Rotation of Coordinated Acetylide Ligands on the Triangular
Surface of Trinuclear Heterometallic Clusters
Der-Kweng Hwang and Yun
Chi'
Department of Chemistry, National Tsing Hua University, Hsinchu 30043, Taiwan, Republic of China
Shie-Ming Pengt and Gene-Hsiang Lee
Department of Chemistry, National Taiwan University, Taipei 10764, Taiwan, Republic of China Received February 26, 1990
Convenient and widely applicable synthetic routes to the trinuclear heterometallic acetylide complexes
LMM',(CO),(C=CR) have been developed. These routes involve the reaction of metal acetylides LM-
(CO),(C=CR) (L
=Cp and Cp*; M
=W and Mo; R
=Ph, C5H4F, C5H,0Me, tBu, and "Pr) with
Os,-(CO),&NCMe), and with RU,(CO)~~.
For the W0s2 derivatives prepared (1-31,
the acetylide ligand adopts
an asymmetric arrangement in which the acetylide C-C vector is coordinated to one of the W-Os bonds.
For all the WRu2 derivatives
(4-91,the acetylide ligand adopts both the asymmetric (with its C-C bond
orthogonal to one of the W-Ru bonds) and the symmetric arrangement (with its C-C bond orthogonal
to the unique Ru-Ru bond) and undergoes rapid interconversion in solution. For the MoRu2 derivatives
(10,
1
l ) ,
the acetylide favors the asymmetric form in both solution and the solid state; however, when the
substituent R and the ligand L are replaced by a bulky tert-butyl group and Cp* ligand, respectively (13),
the symmetric form becomes the dominant species. The dynamic 13C NMR studies suggest that the acetylide
ligand of the W0s2 derivatives is static but, in the asymmetric MoRu2 derivatives (10,
111,the acetylide
is fluxional and undergoes migration from one Mo-Ru edge to the other. The preference of the site selectivity
for the acetylide ligand has also been studied by variation of the transition-metal atoms (M and M'), the
accessory ligand (L), and the substituent (R). The structures of the complexes CpWOs2(CO),(C=CPh)
(l),
CpWRu2(CO),(C*Ph)
(4),and CpMoRu2(CO),(C=CPh) (10) have been determined by single-crystal
X-ray diffraction studies. Crystal data for 1: space group
PZ1/c;
a = 8.332 (3)A,
b
= 14.543 (4)A,
c =17.819 (5)
A,
0
= 94.46 (3)O, 2 = 4;final
R
= 0.068, R, = 0.090,and GOF
= 1.768.Crystal data for
4:space
group
P2,/n;
a = 12.476 (1)A,
b=
13.216 (4)A,
c = 13.395 (4)A, 0
= 97.99 (2)O,2
= 4;final R
= 0.029,R
= 0.027,and GOF
= 1.583.Crystal data for
10:space group P 2 J c ;
a = 12.770 (4)A,
b = 8.188 (4)AYc = 21.313 (4)
A,
0
= 91.26 (2)O, 2 = 4;final R
= 0.030,R,
= 0.031,and GOF =
2.34.Introduction
The C2 hydrocarbyl ligands occupy a key position in the
development of dinuclear, trinuclear, and polynuclear
organometallic chemistry. This position results partially
from the belief that the chemistry of the Cz hydrocarbyl
ligands in the organometallic complexes is analogous to
that of small hydrocarbon intermediates adsorbed on metal
surfaces. Among the many interesting properties of the
C2 ligands is their mobility on the coordination sphere of
the transition-metal complexes. Related studies on the C2
hydrocarbyl moieties have attracted the attention of many
theoretical and synthetic chemists.
Schilling and Hoffmann have reported the extended
Huckel calculation of some hydrocarbons on the face of
trinuclear homometallic transition-metal comp1exes.l
For
the trinuclear
C2
vinylidene complexes, Norton and Mislow
have reported the disrotatory correlated rotation about the
Co3(CO),-C vector
and
C-CHR bond in Co3(CO),(CCHR)+
by variable-temperature 13C NMR studiesS2 The motion
of
the
vinylidene
ligand
in
H30~3(C0)9[C=
CCHzCH2CH2]+
has also been de~cribed.~
For the related
C2 alkyne complexes, the migration of the perpendicular
alkyne ligand
(l.~~-77~-1mode) to the parallel position
( ~ ( ~ - 7 ~ -I(
mode) upon electrochemical reduction has been
documented for some trinuclear c l u ~ t e r s , ~
whereas the
alkyne ligand of the dinuclear heterometallic complexes
CpNiCo(CO),(RC=CR')
undergoes rotational motion
about the Ni-Co bond ~ e c t o r . ~
In addition, both Stone
and co-worker@ and Shapley and co-workers' have de-
scribed the "windscreen-wiper" type of motion for the
coordinated alkyne ligand on a W20s triangular face.
I
'To whom inquiries concerning the X-ray crystallographic work should be addressed.
0276-7333/90/2309-2709$02.50/0
Rosenberg and co-workers have reported the similar free
rotation of the alkyne on the phosphine-substituted tri-
osmium fragments.8
Recently, there has been growing research activity in the
area of syntheses of acetylide cluster c o m p l e x e ~ . ~
How-
ever,
to
our knowledge, only a few papers have focused on
the fluxional behavior of the acetylide ligand.l0 The
reason is that, in the past, only the homometallic trinuclear
acetylide complexes have been prepared. Therefore,
it
is
difficult to distinguish whether the fluxional behavior of
a target molecule is due to the rotation of acetylide or to
(1) Schilling, B. E. R.; Hoffmann, R. J. Am. Chem. SOC. 1979, 101,
(2) Edidan, R. D.; Norton, J. R.; Mislow, K. Organometallics 1982, I ,
4687.
561
--_.
(3) Koridze, A. A.; Kizas, 0. A.; Kolobova, N. E.; Petrovskii, P. V.;
Fedin. E. I. J. Ormnomet. Chem. 1984.265. C33: 1984.272. C31.
(4) 'Osella, D.; Coberto, R.; Montangero, P.; Zanello,
P.;
Cinquantini,(5) Jaouen, G.; Marinetti, A.; Saillard, J.-Y.; Sayer, B. G.; McGlinchey,
A. Organometallics 1986, 5, 1247.
M. J. Organometallics 1982, I, 225.
(6) Busetto, L.; Green, M.; Hesser, B.; Howard, J. A. K.; Jeffery, J. C.;
Stone, F. G. A. J. Chem. SOC., Dalton Trans. 1983, 519.
(7) Shapley, J. R.; Park, J. T.; Churchill, M. R.; Bueno, C.; Wasserrnan,
H. J. J. Am. Chem. SOC. 1981,103, 7385.
(8) Rosenberg,
E.;
Bracker-Novak, J.; Gellert, R. W.; Aime, S.; Go-betto, R.; Osella, D. J. Organornet. Chem. 1989, 365, 163.
(9) (a) Carty, A. J. Pure Appl. Chem. 1982, 54, 113. (b) Aime, S.;
Osella, D.; Deeming, A. J.; Lanfredi, A. M. M.; Tiripicchio, A. J. Orga-
nomet. Chern. 1983,244, C47. (c) Dawoodi, Z.; Maya, M. J.; Henrick, K.
J. Chem. SOC., Dalton Trans. 1984,1769. (d) Deeming, A. J.; Donovan-
Mtunzi, S.; Hardcastle, K. J. Chem. SOC., Dalton Trans. 1986, 543. (e)
Boyar,
E.;
Deeming, A. J.; Kabir, S. E. J. Chem. SOC., Chem. Commun.1986, 577. (f) Nucciarone, D.; MacLaughlin, S. A.; Taylor, N. J.; Carty,
A. J. Organometallics 1988, 7,106. (9) Galindo, A.; Mathieu, R.; Cami-
nade, A.-M.; Majoral, J.-P. Organometallics 1988, 7, 2198. (h) Chi, Y.;
Hwang, D.-K.; Chen, S.-F.; Liu, L.-K. J. Chem. SOC., Chem. Commun.
1989, 1540. (i) Farrugia, L. J. Organometallics 1990, 9, 105. (10) (a) Jangala, C.; Rosenberg, E.; Skinner, D.; Aime, S.; Milone, L.;
Sappa, E. Inorg. Chem. 1980,19, 1571. (b) Predieri, G.; Tiripicchio, A,;
Vignali, C.; Sappa, E. J. Organornet. Chem. 1988, 342, C33.
0
1990American Chemical Society
Downloaded by NATIONAL TAIWAN UNIV on August 12, 2009
2710
Organometallics, Vol. 9, No.
IO,
1990Hwang et al.
the mobility of another accessory ligand, such as inter-
metallic CO scrambling."
In this paper, we report the
preparation and crystal structure of a series of trinuclear
WOs,,
WRu2, and MoRuz acetylide complexes. Varia-
ble-temperature lH and 13C NMR studies indicate that the
acetylide ligand in some complexes undergoes rotation on
the face of the heterometallic triangle. Furthermore,
a
systematic analysis of the preference of site selectivity and
fluxional behavior of the acetylide ligand has been achieved
by varying the transition-metal atom, the accessory ligand,
and the substituent
of
the acetylide ligand.
A portion of
these results has appeared in a preliminary report.12
Experimental Procedure
General Information. Infrared spectra were recorded on a Perkin-Elmer 580 spectrometer or on a Bomen M-100 FT-IR spectrometer. 'H and 13C NMR spectra were recorded with Bruker AM-400 (400.13 MHz) or Varian Gemini-300 (300 MHz) instruments. Mass spectra were obtained on a JEOL-HX110
instrument operating in electron impact or fast atom bombard-
ment modes. All reactions were performed under a nitrogen atmosphere with use of deoxygenated solvents dried by an ap- propriate reagent. The progress of reactions was monitored by analytical thin-layer chromatography (5735 Kieselgel60 F254, E. Merck), and the products were separated on preparative thin-layer
chromatographic plates (Kieselgel60 FaM, E. Merck). Elemental
analyses were performed by the staff of the NSC Regional In- strument Center a t National Cheng Kung University, Tainan, Taiwan.
Materials. Metal carbonyl complexes and pentamethyl- cyclopentadiene were purchased from Strem Chemicals, Inc. Carbon monoxide enriched with 99% 13C was purchased from Cambridge Isotope Laboratories. Phenylacetylene, 1-hexyne, and 1-pentyne were supplied by Aldrich Chemical Co., Inc. (4-
fluoropheny1)acetylene and (4-methoxy)phenylacetylene was
prepared from 4-fluoroacetophenone and 4-vinylanisole, respec-
tively, according to the published p r o ~ e d u r e s . ' ~ J ~ CPW(CO)~H
was prepared by protonation of the sodium salt of the CpW(CO),- anion with acetic acid a t ambient temperature, whereas Cp*W- (CO)3H was prepared by the reaction of pentamethylcyclo- pentadiene with W(CO),(NCEt), in toluene a t 100 OC.15 On the other hand, the molybdenum hydride complexes Cp*Mo(CO),H and CpMo(CO),H were prepared from the reaction of (p-xyl-
ene)Mo(CO), with pentamethylcyclopentadiene and cyclo-
pentadiene monomer, respectively.16 Metal carbonyl chloride complexes LM(CO)&I were generated from the reactions of the
respective hydrides with CCl, under nitrogen." The metal
acetylides LM(CO),C=CR and the triosmium acetonitrile com-
plex Os3(CO)lo(CH3CN)2 were prepared according to literature
13CO-enriched acetylide complexes were prepared by equilibrating the acetylide complexes in a seal tube (25 mL) equipped with a Rotaflo stopcock under approximately 1 atm of 99% 13C0 a t 100 OC in toluene overnight.
Preparation of CpWOs,(CO),(C=CPh). As all the reactions
of the metal acetylides LM(CO),C=CR with O S ~ ( C O ) ~ ~ were
performed under similar conditions, the experimental details of
(11) Rosenberg, E.; Milone, L.; Aime, S. Inorg. Chim. Acta 1975, 15, 33.
(12) Chi, Y.; Liu, B.-J.: Lee, G.-H.; Peng, S.-H. Polyhedron 1989, 8,
2003.
(13) Lambert, J. B.; Larson, E. G.; Bosch, R. J.; TeVrucht, M. L. E.
J. Am. Chem. SOC. 1985, 107, 5443.
(14) (a) Newman, M. S.; Dhawan, B.; Hashem, M. M.; Khanna, V. K.;
Springer, J. M. J. Org. Chem. 1976,41, 3925. (b) Vaughn, T. H.; Vogt,
R. R.; Nieuwland, J. A. J. Am. Chem. SOC. 1934,56, 2120.
(15) Kubas, G. L.; Wasserman, H. J.; Ryan, R. R. Organometallics 1985,4, 2012. (b) Kubas, G. J. Inorg. Chem. 1983,22,692.
(16) Nolan, S. P.; Hoff, C. D. In Organometallic Syntheses; King, R.
B., Eisch, J. J., Eds.; Elsevier: New York, 1989; Vol. 4, p 58.
(17) Piper, T. S.; Wilkinson, G. J. Inorg. Nucl. Chem. 1956, 3, 104.
(18) Bruce, M. I.; Humphrey, M. G.; Matisons, J. G.; Roy, S. K.;
Swincer, A. G. Aust. J. Chem. 1984, 37, 1955.
(19) Johnson, B. F. G.; Lewis, J.; David, A. P. J. Chem. Soc., Dalton Trans. 1981, 407.
only one reaction are reported here.
In a 100-mL round-bottom reaction flask, O S ~ ( C O ) , ~ (456 mg,
0.503 mmol) was treated with sublimed Me3N0 (91 mg, 1.04 mmol) in a mixture of dichloromethane (50 mL) and acetonitrile (20 mL) a t ambient temperature for 60 min. After evaporation of the solvent in vacuo, the acetylide complex CpW(CO),C=CPh (240 mg, 0.553 mmol) was added, and the reaction mixture was
then dissolved in a toluene solution (30 mL) and brought to reflux
for 30 min. Finally the solvent was evaporated, and the residue was separated by thin-layer chromatography (silica gel, di- ch1oromethane:hexane = l:l), giving 90 mg of C ~ W O S ~ ( C O ) ~ ~ - (CECPh) as a red crystalline solid (0.073 mmol, 15%) and 35 mg
of CpWOs2(CO),(C=CPh) (la) as a yellow crystalline solid (0.003
mmol, 8%). Crystals of la suitable for an X-ray diffraction study were obtained from a layered solution of dichloromethane- methanol a t room temperature. Spectroscopic data for complex
la: MS (FAB, '=Os, le4W) m / z 956 (M'); IR (C6H12) u(C0) 2077
(s), 2043 (vs), 2005 (vs), 1995 (s), 1965 (s), 1924 (m) cm-'; 'H NMR
(CD2C12, 294 K) 6 7.68 (d, 2 H), 7.37 (t, 2 H), 7.20 (t, 1 H), 5.29
(s, 5 H); 13C NMR (CD2C12, 294 K) 6 208.3
( J w x
= 165 Hz,W-CO), 204.1
( J w x
= 157 Hz, W-CO), 179.4 (OS-CO), 137.5(CCPh), 73.8
(Jwx
= 16 Hz, CCPh). Anal. Calcd forC21Hlo08WOsz: C, 26.42; H, 1.06. Found: C, 26.40; H, 1.10. Carbonylation of CpWOs3(CO),,(C=CPh). Toluene (40 mL) and the red tetrametallic acetylide complex CpWOs3- (CO)ll(C=CPh) (58 mg, 0.047 mmol) were combined in a 200-mL
pressure bottle. A partial vacuum was drawn over the toluene
solution; then the bottle was charged with carbon monoxide to
a pressure of 30 psi. The bottle was then placed in a preheated
oil bath, and the solution was stirred a t 120 "C for 6 h. After the solvent was evaporated, the residue was separated by thin-layer
chromatography (silica gel, dichloromethane:hexane =
l:l),
giving38 mg of C ~ W O S , ( C O ) ~ ( C = C P ~ ) (0.040 mmol, 85%) in addition
to a trace amount of Os3(CO)12 (not determined).
Preparation of Cp*WOsZ(CO),(C=CPh). The title complex (yield 2%) was prepared under conditions similar to those for complex l a , in addition to 10% of the red tetrametallic acetylide
complex C ~ * W O S , ( C O ) ~ ~ ( C = C P ~ ) . Spectral data for complex
2a: MS (FAB, lg20s, lMW) m / z 1026 (M'); IR (C&12) u(C0) 2076
(s), 2041 (vs), 2004 (vs), 1991 (s), 1963 (m), 1905 (w) cm-l; 'H
NMR
(s, 15 H). Anal. Calcd for C26Hzo08WOs2: C, 30.48; H , 1.97.
Found: C, 30.38; H , 1.99.
Preparation of C ~ W O S ~ ( C O ) ~ ( C = C " B U ) . The title complex
(yield 11%) was prepared under conditions similar to those for complex l a , in addition to 19% of the red tetranuclear acetylide complex CpWOs3(CO)ll(C=CnBu). Spectroscopic data for com-
plex 3a: MS (FAB, 1920s, le4W)
m / z
936 (M'); IR (CsH12) u(C0)2075 (s), 2040 (vs), 2003 (vs), 1991 (s), 1975
(vw),
1961 (m), 1924(w) cm-'; 'H NMR (CDC13, 294 K) 6 5.33 (s, 5 H), 3.04 (m, 2 H),
1.73 (m, 1 H), 1.61 (m, 1 H), 1.39 (m, 2 H), 0.93 (t, 3 H). Anal. Calcd for ClgH,,08WOs2: C, 24.42; H, 1.51. Found: C, 24.33; H, 1.48.
In a 50-mL round-bottom reaction flask, the metal acetylide CpW(CO)3C=
CPh (37 mg, 0.039 mmol) and R u ~ ( C O ) ~ ~ (37 mg, 0.039 mmol) in
toluene (35 mL) were heated to reflux for 30 min. After evapo- ration of the solvent in vacuo, the residue was separated by thin-layer chromatography (silica gel, hexane:dichloromethane = 1:l) and recrystallization, giving 20 mg of C ~ W R U ~ ( C O ) ~ ( C =
CPh) (4) as a n orange crystalline material (0.018 mmol, 46%).
Crystals of 4 suitable for X-ray structural determination were obtained by recrystallization from a layered solution of di- chloromethane-methanol. Spectroscopic data for complex 4: MS
(FAB, lo2Ru, le4W) m / z 778 (M+); IR (C6H12) u(C0) 2076 (s), 2069
(m), 2044 (vs), 2033 (vs), 2010 (vs), 2003 (m), 1994 (m), 1975 (m),
1966 (w), 1952
(vw),
1930(vw),
1918(vw)
cm-'; 'H NMR (CDCl,,294 K) 6 7.64-7.24 (m, P h ) , 5.56 (s, Cp, 4b), 5.25 (s, Cp, 4a); 13C
NMR (THF-d,, 294 K) 6 210.8 (Jw4 = 160 Hz, W-CO, 4a), 210.7
( J w x = 165 Hz, W-CO, 4a), 207.6 ( J w x = 169 Hz, W-CO, 4b),
197.0 (Ru-CO, 4b), 196.6 (Ru-CO, 4a), 168.9 ( J w x = 139 Hz,
CCPh, 4b), 168.6 (CCPh, 4a), 98.3
(Jw.c
= 22 Hz, CCPh, 4b), 85.6(JW4 = 2 1 Hz, CCPh, 4a). Anal. Calcd for C2,Hl0O8WRu2: C ,
32.49; H, 1.30. Found: C, 32.37; H, 1.35.
P r e p a r a t i o n of C p * w R ~ ~ ( C o ) ~ ( c ~ P h ) . The toluene so-
lution of a mixture of Ru3(C0),2 (100 mg, 0.156 mrnol) and
(CDC13, 294 K) 6 7.62 (d, 2 H), 7.31 (t, 2 H), 7.20 (t, 1 H), 1.93
P r e p a r a t i o n of CpWRu,(CO),(C=CPh).
Downloaded by NATIONAL TAIWAN UNIV on August 12, 2009
Cp*W(CO),C=CPh (90 mg, 0.178 mmol) was heated a t reflux for 30 min. After TLC separation and recyrstallization, the
acetylide complex Cp*WRu2(CO)8(C~CPh) (5); 86 mg, 0.10
mmol) was obtained in 57% yield. Spectroscopic data for complex
5: MS (FAB, loZRu, law)
m / z
848 (M'); IR (C6H12) u(C0) 2071(m), 2064 (vs), 2038 (s), 2024 (vs), 2005 (s), 2001 (vs), 1986 (m),
1970 (w), 1965 (w), 1955 (w), 1936
(vw),
1913 (vw) cm-'; 'H NMR(CD2C12, 273
K)
6 7.65-7.27 (m, Ph), 2.30 (s, Me, 5b), 1.93 (s, Me,5a); 13C NMR (THF-d8, 226
K)
6 218.5 (W-CO, 5a), 217.3(W-CO, Sa), 214.5
(Jw4
= 169 Hz, W-CO, 5b), 175.3 (CCPh,5b), 173.0
(Jwc
= 141 Hz, CCPh, 5a), 100.1(Jw4
= 23 Hz, CCPh,5b), 90.2 (CCPh, 5a). Anal. Calcd for C~,J&O~WRU~: C, 36.89;
H, 2.38. Found:
C,
36.86; H, 2.42.P r e p a r a t i o n
of
C p W R u 2 ( C O ) 8 ( C ~ C 6 H , F ) . The toluenesolution of a mixture of R U , ( C O ) ~ ~ (75 mg, 0.117 mmol) and
CpW(C0)3C=CC6H4F (80 mg, 0.177 mmol) was heated a t reflux
for 30 min. After TLC separation and recrystallization, the
acetylide complex C ~ W R U , ( C O ) ~ ( C I C C ~ H ~ F ) (6; 73 mg, 0.091
mmol) was obtained in 52% yield. Spectroscopic data for complex
6: MS (FAB, lo2Ru, le4W) m / z 796 (M'); IR (C6HlZ) u(C0) 2075
(s), 2069 (s), 2042 (vs), 2032 (vs), 2009 (vs, br), 1994 (m), 1979
(vw),
1972 (m), 1963 (w), 1949 (vw), 1928
(vw),
1914(vw)
cm-'; 'H NMR(CDC13, 294 K) 6 7.63 (m, 1 H), 7.52 (m, 1 H), 7.02 (m, 2 H), 5.57
(s, Cp, 6b), 5.25 (s, Cp, 6a); 13C NMR (CDC13, 294
K)
6 211.3(W-CO, 6a), 210.8 (W-CO, 6a), 207.7
( J w x
= 169 Hz, W-CO,6b), 196.9 (Ru-CO, 6b), 196.5 (Ru-CO, 6a), 88.4 (Cp, 6a), 86.2 (Cp, 6b). Anal. Calcd for C2,HSFO8WRu2: C, 31.76; H , 1.14. Found: C, 31.68; H, 1.13.
Preparation
of
C p W R u 2 ( C 0 ) 8 ( ~ C 6 H 4 0 M e ) . The toluenesolution of a mixture of R U , ( C O ) ~ ~ (165 mg, 0.248 mmol) and
CpW(CO),C=CC6H40Me (180 mg, 0.388 mmol) was heated a t reflux for 30 min. After TLC separation and recrystallization,
the acetylide complex C ~ W R U ~ ( C O ) ~ ( C ~ C ~ H ~ ~ M ~ ) (7; 162 mg,
0.200 mmol) was obtained in 52% yield. Spectroscopic data for
complex 7: MS (FAB, lo2Ru, le4W) m / z 808
(M');
IR (C&12)u(C0) 2077 (s), 2070 (s), 2043 (vs), 2033 (vs), 2011 (vs, br), 1991
(m), 1972 (m), 1964 (w), 1948 (vw), 1928 (vw), 1913
(vw)
cm-';'H
NMR (toluene-d8, 244K)
6 7.95 (d, 1.18 H , 7b), 7.84 (d, 0.82H, 7a), 6.90 (d, 0.82 H, 7a), 6.78 (d, 1.18 H, 7b), 4.63 (s, 2.95 H,
Cp, 7b), 4.45 (s, 2.05 H , Cp, 7a), 3.52 (s, 2.05 H , OMe, 7a), 3.41
(s, 2.95 H, OMe, 7b); 13C NMR (CDCl,, 244
K)
6 211.3 (W-CO,7a), 210.8 (W-CO, 7a), 207.7
( J w c
= 178 Hz, W-CO, 7b), 196.7(br, Ru-CO), 166.1 (CCAr, 7b), 165.1 (CCAr, 7a), 98.6 (CCAr, 7b), 84.6 (CCAr, 7a), 88.4 (Cp, 7a), 86.0 (Cp, 7b). Anal. Calcd
for C22H120sWR~2: C, 32.77; H, 1.50. Found: C, 32.72; H, 1.47.
P r e p a r a t i o n
of
C ~ W R U ~ ( C O ) ~ ( C ~ ~ B U ) . The toluene so-lution of a mixture of R u ~ ( C O ) ~ ~ (189 mg, 0.296 mmol) and
CpW(C0)3C=CtBu (184 mg, 0.443 mmol) was heated a t reflux for 40 min. After TLC separation and recrystallization, the
acetylide complex C ~ W R U ~ ( C O ) ~ ( C = C ~ B U ) (8); 223 mg, 0.294
mmol) was obtained in 66% yield. Selected spectroscopic data
for complex 8: MS (FAB, lo2Ru, law) m / z 758 (M'); IR (C6H12)
u(C0) 2074 (w), 2068 (vs), 2039 (w), 2031 (vs), 2005 (vs), 1991 (s),
1981 (w), 1971 (w), 1961 (m), 1928 (w, br) cm-'; 'H NMR (CDCl,,
294
K)
6 5.47 (s, 4.8 H, 8b), 5.38 (s, 0.2 H, 8a), 1.42 (s, 0.36 H,8a), 1.35 (a, 8.64 H, 8b). 13C NMR (CD2C12, 294
K)
6 211.8(W-CO, 8a), 209.9 (W-CO, 8a), 208.0
( J w x
= 172 Hz, W - C O ,8b), 197.6 (Ru-CO, 8b), 196.9 (Ru-CO, 8a), 87.1 (Cp, 8a), 86.1 (Cp, 8b). Anal. Calcd for CI9Hl4O8WRu2: C, 30.17; H, 1.87. Found: C, 30.08; H, 1.86.
P r e p a r a t i o n
of
C~WRU~(CO)~(C=C!"P~). The toluene so-lution of a mixture of R U , ( C O ) ~ ~ (223 mg, 0.349 mmol) and
CpW(CO),C=C"Pr (210 mg, 0.524 mmol) was heated a t reflux for 30 min. After TLC separation and recrystallization, the acetylide complex CpWRuz(CO)8(C=CnPr) (9; 221 mg, 0.298 mmol) was isolated in 57% yield. Selected spectroscopic data
for complex 9 MS (FAB, lo2Ru,
law)
m/z 744 (M'); IR (C6H12)u(C0) 2074 (w), 2068 (s), 2040 (m), 2030 (vs), 2005 (vs), 1998 (m), 1991 (s), 1980 (vw), 1970 (w), 1960 (m), 1924
(vw,
br) cm-'; 'H NMR (CDCl,, 294 K) 6 5.49 (s, 4 H, 9b), 5.31 (s, 1 H , 9a), 2.93 (t, 0.4 H, 9a), 2.84 (t, 1.6 H, 9b), 1.81 (m, 0.4 H, 9a), 1.68 (m, 1.6 H, 9b), 1.00 (t, 0.6 H, 9a), 0.98 (t, 2.4 H, 9b); 13C NMR (CD2C12, 294K)
6 211.6(Jw4
= 163 Hz, W-CO, 9a), 210.1(Jw4
= 166Hz, W-CO, 9a), 208.2 (Jwx = 170 Hz, W-CO, 9b), 197.2
(Ru-CO, 9b), 196.8 (Ru-CO, 9a), 163.1 (CC"Pr, 9a), 161.9 ( J w ~
= 139
Hz,
CCnPr, 9b), 97.9(Jwx
= 22 Hz, CC"Pr, 9b), 87.7 (Cp,9a), 87.0 (CC"Pr, 9a), 86.2 (Cp, 9b). Anal. Calcd for
C18H1208WRu2: C, 29.11;
H,
1.62. Found: C, 29.10; H, 1.59.P r e p a r a t i o n of CpMoRu2(CO)8(C=CPh). The toluene so-
lution of a mixture of R U , ( C O ) ~ ~ (357 mg, 0.559 mmol) and
CpMo(CO),C=CPh (290 mg, 0.838 mmol) was heated a t reflux for 40 min. After TLC separation and recrystallization, the acetylide complex CpMoRu2(CO)&C=CPh) (loa; 244 mg, 0.353 mmol) was isolated in 42% yield. Selected spectroscopic data
for complex loa: MS (FAB, ' q u , %Mol m / z 692 (M');
IR
(C6H12)u(C0) 2078 (vs), 2045 (vs), 2012 (vs), 2005 (s), 1978 (m), 1948 (w),
1920 (vw, br) cm-'; 'H NMR (CDCl,, 294
K)
6 7.64 (d, 2 H), 7.35(t, 2 H), 7.26 (t, 1 H), 5.14 (s, 5 H); 13C NMR (CDC13, 294
K)
6226.8 (Mo-CO, 1 C), 225.3 (Mo-CO, 1 C), 197.9 (Ru-CO, 3 C),
196.4 (Ru-CO, 3C, broad), 91.0 (Cp, 5 C). Anal. Calcd for
C 2 1 H 1 0 0 8 M ~ R ~ 2 : C, 36.64; H, 1.46. Found: C, 36.51; H, 1.46.
P r e p a r a t i o n of C ~ * M ~ R U ~ ( C O ) ~ ( C = C P ~ ) . The toluene
solution of a mixture of Ru,(CO),~ (300 mg, 0.469 mmol) and Cp*Mo(CO),C=CPh (293 mg, 0.704 mmol) was heated a t reflux for 30 min. After TLC separation and recrystallization, the
acetylide complex C ~ * M O R U ~ ( C O ) ~ ( C = C P ~ ) (1 la; 304 mg, 0.399
mmol) was isolated in 57% yield. Selected spectroscopic data
for complex lla: MS (FAB, ' q u , %Mo)
m/z
762 (M+);IR
(C6H12)v(C0) 2072 (vs), 2040 (vs), 2007 (vs), 1999 (s), 1971 (s), 1972 (w,
br), 1885 (w, br) cm-'; 'H NMR (CDCl,, 294 K) 6 7.59 (d, 2 H),
7.31 (t, 2 H), 7.22 (t, 1 H), 1.76 (s, 15 H); 13C NMR (CDCI,, 244
K)
6 230.0 (Mo-CO, 1 C), 229.4 (Mo-CO, 1 C), 201.4 (Ru-CO,1 C), 197.6 (Ru-CO, 3C), 193.7 (Ru-CO, 1 C), 192.8 (Ru-CO,
1 C), 175.3 (CCPh), 94.5 (CCPh), 103.1 (C5Me5). Anal. Calcd for
C26H2008MoRu2:
c,
41.17; H, 2.66. Found:c,
41.13; H, 2.66.P r e p a r a t i o n of C ~ M O R ~ ~ ( C O ) ~ ( C ~ Y B U ) . The toluene
solution of a mixture of Ru,(CO),~ (131 mg, 0.205 mmol) and
C~MO(CO)~C=C*BU (104 mg, 0.319 mmol) was heated a t reflux
for 40 min. After TLC separation and recrystallization, the
acetylide complex C ~ M O R U ~ ( C O ) ~ ( C = C ~ B U ) (12; 80 mg, 0.119
mmol) was isolated in 37% yield. Selected spectroscopic data
for complex 1 2 MS (FAB, '%u, %Mo)
m / z
672 (M'); IR (C6Hldu(C0) 2076 (s), 2070 (w), 2041 (vs), 2032 (m), 2010 (vs), 1999 (s),
1974 (m), 1951
(vw),
1917(vw)
cm-'; 'H NMR (CDCl,, 244K)
6 5.43 (s, 5 H, 12a), 5.34 (s, 12b), 1.38 (s, 12b), 1.33 (s, 9 H, 12a);
13C NMR (CD2C12, 205
K)
6 225.9 (Mo-CO, 1 C, 12a), 225.5(Mo-CO, 1 C, 12a), 223.0 (Mo-CO, 2 C, 12b), 204.0 (Ru-CO,
1 C, 12a), 202.4 (Ru-CO, 1 C, 12a), 200.1 (Ru-CO, 6 C, 12b),
197.4 (Ru-CO, 1 C, 12a), 196.4 (Ru-CO, 1 C, 12a), 194.8
(Ru-CO, 1 C, 12a), 194.3 (Ru-CO, 1 C, 12a). Anal. Calcd for ClSHl4O8MoRu2: C, 34.14; H, 2.11. Found: C, 33.92; H , 2.04.
P r e p a r a t i o n of C ~ * M O R U ~ ( C O ) ~ ( C = C ~ B U ) . The toluene
solution of a mixture of R u ~ ( C O ) ' ~ (43 mg, 0.067 mmol) and
Cp*Mo(CO),C=CtBu (40 mg, 0.101 mmol) was heated a t reflux for 60 min. After TLC separation and recrystallization, the
acetylide complex C ~ * M ~ R U ~ ( C O ) ~ ( C = C ~ B U ) (13b; 27 mg, 0.036
mmol) was isolated in 36% yield. Selected spectroscopic data
for complex 1 3 b MS (FAB, ' q u , %Mo) m / z 742 (M');
IR
(C6H12)u(C0) 2062 (s), 2023 (vs), 1999 (vs), 1986 (s), 1963 (w), 1951 (w),
1912
(vw,
br) cm-'; 'H NMR (CDCl,, 294K)
6 2.08 (s, 15 H), 1.38(s, 9
H);
13C NMR (CDC13, 294K)
6 227.3 (Mo-CO), 199.1(Ru-CO), 186.8 (CCtBu), 115.8 (CCtBu), 103.9 ((&Me5), 35.5
(CMe3), 34.8 (5 Me), 11.6 (3 Me); 13C NMR (CD2C12, 200
K)
6 227.0(Mo-CO, 2 C), 207.0 (Ru-CO, broad, 2 C), 200.6 (Ru-CO,
broad, 2 C), 191.0 (Ru-CO, broad, 2 C). Anal. Calcd for
C 2 4 H 2 4 0 8 M ~ R ~ 2 : C, 39.03; H, 3.28. Found: C, 38.99; H, 3.25.
P r e p a r a t i o n of C ~ M O R U ~ ( C O ) ~ ( C = C C ~ H ~ F ) . The toluene
solution of a mixture of R U ~ ( C O ) ' ~ (193 mg, 0.302 mmol) and
C ~ M O ( C ~ ) ~ C = C C ~ H , F (165 mg, 0.453 mmol) was heated a t reflux
for 30 min. After TLC separation and recrystallization, the
acetylide complex C ~ M O R U ~ ( C ~ ) ~ ( C E C C ~ H ~ F ) (14a; 127 mg,
0.179 mmol) was isolated in 40% yield. Selected spectroscopic
data for complex 14a: MS (FAB, lo2Ru, s8M0) m / z 710 (M'); IR
(C6H12) u(C0) 2077 (vs), 2044 (vs), 2011 (vs), 2004 (S), 1986 (W),
1976 (s), 1948 (m), 1914 (w) cm-I; 'H NMR (CDCl,, 294 K) 6 7.62
(m, 2 H), 7.04 (m, 2 H), 5.15 (s, 5 H); 13C NMR (CDC13, 294 K)
6 226.7 (Mo-CO, 1 C), 225.1 (Mo-CO, 1 C), 197.8 (Ru-CO, 3
C), 196.4 (Ru-CO, broad, 3 C), 171.8 (CCAr), 91.6 (CCAr), 90.8 (Cp, 5 C). Anal. Calcd for C2,HSFO8MoRu2: C, 35.71; H, 1.28. Found: C, 35.63; H, 1.28.
X-ray Crystallography. Diffraction measurements were carried out on a Nonius CAD-4 fully automatic four-circle dif-
Downloaded by NATIONAL TAIWAN UNIV on August 12, 2009
2712
Organometallics, Vol.
9,No.
10, 1990Hwang et al.
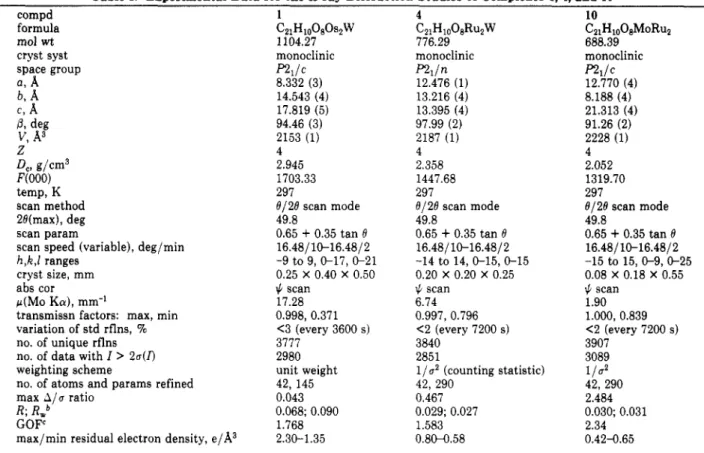
Table I. Experimental Data for the X-ray Diffraction Studies of Complexes 1.4, and 10'
compd 1 4 10
formula C21H10080SZW CZ1H1008RU2W C2IHloOBMoRu2
space group p 2 1 l C R 1 l n R I / C
mol wt 1104.27 776.29 688.39
cryst syst monoclinic monoclinic monoclinic
a,
A
8.332 (3) 12.476 (1) 12.770 (4) b, A 14.543 (4) 13.216 (4) 8.188 (4) c,A
17.819 (5) 13.395 (4) 21.313 (4) P, deg 94.46 (3) 97.99 (2) 91.26 (2)v,
A3 2153 (1) 2187 (1) 2228 (1) Z 4 4 44,
g/cm3 2.945 2.358 2.052 F(000) 1703.33 1447.68 1319.70 temp, K 297 297 297scan method 8/28 scan mode 0/28 scan mode 8/28 scan mode
28(max), deg 49.8 49.8 49.8
scan param 0.65
+
0.35 tan 0scan speed (variable), deg/min 16.48110-16.4812 16.481 10-16.48/2 16.48/10-16.4812
cryst size, mm
abs cor
IC.
scan $ scan $ scantransmissn factors: max, min 0.998, 0.371 0.997, 0.796 1.000, 0.839
variation of std rflns, 90
no. of unique rflns 3777 3840 3907
no. of data with I
>
2 a ( n 2980 2851 3089no. of atoms and params refined 42, 145 42, 290 42, 290
max A / u ratio 0.043 0.467 2.484
R; RWb 0.068; 0.090 0.029; 0.027 0.030; 0.031
GOF' 1.768 1.583 2.34
max/min residual electron density, e/A3 2.30-1.35 0.8W.58 0.42-0.65
[ ~ W ~ F ~ - F , ~ ~ / / ~ W ~ F ~ ~ ~ ) " ~ .
CGOF
= [x:wlF, - Fc12/(No-Nv)]1'2 ( N o = number of observations; N , = number of variables).0.65
+
0.35 tan 0 0.65+
0.35 tan 8h,k,E ranges -9 to 9, 0-17, 0-21 -14 to 14,O-15,0-15 -15 to 15, 0-9,0-25
0.25 X 0.40 X 0.50 0.20 X 0.20 X 0.25 0.08 X 0.18 X 0.55
~ ( M o K a ) , mm-' 17.28 6.74 1.90
<3 (every 3600 s) <2 (every 7200 s) <2 (every 7200 s)
weighting scheme unit weight l / a 2 (counting statistic)
112
OFeatures common to all determinations: X(Mo K a ) = 0.70930
A;
Nonium CAD-4 diffractometer. b R = x l F o - FJ/)JF,I; R, =fractometer. In general, the space group and parameters of unit
cell dimensions were determined and refined from 25 randomly
selected reflections, with a 20 angle about 20°, obtained by using the CAD-4 automatic search, center, index, and least-squares routines. All data reduction and structural refinement were
performed by using the NRCC-SDP-VAX software packages. T h e
structures were solved by the heavy-atom method and refined by least-squares cycles. For complex 1, the tungsten and the osmium metal atoms were refined anisotropically and the rest of the non-hydrogen atoms were refined isotropically. For com-
plexes 4 and 10, all non-hydrogen atoms were refined with an-
isotropic thermal parameters; the hydrogen atoms of the phenyl group and the cyclopentadienyl ligand were added a t the idealized positions and included in the structure factor calculations. The data collection and refinement parameters for complexes 1,4, and
10 are summarized in Table
I.
Atomic positional parameters forcomplex 1 are found in Table 11, whereas some selected bond
angles and lengths are given in Table
V.
The correspondingparameters for complex 4 are given in Tables
I11
and VI and forcomplex 10 in Tables IV and VII, respectively.
Results and Discussion
Preparation
of the WOsz Acetylide Complexes.
Treatment of LW(CO),C=CR (R = Ph,
L
=Cp, Cp*;
R
=
"Bu, L
= C p )with the lightly stabilized triosmium
complex
OS,(CO)~&H~CN)~
in refluxing toluene (110
"C,
30
min) provided a pale yellow trinuclear heterometallic
acetylide complex (1,
L
=Cp,
R
=Ph; 2,
L
=Cp*,
R
=Ph; 3,
L
= Cp, R = "Bu) in low yield (2-ll%), in addition
to the red tetrametallic complex LWOS,(CO)~~(C=CR)
(10-19%
Thermolysis of the tetrametallic acetylide
complexes under a CO atmosphere induced cluster frag-
mentation and produced the trinuclear WOsz acetylide
complexes in high yield (78435%). The latter has been
considered as an alternative, complementary method to
(20) Chi, Y.; Lee, G.-H.; Peng, S.-H.: Wu, C.-H. Organometallics 1989,
8, 1574.
generate large quantities of the WOs, acetylide complexes
in our laboratory. Furthermore, other chemistry of these
WOs3
acetylide complexes, such as the reactions with di-
substituted alkynesz1 and with mononuclear metal ace-
tylide complexes,2z has also been developed.
The structural information of these WOsz acetylide
complexes was initially provided by a 13C NMR study. The
l3CI1H)
NMR spectrum of
1 showed two signals at 6 137.5
and 73.8, reminiscent of those reported for the two ace-
tylide carbon nuclei (6 172.9 and 112.7) in the WFe, ana-
logue CpWFez(CO)8(C=CTol).23 The latter is a hetero-
metallic cluster in which the acetylide ligand functions as
a five-electron donor, a-bonded to the tungsten atom while
employing its two orthogonal alkyne a-bonds to bridge the
unique Fe-Fe edge. However, since the color as well as
the IR spectra of these osmium complexes in the region
of CO absorption differs substantially from those of the
WFe2 derivatives, an X-ray diffraction study was carried
out on complex 1 to determine the location of the acetylide
ligand in the WOsz derivatives.
Description of the Structure of C ~ W O S ~ ( C O ) ~ ( C =
CPh)
(1).
As
indicated in Figure 1, this molecule has
a
triangular WOsz core structure with distances W-Os(1)
=2.830 (2)
A,
W-042)
=2.916 (2)
A,
and Os(l)-Os(2) =
2.814 (2)
A,
in which the tungsten atom is associated with
a Cp ring and two CO ligands, while each of the osmium
atoms is linked to three, mutually orthogonal, terminal CO
ligands. The acetylide moiety is coordinated to the WOsz
triangular face with its a-carbon bound to all three metal
atoms with bond distances W-C(15)
=2.20 (3)
A,
Os-
(21) Wu, C.-H.; Chi, Y.; Peng, S.-H.; Lee, G.-H. Organometallics, in (22) Wu, C.-H.; Chi, Y.; Pew, S.-H.: Lee. G.-H. J. Chem. Soc..Dalton
press.
Trans., in press.
P. J . Chem. Soc., Chem. Commun. 1983, (23) Green, M.; Marsden, K.; Salter, I. D.; Stone, F. 446. G. A.; Woodward,
Downloaded by NATIONAL TAIWAN UNIV on August 12, 2009
Organometallics,
Vol.
9,No.
10, 19902713
Table 11. Atomic Coordinates and Equivalent IsotropicDisplacement Coefficients for CpWOs,(CO),(C=CPh) (1)
X Y z B;,,"
A'
0.48889 (14) 0.26829 (15) 0.22595 (14) 0.416 (4) 0.619 (4) 0.654 (4) 0.105 (5) 0.154 (4) 0.393 (4) 0.314 (4) 0.113 (4) 0.118 (4) 0.188 (4) 0.118 (5) -0.012 (5) -0.014 (4) 0.491 (5) 0.405 (3) 0.589 (4) 0.648 (4) 0.753 (5) 0.793 (6) 0.736 (5) 0.630 (5) 0.360 (3) 0.702 (4) 0.754 (4) 0.002 (3) 0.079 (4) 0.464 (4) 0.358 (3) 0.036 (4) 0.149 0.285 0.151 -0.089 -0.091 0.617 0.786 0.877 0.762 0.587 0,81811 (8) 0.96203 (8) 0.83355 (8) 0.7582 (21) 0.7191 (21) 0.8829 (23) 1.043 (3) 0.9141 (24) 1.046 (3) 0.7106 (21) 0.7723 (23) 0.8041 (24) 0.8884 (24) 0.947 (3) 0.900 (3) 0.8120 (24) 0.891 (3) 0.9351 (19) 0.8897 (22) 0.978 (3) 0.985 (3) 0.908 (4) 0.826 (3) 0.813 (3) 0.7252 (20) 0.6553 (23) 0.9247 (22) 1.0919 (19) 0.8846 (22) 1.0962 (21) 0.6360 (20) 0.7365 (21) 0.748 0.909 1.013 0.928 0.759 1.034 1.049 0.913 0.768 0.752 0.16703 (6) 0.13507 (6) 0.25760 (6) 0.0798 (17) 0.2004 (17) 0.1258 (19) 0.1568 (21) 0.0470 (19) 0.0810 (20) 0.2740 (17) 0.1687 (19) 0.3731 (20) 0.3791 (19) 0.3249 (22) 0.2848 (21) 0.3133 (20) 0.2734 (21) 0.2272 (15) 0.3458 (18) 0.3692 (21) 0.434 (3) 0.475 (3) 0.4554 (23) 0.3874 (21) 0.0233 (16) 0.2167 (19) 0.1005 (18) 0.1733 (16) -0.0045 (18) 0.0514 (17) 0.2923 (16) 0.1219 (17) 0.405 0.415 0.315 0.244 0.296 0.337 0.453 0.520 0.487 0.370 1.35 (5) 1.55 (5) 1.24 (4) 1.6 (5) 1.6 (5) 2.2 (6) 2.9 7) 2.3 (6) 2.6 (6) 1.8 (5) 2.3 (6) 2.4 (6) 2.4 (6) 3.2 (7) 2.9 (7) 2.5 (6) 3.0 (7) 1.0 (4) 2.0 (5) 2.8 (7) 4.1 (9) 4.9 (10) 3.6 (8) 3.0 (7) 3.9 (6) 4.9 (7) 4.5 (6) 3.6 (5) 4.6 (6) 4.2 (6) 3.7 (5) 4.3 (6) 3.6 3.4 3.9 3.6 3.6 3.7 4.9 5.5 4.4 3.7Bi,
is the mean of the principal axes of the thermal ellipsoid./-.
c 2 0
b
\
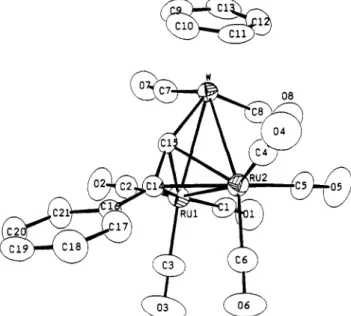
Figure 1. ORTEP diagram of CpWOs2(CO)&C=CPh) (1).
(1)-C(15) = 2.16 (3)
A,
and Os(2)-C(15)
=1.96 (3) 8, and
with its &carbon linked to W and Os(1) atoms with dis-
tances Os(l)-C(14) = 2.17
(4)A
and W-C(14) = 2.36 (4)
A.
The acetylide C-C bond distance (1.23 (5)
A)
in this
compound is only a little longer than the average C-C
Table 111. Atomic Coordinates and Equivalent Isotropic Displacement Coefficients for CpWRu,(CO),(CdPh) (4)
W 0.06565 (3) 0.152711 (25) 0.232915 (24) 2.226 (14) X Y z B b :
A'
Ru(1) 0.01359 (5) Ru(2) -0.06312 (5) C(1) 0.1463 (7) C(2) 0.0384 (7) C(3) -0.0590 (8) C(4) -0.1360 (7) C(5) 0.0559 (7) C(6) -0.1425 (8) C(7) 0.1303 (7) C(8) 0.1938 (8) C(9) 0.0571 (11) C(10) -0.0335 (9) C(l1) 0.0016 (9) C(12) 0.1128 (8) C(13) 0.1507 (9) C(14) -0.1503 (7) C(15) -0.0687 (7) C(16) -0.2617 (6) C(17) -0.3468 (7) C(18) -0.4510 (8) C(19) -0.4724 (8) C(20) -0.3890 (9) C(21) -0.2823 (7) O(1) 0.2222 (6) O(2) 0.0583 (6) O(3) -0.1027 (7) O(4) -0.1778 (6) O(5) 0.1245 (6) O(6) -0.1908 (8) O(7) 0.1733 (6) O(8) 0.2736 (6) H(9) 0.055 H(10) -0.113 H(11) -0.047 H(12) 0.160 H(13) 0.227 H(17) -0.331 H(18) -0.515 H(19) -0.550 H(20) -0.406 H(21) -0.221 0.25009 (5) 0.33911 (5) 0.3217 (6) 0.1407 (7) 0.3353 (8) 0.3451 (7) 0.4236 (6) 0.4459 (7) 0.0634 (7) 0.2410 (7) 0.0034 (7) 0.0565 (10) 0.1431 (8) 0.1409 (8) 0.0560 (9) 0.2249 (6) 0.1789 (5) 0.2087 (6) 0.2556 (7) 0.2338 (8) 0.1642 (8) 0.1175 (8) 0.1390 (7) 0.3666 (6) 0.0838 (5) 0.3884 (7) 0.3511 (6) 0.4739 (5) 0.5078 (6) 0.0066 (5) 0.2861 (6) -0.062 0.038 0.198 0.196 0.032 0.310 0.268 0.149 0.065 0.104 0.42250 (5) 0.24900 (5) 0.4574 (6) 0.5210 (6) 0.5028 (7) 0.1146 (7) 0.2284 (7) 0.3009 (7) 0.3440 (7) 0.2635 (7) 0.1469 (7) 0.1074 (7) 0.0610 (7) 0.0721 (7) 0.1232 (7) 0.3243 (6) 0.2912 (6) 0.3451 (6) 0.2886 (7) 0.3003 (8) 0.3706 (9) 0.4289 (7) 0.4179 (7) 0.4774 (5) 0.5830 (5) 0.5503 (6) 0.0335 (5) 0.2174 (6) 0.3305 (6) 0.3995 (5) 0.2734 (6) 0.186 0.111 0.025 0.046 0.140 0.237 0.257 0.381 0.482 0.464 2.57 (3) 2.53 (3) 3.0 (4) 3.3 (4) 4.4 (5) 3.6 (4) 3.6 (4) 4.3 (5) 3.7 (4) 3.9 (4) 5.0 (6) 5.1 (6) 4.4 (5) 4.1 (5) 4.7 (5) 2.6 (4) 2.4 (3) 2.4 (3) 3.6 (4) 4.6 (5) 4.7 (5) 4.6 (5) 3.8 (4) 5.7 (4) 5.5 (4) 7.6 (5) 5.5 (4) 6.4 (4) 7.6 (5) 5.4 (4) 6.1 (4) 5.1 5.4 4.8 4.6 5.1 4.4 5.2 4.9 5.3 4.5Bi,
is the mean of the principal axes of the thermal ellipsoid.distance of acetylene molecules (1.20
The most salient feature of the structure is the orien-
tation of the acetylide moiety, which
is
a-bonded to an
Os
atom and a t the same time forms a transverse bridge acros9
the second
W-Os
bond. Therefore, the structure of 1 is
related to the CpWFe2(CO)8(C=CTol) by a
120°
rotation
of the acetylide ligand. The heterometallic acetylide
cluster CpNiFe2(CO)6(C=CtBu) also shows a similar
asymmetric
arrange men^?^
Description of the Solution Dynamics of the W 0 s 2
Complexes.
The acetylide ligand of these
WOsp
com-
plexes is static on the time scale of
'3c
NMR spectroscopy.
The
13C
NMR spectrum of a 13CO-enriched sample of
complex 1 exhibits two W-CO signals a t 6 208.6 and 204.0,
a sharp O S ( C O ) ~
signal a t 6 179.7, and two very broad
O s 4 0 signals a t 6 178.8 and 173.4 a t 295
K
in toluene-ds.
When the temperature was increased to 350
K,
the Os(C-
O ) ,
signal a t
6179.7 remained unchanged but the broad
Os-CO signals a t
6178.8 and 173.4 merged into a sharp
signal a t 6 176.5. This behavior is consistent with the
presence of two, independent, localized 3-fold exchanges
of
the CO ligands of the OS(CO)~
unit. One %fold rotation,
( 2 4 ) March, J. Aduanced Organic Chemistry, 3rd ed.; Wiley: New (25) Martinetti, A.; Sappa, E.; Tiripicchio, A.; Camellini, M. T. J .
York, 1985; Chapter 1.
Organomet. Chem. 1980, I97, 335.
Downloaded by NATIONAL TAIWAN UNIV on August 12, 2009
2714 Organometallics, Vol.
9,No.
10, 1990Table IV. Atomic Coordinates and Equivalent Isotropic Disolacement Coefficients for CDMORU.(CO).(C=(
Hwang et al.
Table VII. Relevant Bond Distances
(A)
and Angles (deg) of CDMoRu,(CO).(C=CPh) (10) X Y 2 Bi,," A2 Ru(1) 0.71869 (4) 0.46159 (7) 0.070708 (25) 2.403 (22) Ru(2) 0.87172 (4) 0.22655 (8) 0.09524 (3) 2.97 (3) 0.73598 (5) 0.34163 (8) 0.5996 (6) 0.6026 (10) 0.7169 (6) 0.4012 (9) 0.8218 (6) 0.6254 (10) 0.8887 (6) 0.1398 (9) 0.9978 (6) 0.3537 (12) 0.9385 (6) 0.0524 (11) 0.8640 (6) 0.4783 (10) 0.6627 (6) 0.5566 (10) 0.7400 (12) 0.0920 (12) 0.8106 (7) 0.1975 (14) 0.7576 (9) 0.3200 (12) 0.6509 (9) 0.2983 (16) 0.6412 (9) 0.1552 (17) 0.7203 (5) 0.2079 (8) 0.6274 (5) 0.2643 (8) 0.5142 (5) 0.2296 (8) 0.4394 (5) 0.3283 (9) 0.3339 (5) 0.2930 (10) 0.3015 (6) 0.1626 (10) 0.3744 (6) 0.0641 (10) 0.4796 (6) 0.0917 (9) 0.5275 (5) 0.6812 (8) 0.7181 (4) 0.3595 (7) 0.8822 (5) 0.7242 (8) 0.8967 (4) 0.0877 (7) 1.0699 (5) 0.4355 (9) 0.9347 (4) 0.5666 (8) 0.6222 (5) 0.6797 (7) 0.756 -0.012 0.886 0.181 0.787 0.413 0.589 0.370 0.57s 0.104 0.461 0.420 0.282 0.363 0.22; 0.132 0.352 -0.026 CI.Fix0 0.012the mean of the principal 0.9755 (5) -0.0548 (8) 0.19495 (3) 2.62 (3) 0.0627 (4) 3.8 (4) -0.0146 (3) 3.3 (3) 0.0594 (4) 4.2 (4) 0.0139 (4) 3.6 (4) 0.0937 (4) 5.3 (5) 0.1400 (4) 4.7 (4) 0.1937 (3) 3.9 (4) 0.1956 (3) 4.0 (4) 0.2470 (4) 7.1 (6) 0.2767 (4) 5.4 (5) 0.3024 (4) 5.4 (5) 0.2889 (5) 7.5 (6) 0.2538 (4) 7.4 (6) 0.1044 (3) 2.4 (3) 0.1149 (3) 2.2 (3) 0.1100 (3) 2.4 (3) 0.1362 (3) 3.8 (3) 0.1280 (4) 4.0 (4) 0.0942 (4) 4.1 (4) 0.0681 (4) 4.9 (4) 0.0764 (3) 3.8 (3) 0.0572 (3) 6.6 (3) -0.06550 (23) 5.0 (3) 0.0516 (3) 7.0 (4) -0.0349 (3) 5.5 (3) 0.0903 (3) 8.8 (4) 0.1667 (3) 7.7 (4) 0.2014 (3) 6.2 (3) 0.2035 (3) 6.1 (3) 0.225 6.0 0.278 5.1 0.327 5.5 0.300 6.7 0.238 6.0 0.163 4.1 0.147 4.4 0.089 4.8 0.041 5.2 0.060 4.3
axes of the thermal ellipsoid. Table V. Relevant Bond Distances (A) and Angles (deg) of
(A) Bond Distances
W-OS(l) 2.830 (2) W-Os(2) 2.916 ( 2 )
Os(l)-Os(2) 2.814 (2) W-C(15) 2.20 (3)
O~(l)-C(14) 2.17 (4) W-C(14) 2.36 (4)
W-CO (mean) 1.97 (3) Os-CO (mean) 1.87 (3)
(B) Bond Angles
W-c-0 (mean) 172 (3) Os-C-0 (mean) 176 (3)
Table VI. Relevant Bond Distances (A) and Angles (deg) of CpWOsi(CO)s(Cd!Ph) ( 1 )
Os(l)-C(15) 2.16 (3) 0~(2)-C(15) 1.96 (3)
C(14)-C(15) 1.23 (5) C(16)-C(14) 1.47 (5)
0~(2)-C(15)-C(14) 159 (3) C(15)-C(14)-C(16) 148 (4)
CDWRU,(CO)*(C*Ph) (4) (A) Bond Distances
w-Rul1) 2.998 (1) W-Ru(2) 2.965 (1)
Ru(l)-Ru(2) 2.661 (1) W-C(15) 1.976 (8)
R ~ ( l ) - C ( 1 5 ) 2.128 (8) Ru(2)-C(15) 2.195 (7)
Ru(l)-C(14) 2.297 (8) R~(2)-C(14) 2.188 (8)
C(14)-C(15) 1.31 (1) C(16)-C(14) 1.47 (1)
W-CO (mean) 1.979 (9) Ru-CO (mean) 1.910 (9)
(B) Bond Angles
W-C-0 (mean) 172.1 (8) Ru-C-0 (mean) 177.6 (9)
having a lower energy barrier, produces the sharp signals
a t 6 179.7 a t room temperature; the second, having a rel-
atively greater activation barrier, reaches the limit of fast
W-C(15)-C(14) 162.5 (6) C(15)-C(14)-C(16) 142.5 (8)
(A) Bond Distances
Mo-Ru(l) 2.828 (1) Mo-Ru(2) 2.927 (1)
Ru(l)-Ru(2) 2.784 (1) Mo-C(l4) 2.223 (6)
Ru(l)-C( 15) 2.214 (6) Mo-C(l5) 2.265 (6)
Mo-CO (mean) 1.988 (8) Ru-CO (mean) 1.902 (8)
(B) Bond Angles
Mo-C-0 (mean) 171.0 (7) Ru-C-0 (mean) 177.6 (7)
Ru(l)-C(14) 2.198 (6) Ru(2)-C(14) 1.954 (6)
C( 14)-C( 15) 1.296 (9) C(15)-C(16) 1.475 (9)
Ru(2)-C(14)-C(15) 154.2 (5) C(14)-C(15)-C(16) 144.9 (6)
Table VIII. Numbering Scheme and the Relative Abundance of the Acetylide Derivatives of Type
LMM',(CO)B(C4R)
re1 abundance of each isomer L M M' R c p W os Cp* W
os
Cp Wos
Cp W Ru Cp* W Ru Cp W Ru Cp W Ru Cp W Ru Cp W Ru Cp Mo Ru Cp* Mo Ru Cp Mo Ru Cp* Mo Ru Cp Mo Ru Ph la Ph 2a "Bu 3a Ph 4a Ph 5a CBHIF 6a C6H40Me 7a 'Bu 8a "Pr 9a Ph 1 Oa Ph l l a tBu 12a tBu C6H4F 14a 100% 100% 100% 45 % 15% 45 % 41 % 4% 20 % 100% 100% 95 90 100% 4b 5b 6b 7b 8b 9b 12b 13b 55 % 85% 55% 59% 96% 80 7'0 5 90 100%exchange a t higher temperature. In contrast, rotation of
the CpW(CO), unit cannot be observed because the two
CO ligands on the tungsten atom are diastereotopic. The
coalescences of the two W-CO signals and of the two
O S ( C O ) ~
signals were not observed even a t 370
K,
sug-
gesting that the racemization of 1 has not occurred a t this
temperature.
Preparation a n d Characterization
of the
WRu,
Complexes. In order to investigate the preferred orien-
tation of the acetylide ligand over the triangular face of
the heterometallic complexes, we have carried out the
syntheses of several WRu, derivatives. Complexes
(4,L
=Cp, R
=Ph; 5,
L
=Cp*, R
=Phi 6,
L
=Cp, R = C&&F;
7,
L
=Cp, R
=C&OMe; 8,
L
=Cp, R
=t B ~ ;
9, L
= Cp,
R
=nPr)
were obtained in good yield from the reaction
between Ru,(CO),, and the corresponding tungsten ace-
tylide in a 2:3 molar ratio.
For these WRuz derivatives,
the
'H
NMR spectra and IR spectra in the region of CO
absorption suggest the presence of two isomers in solution
(Table VIII). The assignment of each isomer is further
confirmed by their characteristic 13C NMR data.
In order to prove that the isomerization is due to the
acetylide rotation, we have carried out the structural de-
termination on complex
4.Crystals suitable for X-ray
experiments were obtained by recrystallization from
CH2Clz-hexane a t room temperature. Its molecular
structure is shown in Figure 2, and selected bond angles
and distances are summarized in Table VI. The WRu2
triangle is nearly isosceles with the bond distances W-
Ru(1)
=2.998 (1)
A,
W-Ru(2)
=2.965
(1)A,
and Ru-
(lkRu(2)
=2.661 (1)
A.
The tungsten atom is associated
with two slightly bent CO ligands (LW-C-O(mean)
=172.1
(8)")in addition to a Cp ligand, and each ruthenium atom
islinked to three linear CO ligands (LRu-C-O(mean)
=177.6
(9)").Most interesting, the acetylide moiety is now
u-bonded to the W atom and, quasi-symmetrically,
K -bonded to the two Ru atoms. Therefore, we conclude that
the WRu, derivatives
inthe solid state are isostructural
Downloaded by NATIONAL TAIWAN UNIV on August 12, 2009
- -
I
I
Figure 2. ORTEP
diagram
ofCpWRuz(CO)B(C=CPh)
(4). Scheme II " " " ~ ~ " " ' ~ " " ' " ' ~ " ~ ' 210.0 200.0 1 9 0 . 0
PPm
Figure 3.
Variable-temperature
13CNMR
spectra (CD2C1J of 4,showing
the region of GO resonances.a b
not with the WOsz derivatives but rather with the WFez
derivatives.
The heterometallic acetylide clusters
CpWFe2(CO)8(C=CTol)23
and C O F ~ ~ ( C O ) ~ ( C * S ~ M ~ ~ ) ~ ~
also exhibit a similar symmetric arrangement.
Description of the
13C
N M R
Spectra of the WRu2
Complexes.
Both the lH NMR and IR v(C0) spectra
suggest the presence of two isomers in solution. The so-
lution dynamics of these WRuz complexes are of particular
interest. On the basis of the structural information es-
tablished, we propose that the isomerization is caused by
a 360' rotation of the acetylide ligand over the WRuz
triangle (Scheme
I).
Before we proceed to discuss the
rotation of the acetylide ligands, it is important to un-
derstand the assignment of each isomer and their relative
abundance
a/b
in solution. The 'H NMR and IR spectra
failed to provide adequate information. Fortunately, 13C
NMR spectra in the region of CO resonances can reveal
information on the overall molecular symmetry that allows
us to assign the structure unambiguously.
The 13C NMR spectrum of 4 a t 205
K
exhibits three
W-CO signals a t 6 210.8, 210.5, and 207.3 in the ratio
1:1:2.4 (Figure 3). Therefore, the
first
two resonance lines
are assigned to the "asymmetric" isomer
(4a,
the acetylide
C-C bond bisects the W-Ru bond) and the third one to
the "symmetric" isomer
(4b,
the acetylide C-C bond is
orthogonal to the Ru-Ru bond). This assignment is rea-
(26) Seyferth, D.; Hoke, J. B.; Rheingold, A. L.; Cowie, M.; Hunter, A. D. Organometallics 1988, 7, 2163.
sonable because the CO ligands of the CpW(CO), unit in
the asymmetric isomer
4a
are diastereotopic, whereas in
isomer
4b
the rotation of the CpW(CO)z fragment, having
a relatively smaller activation barrier, would average the
chemical environment of the CO ligands. Furthermore,
five signals a t 6 202.5,200.8, 196.0, 194.2, and 192.4 with
an intensity ratio of 1:1:1:1:2 are assigned to the Ru-CO
resonances of
4a;
the signal at 6 192.4 is double the in-
tensity of the other four signals and therefore corresponds
to two coincident signals. The Ru-CO signals of
4b
were
not observed at this temperature. However, when the
temperature was decreased
to
190
K,
the W-CO signal of
4b
broadened and collapsed slightly, suggesting the slowing
down of the CpW(CO)z rotation, and three very broad
Ru-CO signals at 6 203.0, 198.5, and 189.0 appeared in the
spectrum, consistent with the symmetric nature of
4b.
On
the other hand, when the temperature was increased to
240
K,
isomer 4b showed a broad Ru-CO signal at 6 197.1,
indicating the presence of a rapid 3-fold rotation of the
R u ( C O ) ~
unit. The other three weak Ru-CO signals a t 6
202.9, 194.6, and 192.7 are assigned to isomer
4a.
Again,
the localized Ru(CO)~
rotation in
4a
is responsible for the
observed NMR spectra.
Similar 13C NMR spectra were also observed for other
WRu2 derivatives. The 13C NMR spectrum of 8 at 294
K
exhibits one W-CO signal at 6 208.4 and one Ru-CO signal
at
6198.0 in the ratio 1:3 assigned to isomer
8b,
in addition
to two weak W-CO signals a t
6212.2
and
209.3and one
weak Ru-CO signal a t 6 197.3 assigned to
8a
(Figure 4).
Between 213 and 205
K
the Ru-CO signals assigned to
8b
collapsed to the base line and six relatively weak Ru-CO
signals of equal intensity at 6 204.6, 202.1, 198.0, 196.2,
194.4, and 193.6 emerged from the base line. We assign
these six distinct Ru-CO signals to the asymmetric isomer
8a.When the temperature was decreased to 178
K,
one
broad W-CO signal at
6208.9 and three broad Ru-CO
Downloaded by NATIONAL TAIWAN UNIV on August 12, 2009
2716 Organometallics, Vol.
9,No.
10, 1990Hwang et al.
k
355K--
340K / 337K ---w---,---u- IICK 2i6 2 ; ~ 208 I 20.1 I " 2 0 0 I ' 196 / " ' /1 9 2 " ' l1 8 8 ' ~ ppm ' 'Figure
4. Variable-temperature 13CNMR
spectra(CD2C12)
of8, showing the region of
CO
resonances.signals at 6 205.1,200.2, and 191.0 of isomer
8b
were clearly
observed. Further decreasing of the temperature to 156
K
produced the splitting of these four signals, giving two
W-CO signals at 6 211.6 and 206.7 and six Ru-CO signals
a t 6 207.2, 203.3, 200.8,
200.0,
193.1, and 189.1. We at-
tribute the dynamic motion occurring between 178 and 156
K
to the rotational motion of the CPW(CO)~
unit. This
rotational motion could be of a "pinwheel" type similar to
the localized
R u ( C O ) ~
rotationz7 or a "swinging" motion
with the bulky
Cp
ligand staying away from the acetylide.
Both types of movements would average the environment
of W-CO ligands and exhibit the observed fluxional be-
havior; unfortunately, our data are unable to distinguish
them. From the coalescence temperature (178
K)
of the
W-CO signals, the activation free energy
(AG*)
for the
rotation
of
the CpW(CO)z unit was estimated to be close
to 3 kJ/mol.
Assignment of the 13C NMR data of other WRuz de-
rivatives is
also
based on the generalized experimental
observation that the symmetric isomers
b
give one W-CO
signal but the asymmetric isomers
a
give a pair of dia-
stereotopic W-CO signals. After the major isomer in so-
lution is established from the 13C NMR data, the relative
intensities of the corresponding Cp signals
or
the Cp*
signals in the
'H
NMR spectrum reveal a more accurate
ratio
a/b.
These data are listed in Table
VIII.
Rotation of the Acetylide Ligand
on
the WRu,
Triangle.
The 'H NMR spectrum of 4 in toluene-d,
(Figure 5) shows two Cp signals a t 6 4.78 and 4.58 in the
ratio 1.2:l a t ambient temperature assigned to isomers
4b
and
4a,
respectively. When the system is warmed to 340
K, both Cp signals coalesce to a broad signal at
d4.86,
suggesting the beginning of the interconversion between
4a
and
4b.
The exchange observed is consistent with a
120' rotation
(orthe so-called edge hopping)28
ofthe
(27) Rosenberg, E.; Thoreen-Thorsen, B.; Milone, L.; Aime, S. Inorg. Chem. 1985, 24, 231.
l I l 1 l 1 1 1 1 1 1 1 l
5.00 4.90 4.80 4 . 1 0 4 . 6 0 4 . 5 0 4.40 ppm
Figure 5. Variable-temperature
'H
NMR
spectra (toluene-d8)of 4 in the region of Cp resonances.
Table
IX.
Free Energy(AG')
Data for the AcetylideIsomerization of WRuz Derivatives and Racemerization of
MoRut Derivatives
comdex L R T,. K Av, Hz AG' kJ/mol
WRuz Derivatives 4
CP
Ph 337 80.00 68 6 CP C.5H.J 334 89.0" 67 7 Cp CBH,OMe 300 72.90 61 9 Cp "Pr 353 75.? 72 MoRuz Derivatives 10 Cp Ph 338 163.0b 67 11 Cp* Ph 303 46.4b 63"The chemical shift difference between the Cp signals. 4The chemical shift difference between the diastereotopic W-CO signals.
C h a r t I
acetylide from a W-Ru edge to the Ru-Ru edge
(a
*
b
or
a'
Qb,
Scheme
I).
From the coalescence temperature
of 337
K
for the Cp signals with chemical shift difference
(Av
= 80.0 Hz), an estimate for AG* of 66 kJ/mol is ob-
tained for the barrier of rotation. The kinetic parameters
of other WRu2 derivatives for this process, calculated from
the data of the variable-temperature 'H NMR studies, are
summarized in Table
IX.
However, the second rotational process, racemization of
4a (a
*
a'),
consisting of a 120° rotation of the acetylide
over the tungsten atom, cannot be examined by 'H NMR
studies. On the other hand, the appropriate evidence is
deduced from the variable-temperature 13C NMR data.
The
13CNMR spectrum of 4 in toluene-d, at 310
K
(Figure
S1 of the supplementary material) exhibits two W-CO
signals at 6 212.0 and 211.5 for isomer
4a
and one W-CO
signal a t
6208.5 for isomer 4b. These three signals merge
to
the base line simultaneously on warming to 355
K
and
coalesce to a single line a t 6 209.5 on further warming to
373
K,
indicating that both the racemization
(a
*
a')
and
isomerization
(a
-
b)
occur at the same or about the same
rate.29 These observations indicate that the racemization
may either (i) involve a 240' rotation of the tilted acetylide
(28) Rosenberg, E.; Wang, J.; Gellert, R. W. Organometallics 1988, 7 ,
1093.
(29) The calculated chemical shift of the averaged W-CO signals (based on the chemical shifts at 310 K) for the exchange of the symmetric
and the unsymmetric isomers is at 6 210.0. We believe that the smell
difference (0.5 ppm) is caused by the temperature dependence of the
chemical shifts.
Downloaded by NATIONAL TAIWAN UNIV on August 12, 2009
Vol. 9,
No.
10, 19902717
@
C13un
W
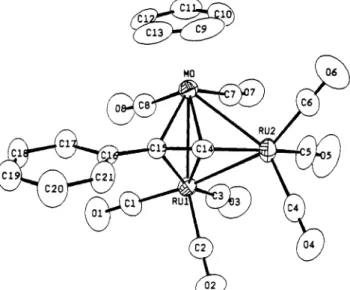
Figure
6. ORTEPdiagram
ofCpMoRu,(CO)B(C=CPh)
(10).(0
<
180") over both ruthenium atoms
(a
Qb
-
a') or
(ii)
involve a common transition state in which the acetylide
C-C bond
is
perpendicular to the WRu2 triangle (Chart
I).
The third possibility involving a direct 120" rotation
of the tilted acetylide
(0
<
180") over the tungsten atom
(a
(=$a')
is presumably a process with a slightly greater
barrier because of the repulsion imposed by the bulky Cp
ligand. Finally, although we are unable to eliminate the
first and third possibilities involving the titled acetylide
moiety, the second pathway involving the vertical acetylide
moiety is preferred. The participation of a similar inter-
mediate containing a vertical C2 vinylidene moiety has
been claimed to account for the isomerization of the eth-
oxyvinylidene ligands of the osmium cluster
H20s3-
Preparation and Characterization of the
MoRu2
Complexes.
The related MoRu2 derivatives
(10,
L
= Cp,
R = Ph;
11,
L
=
Cp*, R = Ph;
12,
L
= Cp, R = tBu; 13,
L
= Cp*, R = tBu; 14,
L
= Cp, R = C6H4F) were syn-
thesized from reactions between R U ~ ( C O ) ~ ~
and the re-
spective molybdenum acetylide under similar conditions.
The X-ray structural determination on phenyl derivative
10
suggests that it has a structure similar to those of
complex
1 and complex 12,28 both possessing the asym-
metric arrangement (Figure 6).
Consistent with the
solid-state structure, its 13C NMR spectrum at 215
K
ex-
hibits two Mo-CO signals at 6 226.4 and 225.3 and six
Ru-CO signals a t 6 202.6, 202.0, 196.6, 196.2, 194.3, and
193.7 (Figure
S2).
When the sample is warmed to 275
K,
the signals at 6 202.6,194.3, and 193.7 and at 6 202.2,196.6,
and 196.2 each coalesce to a singlet a t 6 196.4 and a broad
signal at 6 197.8, indicating the onset of the localized Ru-
(CO), rotation. A similar structure was proposed for the
respective Cp* derivative 11; further support comes from
the 13C NMR spectrum at 244
K,
which shows two Mo-CO
singlets a t
6230.0 and 229.5 in the intensity ratio 1:l and
four Ru-CO signals a t 6 201.4, 197.6, 193.7, and 192.8 in
the ratio 1:3:1:1.
Although the assignment of complexes
10 and 11 insolution is straightforward, the assignment of the tert-butyl
derivatives
12
and 13 is quite different. First, the IR
spectrum of
12
in the region of CO absorptions indicates
the existence of two isomers. The identity of each isomer
is then confirmed by the 13C NMR studies at 205
K,
which
(CO)g(C=CHOEt).30
__cJz___
)k
360K n 338K I " " I " " I ' " I ' " ' I ~ ' ' ~ I ~ ' ' ' 205 200 195 ppm 230 225 220Figure 7.
Variable-temperature
13CNMR spectra (toluene-d8)
of 10 in
the
region ofCO resonances.
Scheme I1
a a'
show the expected eight-line pattern (two Mo-CO and six
Ru-CO signals) at 6 225.9,225.5, 204.0, 202.4, 197.4, 196.4,
194.8, and 194.3 assigned
to
isomer
12a
and the two-line
pattern of isomer
12b
(one Mo-CO and Ru-CO signal) at
6 223.0 and 200.1 in the ratio 1:3. From the intensity ratio
of 13C NMR integration, the abundance
12a:12b
is calcu-
lated to be 19:l. This assignment is in contrast with that
of the recently published report on
12,%
and the incorrect
deduction
was
probably made because of the low concen-
tration of
12b
in solution. Furthermore, the 13C NMR
spectrum
of 13a
at 200
K
exhibits one sharp Mo-CO signal
at 6 227.0 and three broad Ru-CO signals at 6 207.0,200.6,
and 191.9 in the ratio l:l:l:l, consistent with the adoption
of a symmetric arrangement.
Description of the Solution Dynamics of the
MoRu,
Complexes.
The acetylide ligand of the MoRu2 complexes
10
and 11 is associated with one of the Mo-Ru bonds;
therefore, its fluxional motion (Scheme
11)
can be exam-
ined by 13C NMR studies. The 13C NMR spectrum of
10
at 294
K
(Figure 7) exhibits two Mo-CO signals a t
b226.8
and 225.2, a sharp Ru(CO), signal a t 6 198.4, and a broad
Ru(CO), signal at 6 196.9. When the temperature is in-
creased gradually, the sharp Mo-CO signals start to
broaden and the broad Ru(CO), signals start to sharpen.
However, both the Mo-CO and the R~(CO)~.signals
col-
lapse in a pairwise manner at 338
K,
indicating that the
molecule begins to acquire a time-averaged mirror plane.
This observation cannot be explained according to the
concept of intermetallic Ru-CO scrambling proposed
for
the RU,(CO)~(C"L~BU)-
anion.31 Thus, we propose that
a further fluxional process, i.e. migration of the acetylide
(30) Boyar, E.; Deeming, A. J.; Felix, M. S. B.; Kabir, S. E.; Adatia, T.; Bhusate, R.; McPartlin, M.; Powell, H. R. J. Chem. Soc., Dalton Trans. 1989, 5.
(31) Barner-Thonen, C.; Harcastle, K. I.; Rosenberg, E.; Siege], J.; Manotti Landfredi, A. M.; Tiripicchio, A.; Tiripicchio Cammellini, M.
Inorg. Chem. 1981,20, 4306.
Downloaded by NATIONAL TAIWAN UNIV on August 12, 2009