Face selectivity in the photocycloaddition reactions of acrylonitrile to
5-substituted adamantan-2-ones and pyrolysis of the products to
methyleneadamantanes
Wen-Sheng Chung * and Chia-Chin Ho
Department of Applied Chemistry, National Chiao Tung University, Hsinchu, Taiwan, 30050,
ROC
The photocycloaddition of acrylonitrile to 5-substituted adamantan-2-ones (1-X) produces two geometrically isomeric oxetanes in which the oxygen atom and the 5-substituent are in the anti or syn positions. The substituent was varied from fluoro, chloro, bromo, hydroxy to phenyl and the product ratios were similar (ca. 60 : 40) in all instances. Small portions (<5%) of the oxetanes were pyrolysed when
analysed by GC at above 2008C, and the products from the pyrolysis were confirmed to be the
corresponding methyleneadamantanes by independent syntheses. Assignment of the configuration of the
oxetanes was found to be consistent with 13
C additivity scheme and the chemical shifts are more closely predicted using known oxetanes of 1-X with methacrylonitrile. The product formation bias resulting from the attack on the syn-face can be explained using the Cieplak transition state theory.
Introduction
It is now recognized that transition state hyperconjugation, tor-sional and electrostatic effects are important in determining π-facial selectivity.1–6
Although several model studies have clearly demonstrated the importance of long-range electronic effects to be a determining factor in diastereofacial selectivity, the precise nature of the electronic interaction remains controversial.1–6
The symmetry of 5-substituted adamantan-2-ones 1-X and their derivatives makes them useful probes for investigating the electronic effects on transition state energy since both faces of the carbonyl are little affected by steric effects of substitution at C-5.3
Although torsional effects are involved in the transition state of nucleophilic (or electrophilic) addition to 1-X, they are cancelled out due to the symmetry of the system allowing elec-tronic effects to dominate in determining facial selectivity.2b,3
The photocycloaddition of ketones to olefins, also known as the Paterno–Büchi reaction,7 is one of the earliest reported
organic photochemical reactions.8 The reaction was long
neg-lected, as the product oxetanes seemed to be of little interest or value. However, in the past 10 years, this reaction has proved to be of considerable synthetic utility.8c–g Most of the oxetanes
may undergo a thermal retro [2 + 2] cleavage at relatively low temperatures [eqn. (1)]. In some cases this thermal
cyclo-reversion leads to unsaturated long-chain compounds that are otherwise accessible only with much greater difficulty, especially in the case of bicyclic oxetanes derived from carbonyl com-pounds and cyclic alkenes.†
Recently, we have reported9a that photocycloaddition of the
electron-poor olefins (E)-1,2-dicyanoethylene 2, and methacry-lonitrile 3 to 5-substituted adamantanones leads to oxetanes
† In some cases this thermal [2 + 2] cycloreversion is also called a carbonyl–olefin-metathesis (COM) by Jones.8e For leading references see: T. S. Cantrell and A. C. Allen, J. Org. Chem., 1989, 54, 135; 140.
stereoselectively.9a,c Relative fluorescence quenching rates of
1-H by several α,β-unsaturated nitriles 2–4 have been reported10
to be 2 : 3 : 4= 10:1.1:2; however, their products and face selec-tivities have not been fully explored. Thus, we report here our studies on the photocycloaddition of 1-X to acrylonitrile 4 and pyrolysis of the products to methyleneadamantanes in GC at >200 8C.
Results and discussion
The singlet n,π* state of 1-X is trapped by acrylonitrile 4 via a concerted [2 + 2] cycloaddition involving the now occupied π* orbital which lead to a regiospecific reaction. The triplet state of 1-X does not lead to an oxetane but results in olefin dimer-ization (Scheme 1). Exciplex formation has been proposed to be
the initial step of a quenching interaction between ketone n,π* singlet or triplet states and olefins.8a,b The exciplex appears to
have a charge transfer characteristic, with the n,π* state acting as an electron donor to electron-poor olefin (4).8a,b
Once formed, the exciplex is subject to dissociation, or to bond for-mation leading directly to oxetanes.
Irradiation (300 nm) of an acetonitrile solution of 1-F and excess 4 in a Rayonet photoreactor gave two oxetanes in the
Scheme 1
ratio of 60 : 40 (see Scheme 1). Due to poor resolution of the two ABX ring-proton systems (i.e. CH2CHCN) in
1
H NMR spectra, we used GC and 13C NMR spectroscopy to determine the
relative ratio of all oxetanes (see Table 1). Yields of oxetanes were >70%, based on converted 1-X; dimers 6 and polymers of
4 were also produced. An additional peak was observed in the
products of the reaction of each 1-X when analysed by GC; such a phenomenon has not previously been observed in the oxetane products of 1-X with other α,β-unsaturated nitriles9
even at higher analysing temperatures, e.g. >240 8C. However, since 1H NMR spectra of crude reaction products did not show
any new peaks other than oxetanes, we later proved those new peaks in GC to be the pyrolysed products of Xs; i.e. 5-substituted 2-methyleneadamantanes 7-Xs. The structures of
7-X were proved by independent syntheses from the Wittig
reaction of 1-X (see Scheme 2 and Experimental section). The observation of 7-X also implies the formation of 5 rather than those, 8, from the alternative orientation of cycloaddition.
The mass spectra of the oxetanes are also given in the Experimental section. They generally showed weak molecular ions (0–30% by the EI method); the main fragments arise from fission across the oxetane ring. Such ring fission is a known process;9,11,12 thus, the parent oxetane predominantly gives
methyleneadamantanes 7-X via ring cleavage just as that observed in GC analysis (vida supra).12,13 The possible
alterna-tive structures 8 were eliminated from consideration by the lack of a large peak at m/z corresponding to the loss of HCOH from the molecular ion. Peaks at m/z corresponding to the loss of O]]C(CN)H are clearly observed, also supporting the formation of oxetane structure 5.
1H NMR spectra again confirmed the structures 5, rather
than 8. It has been reported14 that hydrogen on the carbon α to
the oxygen of an oxetane ring has a chemical shift of 4–5 ppm, whereas hydrogen on the β-carbon atom has a chemical shift of
Scheme 2
Table 1 anti : syn epimer ratios (%) in the photocycloadditions of 4 to
5-substituted adamantanones (1-X)a in acetonitrile at room temp.
5-Substituent X anti-5-X syn-5-X Analysis
F Cl Br C6H5 OH 60 62 57 62 57 61 40 38 43 38 43 39 GC, NMRb GC, NMR GC, NMR GC, GCMS NMR GC
a Error limit for GC and 13C NMR analysis is ±5% due to pyrolysis at high temperature. Yields are ca. 70–90% based on converted 1-X. b The
inverse gated decoupling method was used for 13C NMR measurement
with a 60 s delay.
2.5–3.6 ppm. In the nine oxetanes we have studied, the oxetane ring proton absorptions fell in two distinct regions, 2.6–2.9 and 5.0–5.1 ppm (see Experimental section), which is in good agreement with the above description and our previous obser-vations in other structural related oxetanes.9 The magnitude of
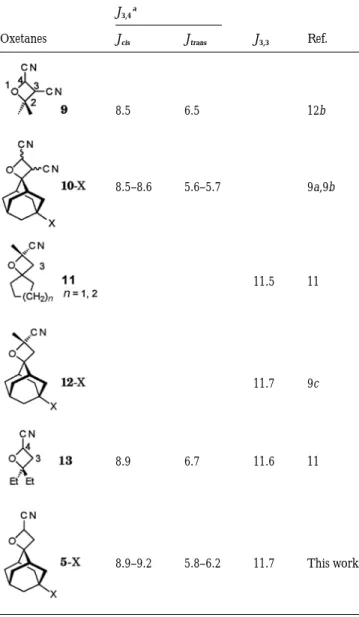
the geminal coupling (ca. 11.7 Hz) of the oxetane ring protons provides further evidence for the oxetane structures 5 (see Table 2). The other regioisomer 8 would have a geminal coupling Jα,α
of 6.0 as described by Lustig et al.15a for the oxetane ring C
α
protons.
Another indication of structure 5 instead of 8 comes from
13
C NMR study, where the chemical shift difference (∆δ) between C-8 and C-10 and between C-4 and C-9 is similar and falls into the range of 0.2 ppm (see Table 3). However, previous studies9b show that a large difference in chemical shifts for
car-bons in proximity to the cyanomethylene group is expected (due to the magnetic anisotropy of the cyano group); i.e. if 8 had been formed, the chemical shift difference (∆δ) would have been larger than 0.5 ppm.
Although a single crystal X-ray structure determination would define the structural assignment, all our attempts to grow a single crystal of compounds 5-X were unsuccessful. Fortu-nately, the configuration assignment of the spiro skeleton was found to be consistent with a 13
C NMR study of the type described by le Noble et al.16 In essence, this is an additivity
scheme in which the chemical shifts are calculated from those of the corresponding carbons in adamantane, 1-fluoroada-mantane and the parent spirooxetane. The C-4,9 and C-8,10
Table 2 Oxetane ring proton coupling constants (Hz)
J3,4a
Oxetanes Jcis Jtrans J3,3 Ref.
8.5 8.5–8.6 8.9 8.9–9.2 6.5 5.6–5.7 6.7 5.8–6.2 11.5 11.7 11.6 11.7 12b 9a,9b 11 9c 11 This work
Table 3 Calculateda and observedb13C chemical shifts in 5-substituted-49-cyanospiro[adamantane-2,29-oxetanes]
Carbon 5-H anti-5-F syn-5-F anti-5-Cl syn-5-Cl anti-5-Br syn-5-Br anti-5-Ph syn-5-Ph
C-1, C-3 C-2 C-4, C-9 C-5 C-7 C-6 C-8, C-10 C-39 C-49 CN C-i C-o C-m C-p 38.87 38.57 91.19 33.03 33.15 26.04 26.16 36.24 31.37 31.43 37.23 59.90 119.74 40.59 (41.47) J 8.8c 40.73 (41.67) J 11.0c 89.12 (89.30) 37.62 (37.94) J 19.8 37.74 (38.06) J 19.8 90.58 (89.94) J 184.6 29.16 (29.26) J 9.2 41.61 (41.15) J 15.4 29.92 (29.48)e 29.92 (29.54)e 36.97 59.96 119.34 41.35 (41.47) J 11.0 41.50 (41.67) J 11.0 89.24 (89.30) 36.30 (36.28)d 36.59 (36.34)d 90.46 (89.96) J 184.6 29.29 (29.14) J 11.0 41.53 (41.15) J 15.4 31.55 (31.14) 31.63 (31.26) 35.92 59.93 119.39 41.02 (41.47) 41.16 (41.67) 88.86 (88.80) 42.76 (42.64) 42.88 (42.76) 65.23 (65.84) 29.39 (29.26) 46.44 (45.85) 29.62 (29.98) 29.68 (29.04) 37.03 59.96 119.31 41.45 (41.47) 41.60 (41.67) 88.42 (88.80) 41.19 (41.04)f 41.19 (40.98)f 65.35 (65.96) 29.42 (29.14) 46.35 (45.85) 31.29 (30.64) 31.37 (30.76) 36.12 59.96 119.37 41.87 41.98 88.74 44.28 44.40 61.44 30.21 47.89 29.60 29.65 37.03 59.98 119.31 42.15 42.30 88.36 42.65 42.71 61.70 30.18 47.83 31.26 31.37 36.27 59.96 119.34 38.95 39.04 90.61 38.48 38.65 34.90 26.80 42.30 30.62 30.67 37.32 60.04 119.69 149.06 124.73 128.28 126.01 39.15 39.24 90.20 36.91 37.14 34.64 26.89 41.63 32.28 32.42 36.91 60.01 119.71 149.49 124.78 128.16 125.83
a Calculated values are in parentheses. b Measured by a VXR-300 NMR spectrometer operated at 75.4 MHz and reported on the δ scale, CDCl
3 (77.00 δ). See Scheme 1 for the numbering system. In the parent compound 5-H, the oxygen is understood to be syn to C-8 and C-10. c C-1 and C-3
were overlapped. d C-4 and C-9 were overlapped so the coupling constant J values were not determined. e Two peaks overlapped at δ 29.92. f Two
peaks overlapped at δ 41.19.
pairs, by far the most informative beacons in this regard, are readily distinguished from one another by virtue of the 13C–19F
splittings, which are ca. 20 and 0 Hz, respectively (Table 3). In the parent compound, they can be recognized by means of their chemical shifts: when one of the two substituents at C-2 is an electronegative atom such as oxygen, the carbon pair ‘below’ it is always shielded compared to the anti pair.9
The chemical shifts thus computed for C-4,9 and C-8,10 agree with the observed values to within a few tenths of 1 ppm; differences of 1 ppm or more result if the opposite configurations are assumed (Table 3).
The experiments with 1-Cl, -Br, -Ph and -OH followed a simi-lar course; i.e. the anti : syn ratio is ca. 60 : 40 in all cases (Table 1). The anti and syn assignments in these instances were also based on the 13
C additivity method. As with the fluoro prod-ucts, the chemical shifts calculated for C-4,9 and C-8,10 agree with the observed values to within a few tenths of 1 ppm (see Table 3). Furthermore, the 13C chemical shifts can be more
closely predicted to within 0.2 ppm using the additivity scheme from known oxetanes (12),9c which is much smaller compared
to those calculated from adamantanes (<0.65 ppm).16
Many of the adamantanone reactions have been interpreted by le Noble3
and Cieplak1
as consistent with the Cieplak hypothesis since the reaction occurs preferentially from the face antiperiplanar to the more electron-rich σ-bonds. Cieplak con-tends this is a result of the preferential donation from the more electron-rich bond to the σ*‡
orbital shown schematically in Fig. 1.1,3 The present results add to the already extensive
evi-dence for the proposition that addition to trigonal carbon occurs at the face antiperiplanar to electron-rich vicinal bond(s). It would seem difficult to find a reason for this prefer-ence in so many reactions as described here and elsewhere with-out involving transition-state hyperconjugation. A recent report by Coxon and Houk2c using ab initio and semiempirical
methods to calculate the transition state energies of syn and
anti face approaches of 1-X by AlH3 also concluded that
hyper-conjugative stabilization dominates the electronic effects. It must be realized, however, that our knowledge of inductive power is based almost exclusively on ground-state chemistry,
and also that with the E/Z ratios generally rather close to unity, it will remain difficult to satisfactorily prove or disprove such a correlation in excited state chemistry.
Experimental
1H NMR spectra were measured on 300 and 400 MHz
spec-trometers. The data reported were recorded at 300 MHz unless otherwise specified. Natural abundance 13C NMR spectra were
measured using pulsed Fourier-transform, on a Varian Unity-300 MHz, high resolution NMR spectrometer, operating at 75.4 MHz. Broad-band decoupling was used to simplify spectra and aid peak identification. δ values are in ppm and J values in Hz for both nuclei, with the solvent (usually CDCl3) peak as an
internal standard. The reference peak for 13C is δ 77.00, which is
set at the centre peak of CDCl3, and for
1H it is δ 7.25 of CHCl 3.
Gas chromatographic analyses were carried out on an instrument equipped with a flame ionization detector and a reporting integrator. The capillary column employed included HP-1 crosslinked methylsilicone (SE-30, 25 m) and Carbowax column (25 m). GC–MS analyses were carried out using EI (at 70 eV).
Materials
All commercially obtained chemicals were reagent or spectro-photometric grade and were not purified prior to use unless
Fig. 1 Cieplak preference for nucleophilic (or electrophilic) addition
anti to electron-donating (left) and syn to electron-withdrawing sub-stituents (right)
otherwise specified. The synthesis of 5-fluoro-adamantan-2-ones (1-F),17,18
5-chloro- (1-Cl),19
5-bromo- (1-Br),20
and 5-phenyladamantan-2-one (1-Ph),18 have all been described. All
the 5-substituted 2-methyleneadamantanes (7-Xs) were pre-pared from the corresponding ketones (1-X) by a standard pro-cedure described by Adcock et al.6a
for the bromo derivative (X= Br).
General procedure for the synthesis of 5-substituted 2-methyleneadamantane (7-X)
By use of a procedure similar to that described by Adcock,6a a
solution of butyllithium (24 ml of 2.5 hexane solution; 60 mmol) was added over a 10 min period to a stirred slurry of methyltriphenylphosphonium bromide (19.6 g, 55 mmol) in dry diethyl ether (100 ml) in a 250 ml twin-necked flask under N2.
The reaction mixture was stirred for 4 h at room temp. before adding dropwise a solution of adamantane-2-one 1-H (7.51 g, 50 mmol) in dry diethyl ether (80 ml). The mixture was stirred overnight then 10 ml water was added and stirred for 10 min. The organic layer was separated and the aqueous layer was washed with diethyl ether (20 ml × 2). The combined extracts were washed with brine, dried over MgSO4, and the solvent was
removed in vacuo to afford a yellow oil. Chromatography over silica gel using hexane as eluent gave the desired compound 7-H as a colourless solid (mp 135–1368C; lit.,6a 135–1368C) 5.28 g
(72%): δH 4.50 (2 H, s), 2.48 (2 H, s) and 1.93–1.76 (12 H, m);
δC 158.41 (Cq), 100.54 (CH2), 39.65 (CH2), 39.06 (CH), 37.29
(CH2) and 28.28 (CH).
7-F was a colourless solid (mp 112–1148C; lit.,6a 110–1128C)
70%: δH 4.57 (2 H, s), 2.69 (2 H, br s), 2.26 (1 H, br s), 1.96–1.91
(6 H, m) and 1.70–1.69 (4 H, m); δC 154.02 (Cq), 103.40 (CH2, d,
J 2.5), 91.70 (Cq, d, J 184.3), 43.63 (CH2, d, J 17.1), 42.41
(CH2, d, J 17.1), 40.65 (CH, d, J 9.8), 37.92 (CH2, d, J 2.5) and
31.47 (CH, d, J 9.8).
7-Cl was a colourless liquid 67%: δH 4.57 (2 H, s), 2.63
(2 H, s), 2.19–2.17 (7 H, m) and 1.80–1.67 (4 H, m); δC 153.63
(Cq), 103.31 (CH2), 67.39 (Cq), 48.68 (CH2), 47.25 (CH2), 41.10
(CH), 37.46 (CH2) and 31.31 (CH).
7-Br was a colourless liquid 70%: δH 4.56 (2 H, s), 2.61
(2 H, s), 2.42–2.39 (6 H, m), 2.14 (1 H, br s) and 1.86–1.73 (4 H, m); δC 157.57 (Cq), 103.51 (CH2), 64.73 (Cq), 50.37 (CH2), 48.54
(CH2), 42.12 (CH), 37.46 (CH2) and 32.38 (CH).
Irradiation of 1-X with 4
A relatively high concentration of 4 (1.7 ) was employed to favour the formation of oxetanes. Thus, a solution of 0.1 g of
1-X and 2.18 ml of 4 in 20 ml of spectrograde acetonitrile
was placed in a Pyrex tube stoppered with a rubber septum. The solution was irradiated at 300 nm in a Rayonet reactor for ca. 6–10 d (>98% conversion) until completion (checked by GC). The solvent was removed from the dark-red solution on a rotary evaporator and the residue was redissolved in ethyl acetate with sonication. The solution was filtered to remove undissolved solids, then concentrated on a rotary evaporator. The residues of all 5-substituted oxetanes were chromatographed over silica gel with ethyl acetate in hexanes (ethyl acetate : hexanes= 1:10; v:v) as eluent. The use of 40– 63 µm silica gel 60 (E. Merck No 9385) and a pressure-driven rate of 1.0 in min21 leads to a successful separation. In every instance, anti-5-X eluted first, followed by syn-5-X. anti- and
syn-5-OH were not separable by either SiO2 or aluminium
oxide.
The peak patterns in the 1H (CDCl
3, 300 MHz) and 13C
NMR spectra for all the oxetanes are very similar (for com-plete assignments of the 13C peaks see Table 2).
49-Cyano-spiro[adamantane-2,29-oxetane] (5-H). A colour-less solid (mp 598C), δH(CDCl3, 300 MHz): 5.07–5.01 (1 H, dd, J 5.79, 8.86), 2.83–2.76 (1 H, dd, J 8.86, 11.72), 2.70–2.64 (1 H, dd, J 5.79, 11.72), 2.36 (1 H, br s), 2.08–1.92 (4 H, m) and 1.81– 1.54 (9 H, m); MS (EI) m/z 203 (M+ , 4%), 150 (M+ 2 4, 100), 148 [M+ 2 O]]C(CN)H, 24], 91 (34) and 79 (56). HRMS (m/z). Calc. for C13H17NO: 203.1310. Found: 203.1309.
anti-49-Cyano-5-fluoro-spiro[adamantane-2,29-oxetane]
(anti-5-F). A colourless solid (mp 85–868C), δH 5.05–5.00 (1 H,
dd, J 6.08, 8.82), 2.86–2.79 (1 H, dd, J 8.82, 11.72), 2.73– 2.67 (1 H, dd, J 6.08, 11.72), 2.55 (1 H, br s), 2.28 (1 H, br s), 2.16 (1 H, br s) and 2.15–1.79 (10 H, m); MS (EI) m/z 221 (M+, 6%), 168 (M+2 4, 100), 166 [M+
2 O]]C(CN)H, 9] and 97 (36). HRMS (m/z). Calc. for C13H16FNO: 221.1216. Found:
221.1205.
syn-49-Cyano-5-fluoro-spiro[adamantane-2,29-oxetane]
(syn-5-F). A colourless solid (mp 78–798C), δH 5.06–5.01 (1 H, dd, J 6.01, 8.89), 2.84–2.77 (1 H, dd, J 8.89, 11.72), 2.70–2.64 (1 H, dd, J 6.01, 11.72), 2.62 (1 H, br s), 2.34 (1 H, br s), 2.23–2.10 (3 H, m), 1.85 (2 H, br s) and 1.74–1.50 (6 H, m); MS (EI) m/z 221 (M+, 19%), 168 (M+2 4, 100), 166 [M+ 2 O]]C(CN)H, 69], 97 (51) and 79 (22). HRMS (m/z). Calc. for C13H16FNO:
221.1216. Found: 221.1219.
anti-49-Cyano-5-chloro-spiro[adamantane-2,29-oxetane]
(anti-5-Cl). A colourless solid (mp 97–988C), δH 5.05–5.00 (1 H,
dd, J 5.75, 9.15), 2.86–2.79 (1 H, dd, J 9.15, 11.72), 2.73–2.67 (1 H, dd, J 5.75, 11.72), 2.21 (1 H, br s), 2.17–1.91 (9 H, m) and 1.87–1.23 (3 H, m); MS (EI) m/z 237 (M+, 2%), 202, (M+2 Cl,
1), 184 (M+2 4, 100), 182 [M+
2 O]]C(CN)H, 2], 91 (17) and 79 (17). HRMS (m/z). Calc. for C13H16ClNO: 237.0920. Found:
237.0915.
syn-49-Cyano-5-chloro-spiro[adamantane-2,29-oxetane]
(syn-5-Cl). A colourless liquid, δH 5.06–5.01 (1 H, dd, J 6.00, 8.88), 2.82–2.76 (1 H, dd, J 8.88, 11.72), 2.69–2.63 (1 H, dd, J 6.00, 11.72), 2.62 (1 H, br s), 2.54–2.31 (3 H, m), 2.23–2.15 (3 H, m), 2.03–1.87 (3 H, m) and 1.74–1.23 (3 H, m); MS (EI) m/z 237 (M+, 3%), 202, (M+2 Cl, 7), 184 (M+2 4, 100), 182 [M+ 2 O]] C(CN)H, 6], 91 (18) and 79 (16). HRMS (m/z). Calc. for C13H16ClNO: 237.0920. Found: 237.0922.
anti-49-Cyano-5-bromo-spiro[adamantane-2,29-oxetane]
(anti-5-Br). A colourless solid (mp 117–1188C), δH 5.50–5.00
(1 H, dd, J 6.16, 9.23), 2.88–2.81 (1 H, dd, J 9.23, 11.72), 2.74– 2.68 (1 H, dd, J 6.16, 11.72), 2.31 (1 H, br s), 2.31–2.18 (9 H, m) and 2.07–1.93 (3 H, m); MS (EI) m/z 283 [(M + 2)+, 0.1%], 281 (M+, 0.1), 230 [(M + 2)+2 4, 1], 228 (M+2 4, 1), 202 (M+2 Br, 100), 146 (76), 91 (50) and 79 (44). HRMS (m/z). Calc. for C13H16 79 BrNO: 281.0415. Found: 281.0422.
syn-49-Cyano-5-bromo-spiro[adamantan-2,29-oxetane]
(syn-5-Br). A colourless liquid, δH 5.06–5.01 (1 H, dd, J 6.00, 8.89), 2.81–2.74 (1 H, dd, J 8.89, 11.72), 2.67–2.62 (1 H, dd, J 6.00, 11.72), 2.56–2.50 (2 H, m), 2.29 (2 H, br s), 2.23–2.09 (3 H, m), 1.99 (1 H, br s) and 1.97–1.58 (5 H, m); HRMS (m/z). Calc. for C13H16 79BrNO: 281.0415. Found: 281.0425. anti-49-Cyano-5-phenyl-spiro[adamantane-2,29-oxetane]
(anti-5-Ph). A colourless solid (mp 107–1088C), δH 7.35–7.17
(5 H, m), 5.10–5.05 (1 H, dd, J 6.07, 8.82), 2.88–2.81 (1 H, dd, J 8.82, 11.72), 2.75–2.69 (1 H, dd, J 6.07, 11.72), 2.53 (1 H, br s), 2.25 (1 H, br s) and 2.13–1.56 (10 H, m); MS (EI) m/z 279 (M+ , 48%), 226 (M+ 2 4, 100), 224 [M+ 2 O]]C(CN)H, 5], 168 (27), 155 (53) and 91 (24). HRMS (m/z). Calc. for C19H21NO:
279.1623. Found: 279.1614.
syn-49-Cyano-5-phenyl-spiro[adamantane-2,29-oxetane]
(syn-5-Ph). A colourless solid (mp 103–1048C), δH 7.36–7.17 (5 H, m), 5.10–5.05 (1 H, dd, J 5.73, 9.16), 2.90–2.83 (1 H, dd, J 9.16, 11.72), 2.78–2.72 (1 H, dd, J 5.73, 11.72), 2.55 (1 H, br s) and 2.33–1.64 (12 H, m); MS (EI) m/z 280 [(M + 1)+, 21%), 279 (M+ , 100%), 226 M+ 2 4, 100), 224 [M+ 2 O]]C(CN)H, 7], 209 (20), 168 (24), 155 (58) and 91 (30). HRMS (m/z). Calc. for C19H21NO: 279.1623. Found: 279.1619.
Acknowledgements
We thank the National Science Council of the ROC for financial support (NSC 85-2113-M-009-002). We also thank one referee for helpful suggestions.
References
1 (a) A. S. Cieplak, B. Tait and C. R. Johnson, J. Am. Chem. Soc., 1989, 111, 8447; (b) A. S. Cieplak, J. Am. Chem. Soc., 1981, 103, 4540.
2 (a) L. Williams and M. N. Paddon-Row, J. Chem. Soc., Chem. Commun., 1994, 353; (b) M. N. Paddon-Row, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 1992, 114, 10 638 and references cited therein; (c) J. M. Coxon, K. N. Houk and R. T. Luibrand, J. Org. Chem., 1995, 60, 418; (d) E. Vedejs, W. H. Dent, III, J. T. Kendall and P. A. Oliver, J. Am. Chem. Soc., 1996, 118, 3556.
3 (a) M. Kaselj and W. J. le Noble, J. Org. Chem., 1996, 61, 4157; (b) J. Lau, E. M. Gonikberg, J.-T. Hung and W. J. le Noble, J. Am. Chem. Soc., 1995, 117, 11 421; (c) E. M. Gonikberg and W. J. le Noble, J. Org. Chem., 1995, 60, 7751; (d) M. Xie and W. J. le Noble, J. Org. Chem., 1989, 54, 3836; (e) H. Li and W. J. le Noble, Tetra-hedron Lett., 1990, 31, 4391; (f) C. K. Cheung, L. T. Tseng, M.-H. Lin, S. Srivastava and W. J. le Noble, J. Am. Chem. Soc., 1986, 108, 1598.
4 (a) B. Ganguly, J. Chandrasekhar, F. A. Khan and G. Mehta, J. Org. Chem., 1993, 58, 1734; (b) V. A. Kumar, K. Venkatesan, B. Ganguly, J. Chandrasekha, F. A. Khan and G. Mehta, Tetrahedron Lett., 1992, 33, 3069.
5 (a) R. L. Halterman, B. A. McCarthy and M. A. McEvoy, J. Org. Chem., 1992, 57, 5585; (b) T. Ohwade, I. Okamoto, N. Haga and K. Shudo, J. Org. Chem., 1994, 59, 3975.
6 (a) W. Adcock, J. Cotton and N. A. Trout, J. Org. Chem., 1994, 59, 1867 and references cited therein; (b) W. Adcock, J. Coupe, V. J. Shiner and N. A. Trout, J. Org. Chem., 1990, 55, 1411; (c) W. Adcock, A. R. Krstic, P. J. Duggan, V. J. Skiner, Jr., J. Coope and M. W. Ensinger, J. Am. Chem. Soc., 1990, 112, 3140; (d) W. Adcock and N. A. Trout, J. Org. Chem., 1991, 56, 3229.
7 (a) L. Paternò and G. Chieffi, Gazz. Chim. Ital., 1909, 39, 341; (b) G. Büchi, C. G. Inman and E. S. Lipinsky, J. Am. Chem. Soc., 1954, 76, 4327.
8 For reviews, see (a) N. J. Turro, Pure Appl. Chem., 1971, 27, 679; (b) N. J. Turro, J. C. Dalton, K. Dawes, G. Farrington, R. Hautala, D. Morton, M. Niemczyk and N. Schore, Acc. Chem. Res., 1972, 5,
92; (c) S. W. Schreiber, Science, 1985, 227, 858; (d) H. A. J. Carless, in Synthetic Organic Photochemistry, ed. W. M. Horspool, Plenum Press, New York, 1984, p. 425; (e) G. Jones, II, in Organic Photo-chemistry, ed. A. Padwa, Wiley, New York, 1981; vol. 5. pp. 1–122; ( f ) M. Demuth and G. Mikhail, Synthesis, 1989, 145; (g) A. G. Griesbeck, in Organic Photochemistry and Photobiology, eds. W. M. Horspool and P.-S. Song, CRC Press, New York, 1994, p. 522; p. 550.
9 (a) W.-S. Chung, N. J. Turro, S. Srivastava, H. Li and W. J. le Noble, J. Am. Chem. Soc., 1988, 110, 7882; (b) W.-S. Chung, N. J. Turro, S. Srivastava and W. J. le Noble, J. Org. Chem., 1991, 56, 5020; (c) W.-S. Chung, N. J. Wang, Y.-D. Liu, Y.-J. Leu and M. Y. Chiang, J. Chem. Soc., Perkin Trans. 2, 1995, 307; (d) W.-S. Chung, Y.-D. Liu and N. J. Wang, J. Chem. Soc., Perkin Trans. 2, 1995, 581.
10 N. J. Turro, C. Lee, N. Schore, J. Barltrop and H. A. J. Carless, J. Am. Chem. Soc., 1971, 93, 3079.
11 J. A. Barltrop and H. A. J. Carless, J. Am. Chem. Soc., 1972, 94, 1951.
12 (a) J. S. Bradshaw, J. Org. Chem., 1966, 31, 237; (b) J. J. Beereboom and M. S. von Writtenau, J. Org. Chem., 1965, 30, 1231.
13 E. J. Gallegos and R. W. Kiser, J. Phys. Chem., 1962, 66, 136. 14 D. R. Arnold, R. L. Hinman and A. H. Glick, Tetrahedron Lett.,
1964, 1425.
15 (a) E. Lustig, E. Ragelis and N. Duy, Spectrochim. Acta A, 1967, 23, 133; (b) D. J. Patel and D. I. Schuster, J. Am. Chem. Soc., 1967, 89, 184; (c) N. J. Turro and J. R. Williams, Tetrahedron Lett., 1969, 321. 16 S. Srivastava, C. K. Cheung and W. J. le Noble, Magn. Reson.
Chem., 1985, 23, 232.
17 M. Xie and W. J. le Noble, J. Org. Chem., 1989, 54, 3839. 18 I. Tabushi and Y. Aoyama, J. Org. Chem., 1973, 38, 3447. 19 H. W. Geluk, Synthesis, 1972, 374.
20 H. Klein and R. Wiartalla, Synth. Commun., 1979, 9, 825.
Paper 6/06039K Received 2nd September 1996 Accepted 22nd October 1996
![Table 3 Calculated a and observed b 13 C chemical shifts in 5-substituted-4 9-cyanospiro [ adamantane-2,2 9-oxetanes ]](https://thumb-ap.123doks.com/thumbv2/9libinfo/7627386.133890/3.918.81.836.85.476/table-calculated-observed-chemical-substituted-cyanospiro-adamantane-oxetanes.webp)
