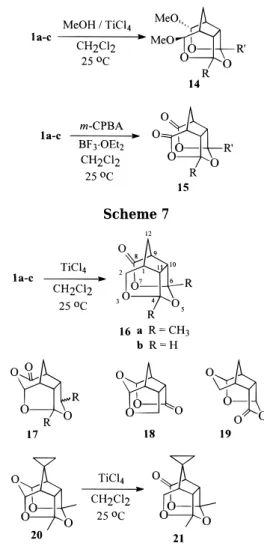
of 23a,c and 16a,b with diisobutylaluminum hydride (DIBAL-H) in dry THF at 0 °C stereoselectively gave compounds 24a-d in 85-90% yields, respectively. The stereochemistry of the hydroxy group and the alkyl substituents on C2of 24 was proven by X-ray analysis of
the crystalline compound 24a (Figure 2).13 Hence, the
stereochemistry of the alkyl substituent on C2of 23a-c was confirmed. Reduction of 21 with DIBAL-H under the same reaction conditions gave 25 in 85% yield.
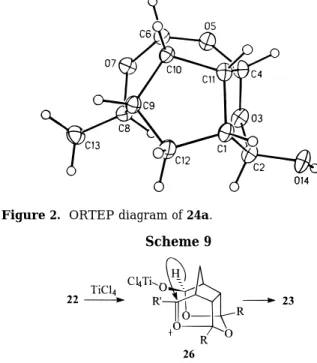
A reaction mechanism is proposed for the hydride rearrangement from 22 to 23 (Scheme 9). Coordination of TiCl4 to the oxygen atom O(4) of 22 followed by
cleavage of the C(3)-O(4) bond gives the oxocarbenium ion 26. Repulsion of the hydride on C5 of 26 by the
alkoxide anion followed by nucleophilic addition of the hydride on the oxocarbenium ion from the inside concave face gives the observed products 23. We propose that the hydride rearrangement is an intramolecular process. From the previous intermolecular nucleophilic substitu-tion reacsubstitu-tions (Schemes 3-6), the nucleophiles attack the oxocarbenium ion 26 from the outside convex face. Nevertheless, more experiments, such as the cross-over rearrangement test, are required before the conclusion can be made.
It is worth to note the stereochemistry of the DIBAL-H reduction of 23a,c and 16a,b. The hydride addition from DIBAL-H to the lactone carbonyl group takes place stereoselectively from the inside concave face. We pro-pose that the aluminum atom of DIBAL-H may coordi-nate to the carbonyl oxygen atom and the oxygen atoms O(3) and O(7) of 23 and 16 in the transition state. Whether these oxa-cages may exhibit such interesting cation-binding properties or not needs to be proven by further extensive studies.
Conclusion
A remarkable regioselective and stereoselective double nucleophilic substitution of the acetal group of tetraacetal tetraoxa-cages with silicon-containing nucleophiles medi-ated by Lewis acids was demonstrmedi-ated. We attribute the highly regioselective nucleophilic substitution of the tetraoxa-cages 1 to the C-O-C bond angle strain of the unusually large bond angle of C(3)-O(4)-C(5) of 1. The stereochemistry of the substitution products was proven by X-ray analysis of the crystalline compound 10a. There are two sequential nucleophilic substitution reaction present in each case and the second nucleophilic substi-tution of the acetal group of 1 is faster than the first one. The Lewis acid-mediated substitution may occur by SN2
or SN1 mechanisms, but a mechanism via double SN2
direct displacement may be excluded. A novel regiose-lective and stereoseregiose-lective hydride rearrangement of acetal group was also discovered. We also attribute the high regioselectivity of the hydride rearrangement to the C(3)-O(4)-C(5) bond angle strain of the tetraoxa-cages. The stereochemistry of the hydride rearrangement is proven by X-ray analysis of the crystalline compound 24a. A mechanism via intramolecular manner is proposed for the stereochemistry of the hydride rearrangement.
Experimental Section
General. Melting points were determined in capillary
tubes with a Laboratory Devices melting point apparatus and uncorrected. Infrared spectra were recorded in CHCl3
solu-tions or on neat thin films between NaCl disks. 1H NMR
spectra were determined at 300 MHz, and13C NMR spectra Scheme 8
Figure 2. ORTEP diagram of 24a. Scheme 9
were determined at 75 MHz, on Fourier transform spectrom-eters. Chemical shifts are reported in ppm relative to TMS in the solvents specified. The multiplicities of13C signals were
determined by DEPT techniques. High resolution mass values were obtained with a high resolution mass spectrometer at the Department of Chemistry, National Tsing Hua University. Elemental analyses were performed at the microanalysis laboratory of National Taiwan University. X-ray analysis were carried out on a diffractometer at the Department of Chem-istry, National Tsing Hua University. For thin-layer chroma-tography (TLC) analysis, precoated TLC plates (Kieselgel 60 F254) were used, and column chromatography was done by
using Kieselgel 60 (70-230 mesh) as the stationary phase. THF was distilled immediately prior to use from sodium benzophenone ketyl under nitrogen. CH2Cl2was distilled from
CaH2under nitrogen.
General Procedure for the Reaction of Tetraacetal Tetraoxa-Cages 1a-c with Triethylsilane in the
Pres-ence of TiCl4. To a solution of tetraacetal tetraoxa-cage 1a
(0.21 g, 1.00 mmol) in dichloromethane (20 mL) were added triethylsilane (0.35 g, 3.00 mmol) and TiCl4 (0.020 g, 0.10
mmol) at-78 °C. The reaction mixture was stirred at-78 °C for 0.5 h. After addition of water (10 mL) and extraction with dichloromethane (3 × 20 mL), the organic layer was washed with brine, dried over MgSO4, and evaporated, and
the residue was purified by column chromatography to give
2a (0.17 g, 0.85 mmol) in 85% yield.
4,6-Dimethyl-3,5,7-trioxatetracyclo[7.2.1.04,11.06,10
]do-decane (2a): white waxy solid; yield 85%; mp 57-58 °C; IR (CHCl3) 2980, 2880, 1060 cm-1 ;1H NMR (300 MHz, CDCl 3) δ 4.01 (dd, J)8.4 Hz, J)2.7 Hz, 4H), 3.08 (dd, J)6.9 Hz, J )3.0 Hz, 2H), 2.98-2.92 (m, 2H), 2.18-2.04 (m, 1H), 1.78 -1.72 (m, 1H), 1.50 (s, 6H);13C NMR (75 MHz, CDCl 3, DEPT) δ 118.46 (2C), 73.91 (2CH2), 60.98 (2CH), 46.79 (2CH), 37.29 (CH2), 24.88 (2CH3); LRMS m/z (rel inten) 196 (M+ , 23), 181 (100); HRMS (EI) calcd for C11H16O3196.1099, found 196.1094.
Anal. Calcd for C11H16O3: C, 67.31; H, 8.22. Found: C, 67.20;
H, 8.30.
4-Methyl-6-benzyl-3,5,7-trioxatetracyclo[7.2.1.04,11.06,10
]-dodecane (2b): white waxy solid; yield 90%; mp 66-67 °C; IR (CHCl3) 2980, 2880, 1600, 1060, 750, 695 cm-1 ;1H NMR (300 MHz, CDCl3) δ 7.33-7.22 (m, 5H), 4.02-3.94 (m, 4H), 3.16 (dd, J)9.6 Hz, J)9.6 Hz, 1H), 3.10, 2.97 (ABq, J) 13.7 Hz, 2H), 2.83-2.72 (m, 3H), 2.15-2.05 (m, 1H), 1.76 -1.72 (m, 1H), 1.31 (s, 3H);13C NMR (75 MHz, CDCl 3, DEPT) δ 136.77 (C), 130.67 (2CH), 127.86 (2CH), 126.43 (CH), 120.74 (C), 118.98 (C), 74.18 (CH2), 74.78 (CH2), 60.75 (CH), 58.37 (CH), 46.39 (CH), 46.23 (CH), 43.59 (CH2), 38.12 (CH2), 24.03 (CH3); LRMS m/z (rel inten) 272 (M+, 15), 195 (100); HRMS
(EI) calcd for C17H20O3272.1412, found 272.1410. Anal. Calcd
for C17H20O3: C, 74.96; H, 7.41. Found: C, 74.84; H, 7.50. 3,5,7-Trioxatetracyclo[7.2.1.04,11.06,10]dodecane (2c): white
waxy solid; yield 85%; mp 53-54 °C; IR (CHCl3) 2980, 2880, 1060 cm-1 ;1H NMR (300 MHz, CDCl 3) δ 5.78 (d, J)3.9 Hz, 2H), 4.07-3.93 (m, 4H), 3.34-3.30 (m, 2H), 2.94-2.87 (m, 2H), 2.28-2.17 (m, 1H), 1.85-1.81 (m, 1H); 13C NMR (75 MHz, CDCl3, DEPT) δ 111.67 (2CH), 73.82 (2CH2), 56.39 (2CH), 45.48 (2CH), 37.60 (CH2); LRMS m/z (rel inten) 168 (M+ , 31), 154 (100); HRMS (EI) calcd for C9H12O3 168.0786, found
168.0781. Anal. Calcd for C9H12O3: C, 64.26; H, 7.20.
Found: C, 64.34; H, 7.16.
General Procedure for the Reaction of Tetraoxa-Cages 1a-c with Cyanotrimethylsilane in the Presence
of TiCl4. To a solution of tetraoxa-cages 1a (0.21 g, 1.00 mmol)
in dichloromethane (20 mL) were added cyanotrimethylsilane (0.30 g, 3.0 mmol) and TiCl4(0.020 g, 0.10 mmol) at 25 °C.
The reaction mixture was stirred at 25 °C for 1 h. After addition of water (10 mL) and extraction with dichloromethane (3× 20 mL), the organic layer was washed with brine, dried over MgSO4, and evaporated, and the residue was purified by
column chromatography to give 6a (0.22 g, 0.90 mmol) in 90% yield.
2β,8β-Dicyano-4,6-dimethyl-3,5,7-trioxatetracyclo-[7.2.1.04,11.06,10]dodecane (6a): white waxy solid; yield 85%;
mp 102-103 °C; IR (CHCl3) 2980, 2250, 1060 cm -1;1H NMR (300 MHz, CDCl3) δ 4.73 (d, J)5.1 Hz, 2H), 3.39-3.26 (m, 4H), 2.48-2.40 (m, 1H), 2.12-2.07 (m, 1H), 1.67 (s, 6H); 13C NMR (75 MHz, CDCl3, DEPT) δ 120.97 (2CN), 118.00 (2C), 71.52 (2CH), 60.31 (2CH), 52.00 (2CH), 37.29 (CH2), 25.25 (2CH3); LRMS m/z (rel inten) 246 (M+, 32), 231 (100); HRMS
(EI) calcd for C13H14O3N2246.1004, found 246.1007.
2β,8β-Dicyano-4-methyl-6-benzyl-3,5,7-trioxatetracyclo-[7.2.1.04,11.06,10]dodecane (6b): white waxy solid; yield 90%;
mp 80-81 °C; IR (CHCl3) 2980, 2880, 2250, 1600, 1060, 745, 700 cm-1 ;1H NMR (300 MHz, CDCl 3) δ 7.35-7.26 (m, 5H), 4.74 (d, J)5.7 Hz, 1H), 4.70 (d, J)5.4 Hz, 1H), 3.42 (dd, J )10.2 Hz, J)10.2 Hz, 1H), 3.23, 3.09 (ABq, J)13.8 Hz, 2H), 3.20-3.12 (m, 2H), 2.99 (dd, J)10.2 Hz, J)9.6 Hz, 1H), 2.43-2.32 (m, 1H), 2.08-2.03 (m, 1H), 1.45 (s, 3H); 13C NMR (75 MHz, CDCl3, DEPT) δ 134.89 (C), 130.61 (2CH), 128.22 (2CH), 127.20 (CH), 122.77 (CN), 121.26 (CN), 118.05 (C), 117.85 (C), 71.76 (CH), 71.20 (CH), 59.66 (CH), 58.18 (CH), 51.83 (CH), 51.59 (CH), 43.38 (CH2), 37.46 (CH2), 24.29 (CH3); LRMS m/z (rel inten) 322 (M+ , 21), 245 (100); HRMS (EI) calcd for C19H18O3N2322.1317, found 322.1312. 2β,8β-Dicyano-3,5,7-trioxatetracyclo[7.2.1.04,11.06,10
]-dodecane (6c): white waxy solid; yield 85%; mp 92-93 °C; IR (CHCl3) 2980, 2880, 2250, 1060 cm-1 ;1H NMR (300 MHz, CDCl3) δ 6.00 (d, J)5.1 Hz, 2H), 4.72 (d, J)5.1 Hz, 2H), 3.63-3.58 (m, 2H), 3.27-3.19 (m, 2H), 2.63-2.51 (m, 1H), 2.13-2.05 (m, 1H); 13C NMR (75 MHz, CDCl 3, DEPT) δ 120.97 (2CN), 112.58 (2CH), 71.61 (2CH), 55.79 (2CH), 51.01 (2CH), 37.61 (CH2); LRMS m/z (rel inten) 218 (M+, 100); HRMS (EI)
calcd for C11H10O3N2218.0691, found 218.0678.
General Procedure for the Reaction of Tetraoxa-Cages 1a,b with Allyltrimethylsilane in the Presence of TiCl4. To a solution of tetraoxa-cages 1a (0.42 g, 2.00 mmol)
in dichloromethane (30 mL) were added allyltrimethylsilane (0.68 g, 6.00 mmol) and TiCl4(0.040 g, 0.20 mmol) at-78 °C. The reaction mixture was stirred at-78 °C for 0.5 h. After addition of water (20 mL) and extraction with dichloromethane (3× 30 mL), the organic layer was washed with brine, dried over MgSO4, and evaporated, and the residue was purified by
column chromatography to give 7a (0.51 g, 1.8 mmol) in 90% yield.
2β,8β-Diallyl-4,6-dimethyl-3,5,7-trioxatetracyclo-[7.2.1.04,11.06,10]dodecane (7a): pale yellow oil; yield 90%; IR
(neat) 2970, 1620, 1070 cm-1;1H NMR (300 MHz, CDCl 3) δ 5.78-5.69 (m, 2H), 5.07-4.98 (m, 4H), 4.27-4.20 (m, 2H), 3.03 (dd, J)7.2 Hz, J)3.0 Hz, 2H), 2.58-2.54 (m, 2H), 2.32 -2.19 (m, 4H), 1.90-1.79 (m, 1H), 1.58-1.54 (m, 1H), 1.43 (s, 6H);13C NMR (75 MHz, CDCl 3, DEPT) δ 134.07 (2CH), 117.12 (2CH2), 116.09 (2C), 84.21 (2CH), 60.58 (2CH), 53.48 (2CH), 39.73 (2CH2), 33.74 (CH2), 26.01 (2CH3); LRMS m/z (rel inten) 276 (M+
, 45), 261 (100); HRMS (EI) calcd for C17H24O3
276.1725, found 276.1719. Anal. Calcd for C17H24O3: C, 73.87;
H, 8.76. Found: C, 73.98; H, 8.68.
2β,8β-Diallyl-4-methyl-6-benzyl-3,5,7-trioxatetracyclo-[7.2.1.04,11.06,10]dodecane (7b): pale yellow oil; yield 90%; IR
(neat) 2980, 1600, 1070, 745, 695 cm-1;1H NMR (300 MHz, CDCl3) δ 7.31-7.21 (m, 5H), 5.83-5.71 (m, 2H), 5.11-5.02 (m, 4H), 4.32-4.24 (m, 2H), 3.22-2.92 (m, 4H), 2.60-2.17 (m, 6H), 1.92-1.82 (m, 1H), 1.72-1.67 (m, 1H), 1.41 (s, 3H); 13C NMR (75 MHz, CDCl3, DEPT) δ 136.76 (C), 134.37 (CH), 134.28 (CH), 130.73 (2CH), 127.76 (2CH), 126.36 (CH), 118.67 (C), 117.18 (C), 117.12 (2CH2), 84.81 (CH), 84.37 (CH), 60.74 (CH), 58.44 (CH), 53.14 (CH), 52.99 (CH), 45.13 (CH2), 40.14 (2CH2), 34.81 (CH2), 26.24 (CH3); LRMS m/z (rel inten) 352 (M+, 64),
275 (100); HRMS (EI) calcd for C23H28O3 352.2038, found
352.2032. Anal. Calcd for C23H28O3: C, 78.36; H, 8.01.
Found: C, 78.45; H, 8.08.
General Procedure for the Reaction of 1a with (Meth-ylthio)trimethylsilane and (Phen(Meth-ylthio)trimethylsilane in the Presence of TiCl4. To a solution of tetraoxa-cage 1a
(0.42 g, 2.00 mmol) in dichloromethane (30 mL) were added methylthiotrimethylsilane (0.72 g, 6.00 mmol) and TiCl4(0.040
g, 0.20 mmol) at-78 °C. The reaction mixture was stirred at -78 °C for 1 h. After addition of water (20 mL) and extraction with dichloromethane (3 × 30 mL), the organic layer was washed with brine, dried over MgSO4, and evaporated, and
the residue was purified by column chromatography to give
2β,8β-Bis(methylthio)-4,6-dimethyl-3,5,7-trioxatetra-cyclo[7.2.1.04,11.06,10]dodecane (10a): white waxy solid; yield
80%; mp 134-135 °C; IR (CHCl3) 2980, 2880, 1380, 1060 cm -1 ; 1H NMR (300 MHz, CDCl 3) δ 5.19 (d, J)6.0 Hz, 2H), 3.20 (dd, J)6.0 Hz, J)2.4 Hz, 2H), 2.80-2.70 (m, 2H), 2.15 (s, 6H), 2.10-2.01 (m, 2H), 1.61 (s, 6H); 13C NMR (75 MHz, CDCl 3, DEPT) δ 119.34 (2C), 90.96 (2CH), 60.68 (2CH), 52.90 (2CH), 37.96 (CH2), 26.65 (2CH3), 13.95 (2CH3); LRMS m/z (rel inten)
288 (M+, 12), 241 (100); HRMS (EI) calcd for C
13H20O3S2
288.0854, found 288.0858.
2β,8r
-Bis(methylthio)-4,6-dimethyl-3,5,7-trioxatetra-cyclo[7.2.1.04,11.06,10]dodecane (11a): white waxy solid; yield
10%; mp 85-86 °C; IR (CHCl3) 2980, 2880, 1380, 1060 cm -1 ; 1H NMR (300 MHz, CDCl 3) δ 5.56 (d, J)9.0 Hz, 1H), 4.98 (d, J)5.4 Hz, 1H), 3.23-3.12 (m, 2H), 3.07-2.96 (m, 1H), 2.64 -2.55 (m, 1H), 2.34-2.30 (m, 1H), 2.27 (s, 3H), 2.24 (s, 3H), 2.05-1.97 (m, 1H), 1.60 (s, 3H), 1.47 (s, 3H); 13C NMR (75 MHz, CDCl3, DEPT) δ 119.77 (C), 115.99 (C), 89.44 (CH), 87.87 (CH), 61.94 (CH), 60.51 (CH), 52.61 (CH), 48.91 (CH), 33.21 (CH2), 26.30 (CH3), 24.00 (CH3), 15.49 (CH3), 14.44 (CH3);
LRMS m/z (rel inten) 288 (M+, 23), 241 (100); HRMS (EI) calcd
for C13H20O3S2288.0854, found 288.0842.
2β,8β-Bis(phenylthio)-4,6-dimethyl-3,5,7-trioxatetra-cyclo[7.2.1.04,11.06,10]dodecane (10b): pale yellow oil; yield
85%; IR (neat) 2980, 1600, 1060, 750, 695 cm-1 ;1H NMR (300 MHz, CDCl3) δ 7.49-7.20 (m, 10H), 5.52 (d, J)5.4 Hz, 2H), 3.25 (dd, J)6.6 Hz, J)3.0 Hz, 2H), 2.90-2.86 (m, 2H), 2.36 -2.12 (m, 2H), 1.69 (s, 6H);13C NMR (75 MHz, CDCl 3, DEPT) δ 134.57 (2C), 130.90 (4CH), 128.81 (4CH), 126.94 (2CH), 119.57 (2C), 92.15 (2CH), 60.42 (2CH), 53.05 (2CH), 37.84 (CH2), 26.68 (2CH3); LRMS m/z (rel inten) 412 (M+ , 56), 303 (100); HRMS (EI) calcd for C23H24O3S2412.1167, found 412.1160. 2β,8r
-Bis(phenylthio)-4,6-dimethyl-3,5,7-trioxatetra-cyclo[7.2.1.04,11.06,10]dodecane (11b): pale yellow oil; yield
8%; IR (neat) 2980, 1600, 1060, 750, 695 cm-1;1H NMR (300 MHz, CDCl3) δ 7.57-7.18 (m, 10H), 5.96 (d, J)8.1 Hz, 1H), 5.23 (d, J)5.4 Hz, 1H), 3.22-3.13 (m, 3H), 2.80-2.74 (m, 1H), 2.48-2.44 (m, 1H), 2.21-2.08 (m, 1H), 1.63 (s, 3H), 1.49 (s, 3H);13C NMR (75 MHz, CDCl 3, DEPT) δ 135.04 (C), 134.31 (C), 131.11 (2CH), 130.79 (2CH), 128.83 (2CH), 128.75 (2CH), 127.00 (CH), 126.79 (CH), 120.12 (C), 116.19 (C), 89.76 (CH), 88.22 (CH), 61.56 (CH), 60.07 (CH), 52.47 (CH), 49.47 (CH), 33.44 (CH2), 26.33 (CH3), 24.03 (CH3); LRMS m/z (rel inten)
412 (M+, 18), 303 (100); HRMS (EI) calcd for C
23H24O3S2
412.1167, found 412.1175.
General Procedure for the Reaction of Tetraoxa-Cages 1a-c with Methanol in the Presence of TiCl4. To a solution of 1a (0.21 g, 1.00 mmol) in dichloromethane (20 mL) were added MeOH (0.13 g, 4.0 mmol) and TiCl4(0.020 g,
0.10 mmol) at 25 °C. The reaction mixture was stirred at 25 °C for 2 h. The reaction mixture was quenched by addition of water (10 mL) and extracted with dichloromethane (3× 20 mL). The organic layer was washed with brine, dried over MgSO4, and evaporated, and the residue was purified by
column chromatography to give 14a (0.21 g, 82%).
2β,8β-Dimethoxy-4,6-dimethyl-3,5,7-trioxatetracyclo-[7.2.1.04,11.06,10]dodecane (14a): white waxy solid; yield 82%;
mp 81-82 °C; IR (CHCl3) 2980, 2880, 1380, 1070 cm -1;1H NMR (300 MHz, CDCl3) δ 4.90 (d, J)1.5 Hz, 2H), 3.35 (s, 6H), 3.22 (dd, J)5.7 Hz, J)3.0 Hz, 2H), 2.75-2.70 (m, 2H), 2.32-2.24 (m, 1H), 2.08-1.97 (m, 1H), 1.57 (s, 6H); 13C NMR (75 MHz, CDCl3, DEPT) δ 119.66 (2C), 111.06 (2CH), 59.69 (2CH), 55.00 (2CH3), 52.70 (2CH), 36.18 (CH2), 27.41 (2CH3); LRMS m/z (rel inten) 256 (M+ , 55), 241 (100); HRMS (EI) calcd for C13H20O5 256.1311, found 256.1307. Anal. Calcd for
C13H20O5: C, 60.91; H, 7.87. Found: C, 60.82; H, 7.95. 2β,8β-Dimethoxy-4-methyl-6-benzyl-3,5,7-trioxatet-racyclo[7.2.1.04,11.06,10]dodecane (14b): white waxy solid;
yield 85%; mp 89-90 °C; IR (CHCl3) 2980, 1600, 1380, 1070, 745, 695 cm-1;1H NMR (300 MHz, CDCl 3) δ 7.33-7.22 (m, 5H), 4.95 (d, J)1.5 Hz, 1H), 4.87 (d, J)1.5 Hz, 1H), 3.41 (s, 3H), 3.31 (s, 3H), 3.27 (dd, J)8.7 Hz, J)8.2 Hz, 1H), 3.22, 2.95 (ABq, J)13.8 Hz, 2H), 2.84 (dd, J)8.7 Hz, J)8.5 Hz, 1H), 2.69-2.62 (m, 2H), 2.29-2.17 (m, 1H), 2.00-1.94 (m, 1H), 1.31 (s, 3H);13C NMR (75 MHz, CDCl 3, DEPT) δ 136.47 (C), 130.73 (2CH), 127.73 (2CH), 126.42 (CH), 121.03 (C), 119.80 (C), 111.15 (CH), 110.92 (CH), 59.26 (CH), 56.98 (CH), 55.41 (CH3), 54.80 (CH3), 52.44 (2CH), 45.42 (CH2), 36.06 (CH2), 26.36 (CH3); LRMS m/z (rel inten) 332 (M+ , 100); HRMS (EI) calcd for C19H24O5332.1624, found 332.1608. Anal. Calcd for
C19H24O5: C, 68.64; H, 7.28. Found: C, 68.72; H, 7.22. 2β,8β-Dimethoxy-3,5,7-trioxatetracyclo[7.2.1.04,11.06,10
]-dodecane (14c): white waxy solid; yield 80%; mp 65-66 °C; IR (CHCl3) 2980, 2880, 1380, 1070 cm-1 ;1H NMR (300 MHz, CDCl3) δ 5.91 (d, J)5.1 Hz, 2H), 4.94 (d, J)2.1 Hz, 2H), 3.48-3.40 (m, 2H), 3.37 (s, 6H), 2.72-2.66 (m, 2H), 2.42-2.30 (m, 1H), 2.02-1.96 (m, 1H); 13C NMR (75 MHz, CDCl 3, DEPT) δ 111.86 (2CH), 111.45 (2CH), 55.30 (2CH3), 55.03 (2CH), 51.36 (2CH), 35.92 (CH2); LRMS m/z (rel inten) 228 (M+ , 66), 213 (100); HRMS (EI) calcd for C11H16O5228.0998, found 228.0991.
Anal. Calcd for C11H16O5: C, 57.87; H, 7.07. Found: C, 57.75;
H, 7.02.
General Procedure for the Reaction of Tetraoxa-Cages 1a-c with m-Chloroperoxybenzoic Acid in the
Presence of BF3‚OEt2. To a solution of 1a (0.21 g, 1,00 mmol) in dichloromethane (20 mL) were added m-CPBA (0.69 g, 4.0 mmol) and BF3‚OEt2(0.010 g, 0.10 mmol) at 25 °C. The reaction mixture was stirred at 25 °C for 1 h. The reaction mixture was quenched by addition of saturated sodium carbonate (20 mL) and extracted with dichloromethane (3× 20 mL). The organic layer was washed with brine, dried over MgSO4, and evaporated, and the residue was purified by
column chromatography to give 15a (0.19 g, 0.85 mmol) in 85% yield. Compounds 15a and 15b were obtained via a different approach, and their spectral data were reported.4a On the
other hand, 15c is a new compound.
2,8-Dioxo-3,5,7-trioxatetracyclo[7.2.1.04,11.06,10
]do-decane (15c): white waxy solid; yield 85%; mp 255-256 °C; IR (CHCl3) 2960, 1767, 1240, 1107 cm-1 ;1H NMR (300 MHz, CD3COCD3) δ 6.19 (d, J)5.7 Hz, 2H), 4.04 (brs, 2H), 3.34 (brs, 2H), 2.67 (brs, 2H); 13C NMR (75 MHz, CD 3COCD3, DEPT) δ 177.26 (2CO), 108.03 (2CH), 52.70 (2CH), 46.70 (2CH), 37.93 (CH2); LRMS m/z (rel inten) 196 (M+ , 5), 97 (78), 152 (100); HRMS (EI) calcd for C9H8O5 196.0372, found
196.0378. Anal. Calcd for C9H8O5: C, 55.11; H, 4.11.
Found: C, 55.25; H, 4.19.
General Procedure for the Hydride Rearrangement of Tetraoxa-Cages 1a,c, 20, and 22a-c. To a solution of
1a (0.21 g, 1.00 mmol) in dichloromethane (40 mL) was added
TiCl4(0.38 g, 2.00 mmol) or BF3‚OEt2(0.20 g, 2.00 mmol) or MeSO3H (0.19 g, 2.00 mmol) at 25 °C. The reaction mixture
was stirred at 25 °C for 12 h. The reaction mixture was quenched by addition of water (30 mL) and extracted with dichloromethane (3× 20 mL). The organic layer was washed with brine, dried over MgSO4, and evaporated, and the residue
was purified by column chromatography to give the hydride rearrangement product 16a (0.19 g, 0.90 mmol) in 90% yield.
2-Oxo-4,6-dimethyl-3,5,7-trioxatetracyclo[7.2.1.04,11.06,10
]-dodecane (16a): white waxy solid; yield 90%; mp 146-147 °C; IR (CHCl3) 2980, 1770, 1240, 1070 cm-1;1H NMR (300 MHz, CDCl3) δ 4.08 (dd, J)9.3 Hz, J)9.3 Hz, 1H), 3.61 (dd, J)9.3 Hz, J)8.7 Hz, 1H), 3.40-3.28 (m, 3H), 3.00-2.93 (m, 1H), 2.37-2.25 (m, 2H), 1.67 (s, 3H), 1.57 (s, 3H); 13C NMR (75 MHz, CDCl3, DEPT) δ 178.54 (CO), 119.39 (C), 115.14 (C), 72.13 (CH2), 61.23 (CH), 55.82 (CH), 48.39 (CH), 45.30 (CH), 35.86 (CH2), 26.13 (CH3), 24.53 (CH3); LRMS m/z (rel inten) 210 (M+
, 16), 166 (100); HRMS (EI) calcd for C11H14O4
210.0892, found 210.0898. Anal. Calcd for C11H14O4: C, 62.83;
H, 6.72. Found: C, 62.94; H, 6.79.
2-Oxo-3,5,7-trioxatetracyclo[7.2.1.04,11.06,10]dodecane
(16b): white waxy solid; yield 90%; mp 112-113 °C; IR (CHCl3) 2980, 1770, 1240, 1070 cm-1 ;1H NMR (300 MHz, CDCl 3) δ 6.04 (d, J)5.7 Hz, 1H), 5.97 (d, J)5.4 Hz, 1H), 4.13 (dd, J )9.6 Hz, J)9.0 Hz, 1H), 3.63-3.58 (m, 2H), 3.50 (dd, J) 9.6 Hz, J) 8.4 Hz, 1H), 3.27-3.21 (m, 1H), 2.96-2.89 (m, 1H), 2.42-2.25 (m, 2H); 13C NMR (75 MHz, CDCl 3, DEPT) δ 179.09 (CO), 112.31 (CH), 106.38 (CH), 71.60 (CH2), 56.72 (CH), 50.72 (CH), 46.88 (CH), 44.85 (CH), 35.35 (CH2); LRMS m/z (rel inten) 182 (M+, 78), 138 (100); HRMS (EI) calcd for
C9H10O4182.0579, found 182.0588. Anal. Calcd for C9H10O4:
2-Oxo-4,6-dimethyl-12-spiroethylene-3,5,7-trioxatet-racyclo[7.2.1.04,11.06,10]dodecane (21): white waxy solid;
yield 90%; mp 155-156 °C; IR (CHCl3) 2980, 1770, 1250, 1070 cm-1;1H NMR (300 MHz, CDCl 3) δ 3.93 (dd, J)9.9 Hz, J) 9.9 Hz, 1H), 3.79 (dd, J)9.9 Hz, J)8.4 Hz, 1H), 3.51-3.35 (m, 2H), 2.51 (d, J)9.3 Hz, 1H), 2.36-2.27 (m, 1H), 1.60 (s, 3H), 1.52 (s, 3H), 1.37-1.31 (m, 1H), 0.61-0.50 (m, 3H); 13C NMR (75 MHz, CDCl3, DEPT) δ 176.74 (CO), 119.86 (C), 114.79 (C), 71.52 (CH2), 59.90 (CH), 55.70 (CH), 53.78 (CH), 53.43 (CH), 30.56 (C), 26.19 (CH3), 24.38 (CH3), 16.22 (CH2), 4.01 (CH2); LRMS m/z (rel inten) 236 (M+ , 82), 192 (100); HRMS (EI) calcd for C13H16O4236.1049, found 236.1041. Anal.
Calcd for C13H16O4: C, 66.07; H, 6.83. Found: C, 66.18; H,
6.89.
2β-Methyl-8-oxo-3,5,7-trioxatetracyclo[7.2.1.04,11.06,10
]-dodecane (23a): white waxy solid; yield 85%; mp 141-142 °C: IR (CHCl3) 2980, 1770, 1240, 1070 cm-1 ;1H NMR (300 MHz, CDCl3) δ 6.03 (d, J)6.0 Hz, 1H), 5.95 (d, J)5.4 Hz, 1H), 3.80-3.74 (m, 1H), 3.66-3.60 (m, 2H), 3.27-3.22 (m, 1H), 2.39-2.27 (m, 3H), 1.28 (d, J)6.0 Hz, 3H); 13C NMR (75 MHz, CDCl3, DEPT) δ 179.28 (CO), 111.28 (CH), 106.21 (CH), 78.92 (CH), 57.19 (CH), 52.59 (CH), 50.59 (CH), 47.04 (CH), 34.87 (CH2), 19.55 (CH3); LRMS m/z (rel inten) 196 (M+, 43), 152
(100); HRMS (EI) calcd for C10H12O4196.0736, found 196.0730.
Anal. Calcd for C10H12O4: C, 61.20; H, 6.17. Found: C, 61.28;
H, 6.25.
2β-Isopropyl-4,6-dimethyl-8-oxo-3,5,7-trioxatetracyclo-[7.2.1.04,11.06,10]dodecane (23b): white waxy solid; yield 90%;
mp 120-121 °C; IR (CHCl3) 2980, 1770, 1380, 1240, 1070 cm -1 ; 1H NMR (300 MHz, CDCl 3) δ 3.51 (dd, J)8.7 Hz, J)6.6 Hz, 1H), 3.36-3.24 (m, 3H), 2.65-2.57 (m, 1H), 2.38-2.20 (m, 2H), 1.80-1.68 (m, 1H), 1.65 (s, 3H), 1.57 (s, 3H), 0.95 (d, J)6.6 Hz, 3H), 0.91 (d, J)6.6 Hz, 3H); 13C NMR (75 MHz, CDCl 3, DEPT) δ 178.86 (CO), 118.46 (C), 114.91 (C), 88.39 (CH), 62.23 (CH), 55.93 (CH), 49.12 (CH), 48.45 (CH), 36.41 (CH2), 33.03 (CH), 26.42 (CH3), 25.81 (CH3), 18.81 (CH3), 18.12 (CH3);
LRMS m/z (rel inten) 252 (M+, 47), 208 (100); HRMS (EI) calcd
for C14H20O4 252.1362, found 252.1368. Anal. Calcd for
C14H20O4: C, 66.63; H, 7.99. Found: C, 66.75; H, 7.95. 2β-Benzyl-4,6-dimethyl-8-oxo-3,5,7-trioxatetracyclo-[7.2.1.04,11.06,10]dodecane (23c): white waxy solid; yield 90%;
mp 112-113 °C; IR (CHCl3) 2980, 1770, 1600, 1240, 1070, 740, 700 cm-1 ;1H NMR (300 MHz, CDCl 3) δ 7.34-7.18 (m, 5H), 4.05-4.00 (m, 1H), 3.34-2.98 (m, 4H), 2.80-2.50 (m, 2H), 2.05-1.78 (m, 2H), 1.63 (s, 3H), 1.58 (s, 3H); 13C NMR (75 MHz, CDCl3, DEPT) δ 178.60 (CO), 136.64 (C), 129.48 (2CH), 128.34 (2CH), 126.53 (CH), 118.37 (C), 114.82 (C), 83.12 (CH), 61.94 (CH), 55.61 (CH), 51.16 (CH), 48.42 (CH), 40.64 (CH2), 35.10 (CH2), 26.51 (CH3), 25.69 (CH3); LRMS m/z (rel inten)
300 (M+, 45), 256 (100); HRMS (EI) calcd for C 18H20O4
300.1362, found 300.1366. Anal. Calcd for C18H20O4: C, 71.97;
H, 6.72. Found: C, 71.90; H, 6.78.
General Procedure for the Reduction of Compounds 23a,c 16a,b and 21 with Diisobutylaluminum Hydride (DIBAL-H). To a solution of 16a (0.42 g, 2.00 mmol) in dry
THF (40 mL) was added DIBAL-H (1.70 g, 2.40 mmol, 20% in
n-hexane) at 0 °C. The reaction mixture was stirred at 0 °C
for 0.5 h. The reaction mixture was quenched by slow addition of water (30 mL) at 0 °C and extracted with ether (5× 30 mL). The organic layer was washed with saturated sodium bicarbonate and brine, dried over MgSO4, and evaporated, and
the residue was purified by column chromatography to give the reduction product 24c (0.38 g, 1.8 mmol) in 90% yield.
2 β M e t h y l 8β h y d r o x y 3 , 5 , 7 t r i o x a t e t r a c y c l o -[7.2.1.04,11.06,10]dodecane (24a): white waxy solid; yield 90%;
mp 95-96 °C; IR (CHCl3) 3500-3300, 2970, 1110, 1070 cm -1 ; 1H NMR (300 MHz, CDCl 3) δ 5.93 (d, J)5.4 Hz, 1H), 5.91 (d, J)5.4 Hz, 1H), 5.51 (d, J)1.5 Hz, 1H), 4.09-4.00 (m, 1H), 3.89 (brs, 1H), 3.50-3.44 (m, 2H), 2.84-2.78 (m, 1H), 2.34 -2.15 (m, 2H), 1.94-1.89 (m, 1H), 1.25 (d, J)6.0 Hz, 3H); 13C NMR (75 MHz, CDCl3, DEPT) δ 112.30 (CH), 110.69 (CH), 106.11 (CH), 79.30 (CH), 57.87 (CH), 54.25 (CH), 53.21 (CH), 52.50 (CH), 34.86 (CH2), 19.65 (CH3); LRMS m/z (rel inten)
198 (M+, 36), 181 (100); HRMS (EI) calcd for C 10H14O4
198.0892, found 198.0898. Anal. Calcd for C10H14O4: C, 60.58;
H, 7.12. Found: C, 60.65; H, 7.18.
2β-Benzyl-8β-hydroxy-4,6-dimethyl-3,5,7-trioxa-tetracyclo[7.2.1.04,11.06,10]dodecane (24b): white waxy solid;
yield 90%; mp 82-83 °C; IR (CHCl3) 3500-3300, 2970, 1600, 1070, 750, 695 cm-1 ;1H NMR (300 MHz, CDCl 3) δ 7.32-7.20 (m, 5H), 5.32 (s, 1H), 4.26-4.20 (m, 1H), 3.25-3.04 (m, 4H), 2.75-2.63 (m, 2H), 2.53-2.42 (m, 1H), 1.92-1.80 (m, 1H), 1.60 (s, 3H), 1.55 (s, 3H), 1.30-1.26 (m, 1H); 13C NMR (75 MHz, CDCl3, DEPT) δ 137.60 (C), 129.39 (2CH), 128.28 (2CH), 126.27 (CH), 119.10 (C), 118.40 (C), 105.82 (CH), 83.44 (CH), 62.87 (CH), 58.70 (CH), 53.66 (CH), 52.26 (CH), 41.05 (CH2), 35.28 (CH2), 28.28 (CH3), 25.92 (CH3); LRMS m/z (rel inten) 302 (M+
, 52), 285 (100); HRMS (EI) calcd for C18H22O4
302.1518, found 302.1523. Anal. Calcd for C18H22O4: C, 71.49;
H, 7.34. Found: C, 71.58; H, 7.40.
2β-Hydroxy-4,6-dimethyl-3,5,7-trioxatetracyclo-[7.2.1.04,11.06,10]dodecane (24c): white waxy solid; yield 90%;
mp 103-104 °C; IR (CHCl3) 3500-3300, 2970, 1070 cm -1 ;1H NMR (300 MHz, CHCl3) δ 5.50 (s, 1H), 4.00 (dd, J)9.0 Hz, J)7.8 Hz, 1H), 3.81 (dd, J)9.0 Hz, J)8.7 Hz, 1H), 3.46 (brs, 1H), 3.26-3.06 (m, 2H), 2.92-2.80 (m, 2H), 2.26-2.12 (m, 1H), 1.94-1.89 (m, 1H), 1.63 (s, 3H), 1.53 (s, 3H); 13C NMR (75 MHz, CDCl3, DEPT) δ 119.41 (C), 119.18 (C), 105.65 (CH), 72.44 (CH2), 62.05 (CH), 59.00 (CH), 54.08 (CH), 46.38 (CH), 35.60 (CH2), 28.19 (CH3), 25.27 (CH3); LRMS m/z (rel inten) 212 (M+
, 41), 195 (100); HRMS (EI) calcd for C11H16O4
212.1049, found 212.1063. Anal. Calcd for C11H16O4: C, 62.23;
H, 7.60. Found: C, 62.35; H, 7.68.
2β-Hydroxy-3,5,7-Trioxatetracyclo[7.2.1.04,11.06,10
]do-decane (24d): white waxy solid; yield 85%; mp 121-122 °C; IR (CHCl3) 3500-3300, 2970, 1070 cm -1;1H NMR (300 MHz, CDCl3) δ 5.94 (d, J)5.4 Hz, 1H), 5.90 (d, J)5.4 Hz, 1H), 5.51 (s, 1H), 4.02 (ddd, J)10.2 Hz, J)9.0 Hz, J)1.2 Hz, 1H), 3.75 (dd, J)10.2 Hz, J)7.2 Hz, 1H), 3.61 (brs, 1H), 3.52-3.38 (m, 2H), 2.86-2.77 (m, 2H), 2.28-2.20 (m, 1H), 1.98-1.93 (m, 1H); 13C NMR (75 MHz, CDCl 3, DEPT) δ 113.01 (CH), 110.81 (CH), 105.83 (CH), 72.34 (CH2), 57.17 (CH), 54.33 (CH), 52.59 (CH), 45.23 (CH), 35.43 (CH2); LRMS m/z (rel inten) 184 (M+
, 61), 167 (100); HRMS (EI) calcd for C9H12O4
184.0736, found 184.0732. Anal. Calcd for C9H12O4: C, 58.67;
H, 6.57. Found: C, 58.79; H, 6.75.
2β-Hydroxy-4,6-dimethyl-12-spiroethylene-3,5,7-tri-oxatetracyclo[7.2.1.04,11.06,10]dodecane (25): white waxy
solid; yield 85%; mp 89-90 °C; IR (CHCl3) 3500-3300, 2970, 1070 cm-1;1H NMR (300 MHz, CDCl 3) δ 5.58 (s, 1H), 3.95 (dd, J)9.0 Hz, J)8.7 Hz, 1H), 3.83 (dd, J)9.0 Hz, J)8.1 Hz, 1H), 3.44 (brs, 1H), 3.31 (dd, J)10.5 Hz, J)8.7 Hz, 1H), 3.20 (dd, J)10.5 Hz, J)9.3 Hz, 1H), 2.28-2.19 (m, 1H), 2.15 (d, J)8.7 Hz, 1H), 1.56 (s, 3H), 1.46 (s, 3H), 0.87-0.68 (m, 2H), 0.58-0.45 (m, 2H); 13C NMR (75 MHz, CDCl 3, DEPT) δ 119.37(C), 118.99 (C), 104.30 (CH), 71.41 (CH2), 61.33 (CH), 60.74 (CH), 58.76 (CH), 54.19 (CH), 29.54 (C), 27.76 (CH3), 25.05 (CH3), 17.79 (CH2), 5.47 (CH2); LRMS m/z (rel inten) 238
(M+, 61), 221 (100); HRMS (EI) calcd for C
13H18O4238.1205,
found 238.1202. Anal. Calcd for C13H18O4: C, 65.51; H, 7.62.
Found: C, 65.43; H, 7.68.
Acknowledgment. We thank the National Science
Council of the Republic of China for financial support
(Grant No. NSC85-2113-M009-004), and Miss F. L. Liao
and Dr. S. L. Wang at the Department of Chemistry,
National Tsing Hua University, for carrying out the
X-ray crystallographic analysis.
Supporting Information Available:1H and 13C NMR
spectra of 2a, 6a, 7a, 14a, and 16a (10 pages). This material is contained in libraries on microfiche, immediately follows this article in the microfilm version of the journal, and can be ordered from the ACS; see any current masthead page for ordering information.