A New Trans-to-Cis Photoisomerization Mechanism of Azobenzene on the S
1(n,
π*) Surface
Eric Wei-Guang DiauDepartment of Applied Chemistry and Center for Interdisciplinary Molecular Science, National Chiao Tung UniVersity, Hsinchu, Taiwan 30050, Republic of China
ReceiVed: October 9, 2003
The rotation-inversion controversy for the photoisomerization mechanism of azobenzene has been addressed on the basis of the CASSCF calculations for the excited-state relaxed surface scan along the CNN bending (inversion), the CNNC torsion (rotation), and the concerted CNN bending (concerted inversion) reaction pathways. According to our calculated results, the inversion channel involves a substantial energy barrier and a large S0-S1energy gap so that it is a highly unfavorable channel to be considered for an efficient electronic relaxation. The rotation channel is essentially barrierless with a conical intersection close to the midpoint of the pathway to reasonably account for the subpicoseond to picosecond relaxation times observed in recent ultrafast experiments. Along the concerted inversion reaction path, a sloped conical intersection (S0/S1CI_inv) was found to have the geometry close to a linear CNNC configuration. When the photoexcitation occurs in the S2state, the concerted inversion channel may be open and the S0/S1CI_inv would become energetically accessible to produce more trans-S0isomers. A new mechanism is proposed to rationalize the early experimental results for the following two cases: (1) the trans-to-cis photoisomerization quantum yields on S2excitation are much lower than the yields on S1excitation; (2) the yields from both the S2and the S1excitations become essentially equal when the rotation channel is blocked by chemical modification or in an inclusion complex.
1. Introduction
The mechanism of the photoisomerization of trans-azoben-zene has been a fundamental subject for many years not only for its potential applications in industry1-7but also for its long-standing debate on the isomerization process occurring along either the inversional or the rotational pathway (Scheme 1). In solutions, the UV-visible spectra of the molecule are featured with two major absorption bands corresponding to the S0f S1 and the S0f S2transitions. The former transition relates to a symmetry-forbidden n-π* transition with the maximum inten-sity near 440 nm whereas the latter transition corresponds to a symmetry-allowedπ-π* transition with the maximum intensity near 320 nm.7,8Early steady-state measurements show that the quantum yields of the trans-to-cis isomerization are wavelength-dependent: The yield measured in hexane solvent is 0.20-0.27 upon excitation to the S1(n,π*) state but it drops dramatically to 0.09-0.12 for the S2(π,π*) excitation.7-9 However, the quantum yields become almost wavelength-independent when rotation about the NN double bond is restricted by blocking the rotation with a cyclophane structure,10with a crown ether,11 or with the inclusion in the cyclodextrin cavity.12 These observations imply that two different isomerization mechanisms are operated on excitation into these two electronic excited states; i.e., visible excitation to the S1(n,π*) state leads to the isomerization via inversion around one nitrogen atom in the same molecular plane whereas UV excitation to the S2(π,π*) state results in the isomerization via the rotation around the NN double bond.7This conclusion is supported by early ab initio work of Monti et al.13based on minimal basis set CI calculations and has been widely adopted in recent time-resolved studies using the technique of femtosecond transient absorption spectroscopy.14-18
Moreover, recent time-resolved Raman study on the S2(π,π*) excitation has demonstrated the similarity of the NN stretch
frequency between the S1and the S0states and suggested that the NN bonding retains a double bond character and thus the S1species has a planar structure.19This evidence partly supports the previous conclusion that the isomerization of azobenzene on the S1surface is via an inversion mechanism. However, the subsequent time-resolved fluorescence studies20,21of the same laboratory have made a conclusion that the rotational isomer-ization mechanism starting directly from the S2state does not exist and the isomerization of azobenzene takes place exclusively on the S1surface by an in-plane inversion mechanism regardless of the initial excitations. Apparently, such a consecutive model (S2f S1f S0via inversion only) cannot account for the early spectroscopic data. To rationalize for the smaller isomerization quantum yield previously observed from the S2 excitation, a new relaxation channel in the vibrationally hot S1state that is open to form only the trans isomer has been proposed.20,21
On the other hand, the aforementioned inversion photo-isomerization mechanisms have been challenged by recent high-SCHEME 1
10.1021/jp031149a CCC: $27.50 © 2004 American Chemical Society Published on Web 01/08/2004
level theoretical calculations. Based on the multiconfiguration self-consistent field (MCSCF) approach using the complete active space (CAS) SCF wave functions, both recent ab initio results22,23have shown that the S1potential energy surface (PES) of trans-azobenzene involves substantial energy barrier along the inversion coordinate, but the surface is essentially barrierless along the rotation coordinate. Furthermore, the results of Ishikawa et al.23also indicate that a conical intersection (CI) between the S0 and the S1 states may be located near the midpoint of the rotation pathway, suggesting that the photo-isomerization of azobenzene on the S1 surface may favor a rotation channel. The rotation mechanism reported for azoben-zene is also consistent with the topological feature of azomethane on the S1surface.24
In a previous study,25 we reported on the femtosecond fluorescence dynamics of trans-azobenzene in hexane with excitation directly into the S1(n,π*) state. The observed fluorescence temporal profiles feature two components and our assignments are based on the rotation mechanism supported by recent ab initio calculations:22,23The fast-decay component is due to a structural relaxation from the Franck-Condon region toward the rotational reaction path and the slow-decay compo-nent is arisen from the other nuclear motions along the rotation coordinate to search for the S0/S1CI on the multidimensional PES. However, such an energy-favorable rotational isomeriza-tion channel in azobenzene is not feasible for the rotaisomeriza-tion- rotation-blocked azobenzene derivatives and a new relaxation channel must be open to account for the observed quantum yield discrepancies previously reported.7To shed light on the issue for the rotation-inversion controversy, we have carried out CASSCF calculations for azobenzene to follow the minimum-energy pathways on the S1surface along three different reaction coordinatessthe CNN bending coordinate, the CNNC torsional coordinate, and the concerted CNN bending coordinate. Our results on the CNN bending angle scan and the CNNC torsional angle scan are similar to those of recent studies,22,23but we have found a new conical intersection along the concerted CNN bending coordinate responsible for the new relaxation channel to rationalize the old quantum yield results. A new mechanism is presented in this paper to provide a solution for the long-standing controversy on the subject of photoisomerization of azobenzene.
2. Method of Calculations
Our strategy for the CASSCF calculations is aimed at finding a proper active space that is capable of describing the trans-to-cis isomerization processes of azobenzene on the S1PES. According to the previous calculations of azomethane,24 the active space should at least include six electrons distributed among four orbitals (n-,π, n+, andπ*), abbreviated by CAS-(6,4), to reasonably describe the PES along the rotational isomerization coordinate. Because there are some delocalized π* and σ* orbitals that are relevant to better describe the PES of azobenzene along the three coordinates of interest, we have included two more virtual orbitals (either twoπ* for the rotation channel orπ* + σ* for the inversion channels) in the active space for the calculations at the CAS(6,6) level. The selected orbitals in the active space are 3a + 3b for the CNNC torsional angle scan (C2), 3a′ + 3a′′for the CNN bending angle scan (Cs), and ag+ au+ 2bg+ 2bufor the concerted CNN bending angle scan (C2h).
The geometry of each point along three different reaction coordinates was optimized according to a relaxed surface scan procedure at the state-specific CAS(6,6)/4-31G level whereas
the geometry of a conical intersection between the S0and the S1states was fully optimized at the state-averaged CAS(6,6)/ 4-31G level. For all the stationary points optimized on the potential energy surfaces, force constant calculations were performed at the CAS(6,6)/4-31G level, the same as geometry optimization, and single point energy calculations were further carried out at the CAS(6,6)/cc-pVDZ level of theory. All the electronic structure calculations were performed using the Gaussian software package.26
Because trans-azobenzene does not have a permanent dipole moment, the effect of the solvents affecting the PES is not expected to be significant. In fact, we have checked such an effect on the basis of the time-dependent density functional theory (TDDFT) calculations with the polarizable continuum model (PCM) implemented in Gaussian 03. In comparison with the isolated molecule in the gas phase, the vertical excitation energy of azobenzene is lower by only 0.01 and 0.05 eV in hexane and DMSO, respectively, predicted at the TD-B3LYP/ 6-311++G(d,p) level of theory. The solvation energies of trans-azobenzene (<0.05 eV) were computed to be much smaller than the uncertainties of the excitation energies (typically∼0.2 eV; see below) from the theoretical calculations currently employed. Therefore, all the surface-scan calculations reported in this article were carried out in the gas-phase environment without consider-ing the negligible solvent effect.
3. Results and Discussions
Three minimum-energy pathways on the S1PES of azoben-zene were characterized by optimizing the geometries along the CNN bending coordinate (the inversion channel), the CNNC torsional coordinate (the rotation channel), and the concerted CNN bending coordinate (the concerted inversion channel); the corresponding potential energy curves are shown in Figures 1-3, respectively. The key structural parameters and the relative energies of the relevant stationary points and conical intersec-tions are summarized in Table 1. There are two major points worth mentioning to justify the accuracy of our calculations. First, we found that the calculated S1excitation energy at the Franck-Condon geometry (S1•FC) is 59.6 kcal mol-1 at the CAS(6,6)/4-31G level, and it slightly increases to 63.4 kcal mol-1with further single-point energy calculation at the CAS-(6,6)/cc-pVDZ level of theory. Note that the observed absorption spectrum of trans-azobenzene has the maximum intensity at 450 nm ()63.5 kcal mol-1 in energy), which is in excellent agreement with our theoretical prediction at the CAS(6,6)/cc-pVDZ level. Second, the S1minimum, S1(C2), was calculated to be 47.7 kcal mol-1in energy above the S0minimum at the level of geometry optimization.27Further single-point energy calculation at the CAS(6,6)/cc-pVDZ level gives the relative energy of S1(C2) with respect to S0(C2h) being 50.1 kcal mol-1, which is again in excellent agreement with the experimental observation that the red edge of the S1absorption band ends at
∼560 nm (∼51 kcal mol-1 in energy). Therefore, we expect that the energetic uncertainties for the results of the surface-scan calculations shown in Figures 1-3 are within ∼4 kcal mol-1(or∼0.2 eV). The results and their dynamical significance for the photoisomerization of azobenzene are discussed in the following.
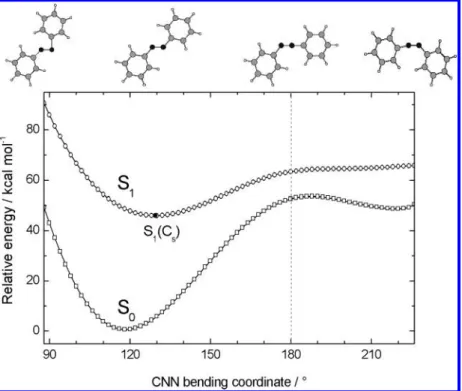
3.1. Inversion Channel. The potential energy curves shown in Figure 1 were obtained from scanning one of the CNN bending reaction coordinate (RC) when optimizing the other structural parameters on the S1PES with Cssymmetry. The S1
potential energy significantly increases along the CNN bending RC toward the smaller bending angle direction. On the other Trans-to-Cis Photoisomerization of Azobenzene J. Phys. Chem. A, Vol. 108, No. 6, 2004 951
hand, following the in-plane CNN bending RC toward the larger bending angle direction, the S1potential energy curve also goes uphill and reaches a plateau area at the RC greater than 180°. The results are similar to those of Cattaneo and Persico22and Ishikawa et al.23and this topological picture may provide a theoretical description for the inversion mechanism shown in Scheme 1.
The minimum-energy point of the S1curve, S1(Cs), is located
above the ground-state minimum by 45.0 kcal mol-1(with ZPE corrections) at 129.4°;27the other CNN bending angle is 127.8°.
Following the in-plane CNN bending reaction path from S1-(Cs) toward the larger bending angle direction, the S1and the
S0potential energy curves do come closer but there are two factors affecting the electronic relaxation through this channel. First, the energy difference between the S1(Cs) and the S1species
at 180°, S1•180(Cs), is close to ∼20 kcal mol-1, which is a
substantial energy barrier to overcome along this reaction path. Second, the energy gap between the two states is larger than 10 kcal mol-1at the bending angles greater than 180°(toward the cis isomer). In fact, a state-averaged calculation at the
CAS-Figure 1. Minimum-energy pathway of azobenzene along the CNN bending coordinate optimized for the second root (the S1state) at the
state-specific CAS(6,6)/4-31G level of theory. On top, the corresponding optimized structures are shown from left to right for the bending angles of 90°, 130°, 180°, and 226°, respectively.
Figure 2. Minimum-energy pathway of azobenzene along the CNNC torsional coordinate optimized for the second root (the S1 state) at the
state-specific CAS(6,6)/4-31G level of theory. On top, the corresponding optimized structures are shown from left to right for the torsional angles of 180°, 90°, and 0°, respectively. The optimized structure of the S0/S1CI•rot is shown in the inset.
(6,6)/cc-pVDZ level indicates that the energy difference between the two states is 21.2 kcal mol-1at this semilinear geometry. Such a large energy gap between the S0 and the S1states is inconsistent with recent real-time observations that the electronic relaxation of azobenzene on the S1 surface occurs on the subpicosecond to picosecond time scale.14-21,25 Furthermore, the force constant calculation at the optimized semilinear geometry (Table 1) gives three imaginary frequencies, indicating that the semilinear azobenzene corresponds to a high-order saddle point on the S1surface along the inversional pathway. This is another theoretical evidence for the photoisomerization of azobenzene following the in-plane inversion channel to be highly unfavorable.
Fujino and Tahara have recently reported on the NN stretch frequency observed in the transient Raman spectra to be close
to that of the ground state (1428 cm-1vs 1440 cm-1).19This observation leads to a conclusion for the excited-state species having a double bond character that supports an inversion mechanism for photoisomerization of azobenzene. However, the NN bond order throughout the entire inversional pathway was calculated to be∼1.0,23which is inconsistent with the conclu-sion of the time-resolved Raman study.19Our results support the argument of Ishikawa et al.23 but disagree with the conclusion of Tahara and co-workers.19-21
3.2. Rotation Channel. The potential energy curves shown in Figure 2 were obtained from scanning the CNNC torsional angle when the other structural parameters were optimized on the S1PES with C2symmetry. Once again, the results are similar to those of Cattaneo and Persico22and Ishikawa et al.23 and this topological picture may provide a theoretical description
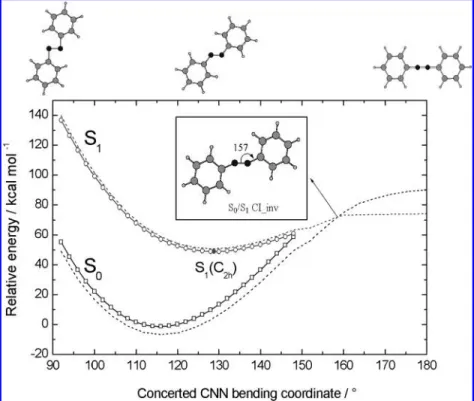
Figure 3. Minimum-energy pathway of azobenzene along the concerted CNN bending coordinate optimized for the second root (the S1state) at
the state-specific CAS(6,6)/4-31G level of theory. On top, the corresponding optimized structures are shown from left to right for the two equivalent bending angles equal to 92°, 128°, and 180°, respectively. The optimized structure of the S0/S1CI•inv is shown in the inset. The dashed curves
are the corresponding state-averaged results for the S0and the S1states.
TABLE 1: CASSCF-Optimized Structural Parameters and High-Correlated Energies of Relevant Stationary Points on the S0
and the S1PESs of Azobenzenea
distance/Å angle/deg relative energyb/kcal mol-1 species
Vimaginary/ i cm-1
ZPE/
kcal mol-1 r(NN) r(CN) θ(CNN) δ(CNNC) φ(NNCC) ∆E(4-31G)c ∆E(pVDZ)d ∆E(exp)e
S0(C2h),1Ag f 130.1 1.253 1.428 116.8 180.0 0.0 0.0 0.0 0.0 S1•FC g 130.1h 1.253 1.428 116.8 180.0 0.0 59.6 63.4 63.5 S1(C2h),1Bg 50(bu), 22(au) 128.2 1.270 1.384 128.7 180.0 0.0 47.1 51.1 S1(C2),1B f 129.1 1.263 1.398 127.9 128.5 -5.7 47.7 50.1 ∼50 S1(Cs),1A′′ 21(a′′) 129.1 1.269 1.387 129.4 180.0 0.0 45.0 50.7 1.390 127.8 0.0
S1•180(Cs),1A′′ 290(a′′), 227(a′), 17(a′′) 127.7 1.249 1.400 127.3 i 0.0 61.0 68.1
1.362 180.0 i
S0/S1CI•rot g 129.1j 1.291 1.391 129.2 92.1 -0.4 42.2 47.8
1.421 120.2 0.9
S0/S1CI•inv g 128.2k 1.211 1.349 157.3 180.0 0.0 68.9 77.5
1.346 156.0 0.0
aGeometries and zero-point energies (ZPEs) were determined at the CAS(6,6)/4-31G level of theory.bValues are relative to the ground-state minimum, S0(C2h),1Ag, including the ZPE corrections.cCAS(6,6)/4-31G; the reference total energy is -568.30919 hartree.dCAS(6,6)/cc-pVDZ;
the reference total energy is -569.18400 hartree.eBased on the absorption spectrum shown in ref 16 that the maximum S
1absorption band locates
at 450 nm and the tail of the absorption at∼560 nm.fAll vibrational frequencies are positive values.gVibrational frequency calculations were not performed.hZPE is taken from the S
0(C2h) species.iδ(CNNC) or φ(NNCC) is not defined for a linear CNN configuration.jZPE is taken from the
S1(C2) species.kZPE is taken from the S1(C2h) species.
for the rotation mechanism shown in Scheme 1. The molecule is planar with C2hsymmetry at the torsional angle of 180°, but the rotational motion along the CNNC torsional coordinate reduces the molecular symmetry to C2. Along the rotational reaction path, a local minimum, S1(C2), is found at the torional angle of 128.5°. According to our calculations, the S1surface along the rotational pathway is rather flat (Figure 2) and S1-(C2) is a true S1minimum with all the vibrational frequencies being positive; the frequency corresponding to the CNNC torsional motion is 15 cm-1. Following the CNNC out-of-plane torsional reaction path from S1(C2) toward the smaller torsional angle direction, the S1curve still remains flat but the S0curve increases drastically so that a surface touching is expected at a perpendicularly twisted configuration (torsional angle∼90°) as in the case of azomethane recently reported.24According to the two-dimensional surface-scan calculation reported by Ishikawa et al.,23a conical intersection was evident at the location with the CNNC torsional angle of 88°and the CNN bending angle of 130°. In the present study, this conical intersection (S0/S1 CI•rot) has been fully optimized to locate at the CNNC torsional angle of 92.1°with two CNN bending angles equal to 129.2°and 120.2°, respectively; the optimized structure is shown in the inset of Figure 2 and its key structural parameters are summarized in Table 1. The structure of CI•rot has C1 symmetry and the energy relative to the S0minimum is 47.8 kcal mol-1 predicted at the CAS(6,6)/cc-pVDZ level with the ZPE corrections according to the S1(C2) species.
According to our calculated results, there are two important factors affecting the electronic relaxation through this rotation channel. First, there is no energy barrier involved along the rotational pathway from the Franck-Condon region (torsional angle ) 180°) down to the S1(C2) minimum area (torsional angle
∼130°). This feature provides an energy-favorable reaction path for the excited molecules to follow upon initial excitation to the S1(n,π*) state. Second, the S0/S1 CI•rot can be easily reached at the middle point of the rotational pathway without involving a significant energy barrier. Funneling through the CI, different reaction pathways on the ground-state surface may be predicted by following the gradient difference vector (x1) or
the derivative coupling vector (x2) direction.28The calculated
results of azobenzene are similar to those of azomethane:24The x1direction gives the guidance for a combined motion involving
the NN stretch, the asymmetric CN stretch and the asymmetric CNN bending modes whereas the x2direction is mainly related
to the CNNC torsional mode that gives the trans and the cis isomers as final products on the S0PES. Therefore, the rotational reaction pathway is not only energetically but also dynamically a favorable electronic relaxation channel for the photoisomer-ization of azobenzene to occur from the S1surface going through the S0/S1CI•rot and eventually forming both the trans-S0and the cis-S0 species via the x2 direction of the CI. When this
channel is blocked by structural10,11or environmental12 modi-fication, another relaxation pathway must be considered.
3.3. Concerted Inversion Channel. The potential energy curves shown in Figure 3 were obtained from synchronously scanning the two CNN bending angles when the other structural parameters on the S1PES with C2hsymmetry were optimized. The minimum-energy point of the S1curve, S1(C2h), is located above the ground-state minimum by 51.1 kcal mol-1(including ZPE corrections) predicted at the CAS(6,6)/cc-pVDZ level with the two equivalent CNN bending angles equal to 128.7°(Table 1). Following the concerted CNN bending reaction path from S1(C2h) toward the larger bending angle direction, the S1and the S0potential energy curves come closer quickly and the two states tend to touch at bending angles greater than 148°. Further state-averaged CASSCF calculations have indicated that the two states interact strongly at larger angles and a surface crossing occurs at ∼157°; the results are shown as dashed curves in Figure 3. The attempt to optimize the geometry for the conical intersection (S0/S1CI•inv) with C2hsymmetry was unsuccess-ful, but the geometry was successfully optimized without imposing a symmetry constraint. The optimized structure of S0/ S1CI•inv has Cssymmetry with two different CNN bending
angles equal to 157.3°and 156.0°, respectively. The energy of S0/S1 CI•inv with respect to S1(C2) is 21.2 kcal mol-1 calculated at the CAS(6,6)/4-31G level, but the relative energy increases to 27.4 kcal mol-1when calculated at the CAS(6,6)/ cc-pVDZ level of theory (Table 1). This theoretical finding suggests that another photochemical funnel (S0/S1CI•inv) may be accessible along the concerted CNN bending coordinate when sufficient internal energy is provided upon excitation to the hot S1state or higher electronic excited states.
According to the results demonstrated in Figure 4, funneling through the S0/S1CI•inv may lead to two different reaction
Figure 4. Structure and the corresponding gradient difference vector (x1) and the derivatives coupling vector (x2) of the S0/S1CI•inv obtained
from the optimization at the state-averaged CAS(6,6)/4-31G level of theory. The right panel shows the plausible reaction paths on the S0PES after
funneling through the S0/S1CI•inv: Following the gradient difference vector x1either a hot trans-azobenzene or the phenyl radical pair/N2is
paths to be followed on the ground-state surface via either the gradient difference vector (x1) or the derivative coupling vector
(x2) direction.28 Basically, the x1 vector corresponds to a
combined in-plane motion whereas the x2vector relates to an
out-of-plane bending motion. Following the x1 vector in the
forward direction, the NN bond is stretched but the two CN bonds and the two CNN angles are simultaneously compressed and bent. Because breaking the NN double bond on the S0PES requires overcoming a substantial energy barrier (112.6 kcal mol-1),29we expect the molecule would become vibrationally hot at the trans-S0configuration. If the reverse x1vector direction
is followed, the NN bond is compressed but the two CN bonds are stretched together with the two CNN bending angles bent in a concerted manner. This motion allows a concerted CN bond-breaking process to occur on the S0PES if the available internal energy is sufficient (65.8 kcal mol-1).29On the other hand, following both forward and reverse directions of x2, the
concerted CNN out-of-plane bending motion becomes active and the two N atoms are moving in the opposite direction perpendicular to the original molecular plane to form a hot twisted trans-S0species. Even though this hot trans-S0species has the CNNC torsional angle of∼180°, its two phenyl rings are perpendicularly twisted with respect to the new molecular plane; i.e., the two NNCC torsional angles are∼90°. Therefore, we expect that eventually a planar trans-S0isomer would be formed via twisting the two phenyl rings on the ground-state surface because the twisted species is unstable and it corresponds to a second-order saddle point on the S0PES with the energy higher than the trans-S0minimum by 12.2 kcal mol-1.29
Early experimental measurements indicate that the trans-to-cis photoisomerization quantum yield is∼0.25 on S1excitation but it drops to only∼0.1 on S2excitation.7The quantum yields are independent of the wavelengths on both excitations with a critical transition point occurring at∼370 nm.7The reduction of the quantum yield on S2excitation has been proposed to be due to the opening of a new relaxation channel that produces only the trans-S0 isomer.19-21On the basis of our theoretical analysis aforementioned, following the concerted CNN bending reaction path (concerted inversion channel) to funnel through the S0/S1CI•inv would give either a phenyl radical pair and a N2 molecule or a hot trans-azobenzene on the ground-state surface. Because the production of a cis isomer is less favorable for the electronic relaxation along this channel, the trans-to-cis quantum yield of azobenzene is expected to be lower if this channel is open upon excitation. Furthermore, the calculated relative energy of the S0/S1CI•inv is 77.5 kcal mol-1(Table 1), which is in excellent agreement with the transition point being observed at ∼370 nm (∼77 kcal mol-1 above S0 minimum). These theoretical findings suggest that the new electronic relaxation channel may be the concerted CNN bending reaction path with a photochemical funnel (S0/S1 CI•inv) located at the geometry close to a linear CNNC configuration. Therefore, our results may provide a theoretical foundation to rationalize the early steady-state observations: The rotation channel is dominant on S1excitation to give a higher trans-to-cis quantum yields whereas the concerted inversion channel is involved for lower values being observed when this channel is open upon excitations with sufficient energy (i.e.,λ
< 370 nm).
When the rotation channel is blocked by putting the molecule in a cyclodextrin cavity, only this new relaxation channel is feasible to give more trans isomers so that the values of the photoisomerization yields obtained from both the S2and the S1 excitations become equally small (∼0.13).12 The
rotation-blocked azobenzene derivatives via chemical modification also show equal photoisomerization yields from the two excitations but with higher values (∼0.25).10,11Higher trans-to-cis quantum yields means more cis isomers were produced via the new relaxation channel. For a rotation-blocked azobenzene derivative, the two phenyl rings are structurally restricted so that they cannot twist freely to form more stable trans-S0isomers when following both forward and reverse x2directions of the S0/S1CI•inv along
the new relaxation pathway. Thus, relatively more cis-S0isomers will be produced in the case of rotation-blocked azobenzene derivatives10,11 than in the case of azobenzene-cyclodextrin inclusion complexes.12
Further experimental evidence for the involvement of the S0/ S1CI•inv was found in recent time-resolved studies, where the azobenzene derivative (methyl orange) in different cyclo-dextrin environments was investigated using the ultrafast transient lens technique.30,31 The trans-to-cis isomerization quantum yield of methyl orange on S2excitation (λ ) 400 nm)32 was determined to be 0.1 in pure water, and the yield remains the same in the 1:1 methyl orange/R-cyclodextrin inclusion complex. This is because only the dimethylaniline moeity of the molecule is included in the nanocavity and the azo functional group is outside the cavity so that it can still move freely; either via the CNNC torional motion or through the concerted CNN bending motion. However, the quantum yield only reduces to 0.07 in the 1:2 complex where the molecule was completely included in two R-cyclodextrins and the rotational motion about the NN double bond was completely inhibited. This experi-mental evidence indicates that the rotational reaction pathway is only a minor photoisomerization channel to be considered upon S2excitation of an azo compound. The observed subpi-cosecond to pisubpi-cosecond relaxation times in the inclusion complexes support the mechanism that the electronic relaxation of the azobenzene derivative mainly proceeds through the concerted inversion channel with the involvement of the S0/S1 CI•inv as an efficient photochemical funnel. The observed quantum yield of 0.07 in the 1:2 inclusion complex may be rationalized by the fact that the azo group inside the nanocavity still has room to adjust its position to produce some cis isomers on the hot ground-state PES, and this is the reason the yield in 1:1 complex is very similar to the yield (0.1) in 1:1 complex and in aqueous solution. On the other hand, in a 2:2 complex when two molecules were included in parallel in two γ-cyclo-dextrins, not only the observed time constants were remarkably elongated but also the quantum yield was further reduced to zero. The interaction between the two monomers should be considered in this case and this could effectively hinder the motion of the azo group along the concerted inversion channel. If this channel is only partly followed, the electronic relaxation may occur at the early stage without the involvement of the photochemical funnel (S0/S1CI•inv), which is the result for the much slower relaxation rates being observed. Moreover, the two CNN bending angles do not have to change from the Franck-Condon structure too much, which eventually leads to the production of only the trans isomers as final products being observed.
4. Conclusion
Our results for the photoisomerization mechanism of trans-azobenzene from the S1 state down to the S0 surface are summarized in Scheme 2. According to the present study, the widely accepted inversion channel has been ruled out because a substantial energy barrier is involved and the energy gap between the two electronic states is too large to allow an efficient Trans-to-Cis Photoisomerization of Azobenzene J. Phys. Chem. A, Vol. 108, No. 6, 2004 955
electronic relaxation to occur. On the other hand, a photochemi-cal funnel (S0/S1 CI•rot) is located on the midpoint of the rotational pathway that produces both trans and cis isomers on the S0 PES. Therefore, the conventional rotation channel is responsible for a cold S1species to proceed with zero or little available internal energy. Upon excitation to the S2state, a hot S1species is produced so that the new relaxation channel can be open via the concerted CNN bending motion to reach another photochemical funnel (S0/S1 CI•inv). Funneling through the S0/S1 CI•inv gives either a trans isomer or the other bond-breaking side products, which may be the reason for the observed trans-to-cis photoisomerization quantum yields on S2 excitation being much lower than the S1excitation. When the rotation channel is blocked by chemical modification10,11or an inclusion complex,12only the new relaxation channel is feasible so that the values of the photoisomerization yields obtained from both the S2and the S1excitations become equal. The present study provides solid theoretical evidence to address the issue for the long-standing controversy on the photoisomerization mechanism of azobenzene. The new relaxation channel, hitherto unreported, is intrinsically an inversion channel involving the concerted CNN bending motion in the same molecular plane to approach a linear CNNC configuration to reach the conical intersection for an efficient electronic relaxation. We notice that the semilinear (inversion), the skew (rotation), and the linear (concerted inversion) isomerization channels are three typical photochemical processes being considered for azomethane since the 1970s.33For azobenzene, the photochemical behavior of the concerted inversion channel has not been discussed until now, which indeed has led to the rotation-inversion controversy on the photoisomerization mechanism of azobenzene and its derivatives for more than 20 years.
Acknowledgment. This work was supported by the National Science Council of Republic of China with the project contract number 90-2119-M-009-001. I thank Prof. M. C. Lin, Prof. Wen-Sheng Chung, Prof. Robert S. H. Liu, and Prof. Maurizio Persico for many helpful discussions. The suggestions and comments made by the reviewers are much appreciated. References and Notes
(1) Berg, R. H.; Hvilsted, S.; Ramanujam, P. S. Nature 1996, 383, 505-508.
(2) Ikeda, T.; Tsutsumi, O. Science 1995, 168, 1873-1875. (3) Willner, I.; Rubin, S.; Riklin, A. J. Am. Chem. Soc. 1991, 113, 3321-3325.
(4) Liu, Z. F.; Hashimoto, K.; Hujishima, A. Nature 1990, 347, 658-660.
(5) Sudesh, G.; Neckers, D. C. Chem. ReV. 1989, 89, 1915-1925. (6) Tamai, N.; Miyasaka, H. Chem. ReV. 2000, 100, 1875-1890. (7) Rau, H. In Photochromism: Molecules and Systems; Durr, H., Bouas-Laurent, H., Eds.; Elsevier: Amsterdam, 1990; pp 165-192 and references therein.
(8) Zimmerman, G.; Chow, L.-Y.; Paik, U.-J. J. Chem. Phys. 1958, 80, 3528-3531.
(9) Bortollus, P.; Monti, S. J. Phys. Chem. 1979, 83, 648-652. (10) Rau, H.; Lu¨ddecke, E. J. Am. Chem. Soc. 1982, 104, 1616-1620. (11) Rau, H. J. Photochem. 1984, 26, 221-225.
(12) Bortolus, P.; Monti, S. J. Phys. Chem. 1987, 91, 5046-5050. (13) Monti, S.; Orlandi, G.; Palmieri, P. Chem. Phys. 1982, 71, 87-99. (14) Lednev, I. K.; Ye, T.-Q.; Hester, R. E.; Moore, J. N. J. Phys. Chem. 1996, 100, 13338-13341.
(15) Na¨gele, T.; Hoche, R.; Zinth W.; Wachtveitl, J. Chem. Phys. Lett. 1997, 272, 489-495.
(16) Lednev, I. K.; Ye, T.-Q.; Matousek, P.; Towrie, M.; Foggi, P.; Neuwahl, F. V. R.; Umapathy, S.; Hester, R. E.; Moore, J. N. Chem. Phys. Lett. 1998, 290, 68-74.
(17) Lednev, I. K.; Ye, T.-Q.; Abbott, L. C.; Hester, R. E.; Moore, J. N. J. Phys. Chem. A 1998, 102, 9161-9166.
(18) Hirose, Y.; Yui, H.; Sawada, T. J. Phys. Chem. A 2002, 106, 3067-3071.
(19) Fujino, T.; Tahara, T. J. Phys. Chem. A 2000, 104, 4203-4210. (20) Fujino, T.; Arzhantsev, S. Y.; Tahara, T. J. Phys. Chem. A 2001, 105, 8123-8129.
(21) Fujino, T.; Arzhantsev, S. Y.; Tahara, T. Bull. Chem. Soc. Jpn. 2002, 75, 1031-1040.
(22) Cattaneo, P.; Persico, M. Phys. Chem. Chem. Phys. 1999, 1, 4739-4743.
(23) Ishikawa, T.; Noro, T.; Shoda, T. J. Chem. Phys. 2001, 115, 7503-7512.
(24) Diau, E. W.-G.; Zewail, A. H. Chem. Phys. Chem. 2003, 4, 445-456 and references therein.
(25) Lu, Y.-C.; Chang, C.-W.; Diau, E. W.-G. J. Chin. Chem. Soc. 2002, 49, 693-701.
(26) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; et al. Gaussian98, revision A.11; Gaussian, Inc.: Pittsburgh, PA, 1998.
(27) Based on the similar CAS orbitals, the total energies of the S1(Cs)
and the S1(C2h) species are -568.23581 and -568.23121 hartree,
respec-tively, calculated at the CAS(6,6)/4-31G level of theory. However, due to the convergence problem for describing the entire rotational reaction path, the S1(C2) species was calculated at the same level using the slightly different
CAS orbitals, which gives -568.23152 hartree. Force-constant calculations indicate that both the S1(Cs) and the S1(C2h) species involve imaginary
frequencies whereas the S1(C2) species is a true local minimum with all
frequencies being positive (Table 1).
(28) Bernardi, F.; Olivucci, M.; Robb, M. A. Chem. Soc. ReV. 1996, 25, 321-328, and references therein.
(29) Based on the results calculated at the UB3LYP/6-31G(d) level of theory with zero-point energy correction at the same level.
(30) Takei, M.; Yui, H.; Hirose, Y.; Sawada, T. J. Phys. Chem. A 2001, 105, 11395-11399.
(31) Yui, H.; Takei, M.; Hirose, Y.; Sawada, T. ReV. Sci. Instrum. 2003, 74, 907-909.
(32) Deffering from the case of trans-azobenzene, the S2state of methyl
orange is hit by the 400 nm excitation due to a substantial red shift of the S2band in absorption spectrum upon substitution of azobenzene with the
dimethyl amino group.
(33) Camp, R. N.; Epstein, I. R.; Steel, C. J. Am. Chem. Soc. 1977, 99, 2453-2459.