C H I N E S E 30URNAL OF PHYSICS VOL. 33, NO. 2 APRIL 1995
Black-Body Radiation Results in the Activation Theory
Der-Ruenn Su
Department of Physics, National Tazwan University, Tazpei, Taiwan 106, R.O. C.
(Received December 14, 1994)
Activation energy (A-W) is regarded as a requirement for an energy-conserved process or reaction to initiate atomic exchanges. Stimulation of this kind, with the requirement of such an extra type of energy, occurs in many situations. We give a simple review with emphasis on hydrogen chemisorption on a solid surface. 1Ve attribute the extra energy as being used to activate the atom or molecule to pass through an AV static barrier. In a statistical approach, we find that XV can exist only if there are tunnelling states. AV is calculated to have discrete increases as the temperature increases. \Ve conclude that for high temperatures, black-body radiation occurs for this atomic desorption. PACS. 79.20.Rf - Atomic, molecular, and ion impact beam interactions with surfaces P A C S . 05.4O.+j - Fluctuation phenomena, random processes, and Brownian motion. PACS. 73.40.Gk - Tunnelling.
In the theory of chemical reaction, a formalism of potential-energy-surface in terms of relative distances between reactant atoms is accepted for an interpretation of the reaction path. On such a potential-energy-surface, we discuss the activated complex on a plot in which the maximum energy on the reaction path is the activation energy (AV). This AV is an energy in nature. In our opinion, it is a kind of thermodynamic potentials. Some conclusions based on the theoretical criteria on the potential-energy-surfaces are made in Ref. [l]. They are given as follows. Because the usual treatment of AV is an ab initio calculation, it usually does not provide potential energies including AV. When we investigate the dynamics or thermodynamics of chemical reactions, the explicit calculation of all kinds of energies at every point is rarely feasible. They are interesting for computation but not realistic in theory. What we can obtain effectively are the global surfaces. Furthermore, these surfaces are known only to “chemical accuracy”, which is about a few tens of meV (say 0.05 eV).
In this paper, we investigate AV from a statistical approach. We find that we need a combination of the tunnelling mechanism and a static AV barrier. This kind of approach was developed and applied to many physical problems by Azbel [2]. 1Ve believe that our 1 8 1 @ 1995 THE PHYSICAL SOCIETY OF THE REPUBLIC OF CHINA
results have rather good reliability for a global understa.nding of the problems in systems
of atoms, small molecules, and large global clusters, etc.
For simplicity, we consider a system of vibrating ad-hydrogen-atoms on a surface. For
single-atomic adsorption, the standard electron configuration can be found for H/Al(lll)
in Borisov’s paper [3]. The electron distribution is continuous ‘from far-from-the-surface
to near-surface’. This is a typical chemisorption case. When the coverage of ad-H-atoms
increases, the energy-band structure is finally formed [4,5] for H/Cu(llO), etc. In these
references, the authors call it the “protonic” two-dimensional energy band; the reason being
obvious from the above chemisorption description. They also describe the phenomenon as
H “quantum delocalization”.
Another interesting paper is Ref. [G], in which quantum
fluid is formed from ad-H-atoms.
The coverage problem was studied. A further step
in the study of the vibrational and phonon effects on core-levels can be found in Refs.
[?,8] for Si(lll)-(1 x l):H etc. For the H/W-surface, core level shifts in W can be found
in Ref. [9]. A n investigation resembling the inner-shell electron promotion study of the
bombardment atom can be found in Ref. [lo]. Hydrogen-elimination reactions (in parallel
to desorption cases) with low AV for amorphous silicon are reported by Sato et al. [II].
For the case of GaAs heterostructures, AV was reported in Ref. [12]. On the other hand,
for collisions of an H-atom with an Al(lll)- surface, we have H- formation which exhibits
a charge transfer [3]. The detachment dissociation for H-atom(s) from an Al(llO)-surface
has been reported in Ref. [13]. Many h ave considered only the initial and final energies,
together with momenta at most. From only energy considerations, the potential diagram
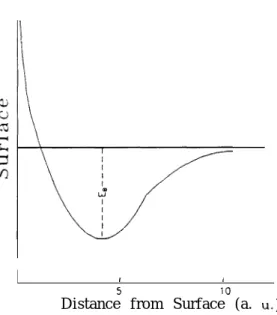
is shown in Fig. 1 [14]. If we do not take the initial and final states into account, or if
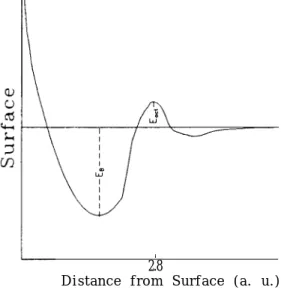
we concerntrate on the dynamical process, we have an AV
Eact asshown in Fig. 2. In
Liith’s opinion [14], this AV comes from the recombination of two H-atoms to form an
Hz-molecule. Actually, if we consider the chemical physics point of view, the activated
complex in the theory of the potential-energy-surface must be introduced. Then this part
of the recombinative contribution is not all that we must consider. There are very active
recent developments in the same spirit with Hamiltonians without the kinetic energy, e.g.,
the Hubbard model investigations for electrons are in this category. The kinetic energy
measurements in the processes of interest in this paper use the time-of-flight technique.
A report of Si+ from Si(lO0) is given by Liu and Wu [15]. This report includes a charge
transfer process. Their experimental results also exhibit the strength of bonding, where we
have the surface dangling bond of the Si/Si-surface. For H from W(211), the mechanism
of thermally activated vibrational motions is discussed in Ref. [lG]. AV of Si on a solid
surface was also investigated and recently a measurement was attempted in Ref. [17]. The
recombination to form Hz molecules is also an active topic. In a paper of Eenshuistra et al.
[18], the “wall” recombination of II* molecules from the tungsten wall is observed to have
VOL.33
DER-RUENN SU
183
, t
5 10
Distance from Surface (a. u.)
FIG. 1. A schematic plot of the potential energy of an atom or molecule in front of a solid surface when only the initial and final states are taken into account.
EB
stands for the binding energy which has a magnitude on the order of 1 eV. The distance from the surface is estimated from Ref. (31 for the embedded H-atom on an AI(111) surface. But from Ref.[14], the scale of the distance is smaller approximately by a factor of l/7.
vibrational levels up to v = 5, which is usually v = 3. Another report [19] gives the wall recombination of Hz molecules from a stainless steel wall with 1/ up to u = 9 (the usual Boltzmann distribution value near 3000 I< is v = 3). A calculation by MiiIIer [20] s h o w s that the H-H bond is weakened for recombinative desorption of Hz from a Pt surface. All of these experimental and computational results hint that in the final state after the formation of Hz molecules from a recombinative desorption process, the H-H bond is weakened by the assumption that it is formed. In the author’s opinion, recombinations of this kind can hardly occur near the surface. They can be excluded from the standard surface processes because the desorbed H-atoms have to move far enough outside the “surface region” before the recombination process occurs. Particularly, we consider a local region of the vibration mechanism. Apart from the surface system, this problem has been also studied using clusters [21]. Vibrational excitations are ensured for H3f in this process. The collapse of surfaces and clusters is studied in Refs. [11,22-26). Al so, influences of the bonding of H-atoms, and thus its AV in chemical reactions have been reported in Ref. [27]. Consequently, we have to temporarily neglect many processes such as the recombination process, etc. in this paper in our theoretical considerations. We still keep in mind the activation, such as
in Fig. 2, but the formation concept is not attributed to Hz recombination. There are also ma.ny other interesting views a.bout the sequential dynamics of adsorption [2S]. A paper of Tsong [29] states that, for a tip near a metal, AV comes from the superposition of potentials from two sides. In the sense of magnetism [30] of ferromagnets, from a statistical viewpoint, the potential diagram should be like Fig. 2 with an electromagnetic polarizability. This assignment is of course, a physical continuation of the oscillating dipole-dipole interaction which implies that the van der M’aals force is its time average. Hence it is a kind of electromagnetic interaction, a derived coulombic interaction, and subject to electromagnetic symmetries. Close to atomic systems, the oscillating emission of Us-clusters etc. was studied in Ref. [31]. For the magnetic effect, the surface barrier was found by Watanabe and Iwata [32]. This point is different from the kinetic-less potential-energy-surface mentioned above. On the contrary, kinetic energy-emphasized studies can also be found for II reactive collisions induced by solid-liquid transitions on targets [2G]. Other research on the vibration and oscillating dipoles for II/Si(lll) ca.n be found in Refs. [34,35]. For noble metals and inert ga.ses, the physisorption implies that adatoms may be taken into account individually, each atom by itself. For chemisorption such a.s the II-a,tom adsorptions mentioned above [3], charge transfers can be a dominating fa.ctor, or at least an influential part when the
4
2.8
D i s t a n c e f r o m S u r f a c e ( a . u . )
FIG. 2. A schematic plot of t.he potential energy of an atom or molecule in front of a solid surface when the dynamical process .is considered. For the barrier, E,,, denotes the activation energy which is calculated in this paper and for the well, ED denotes the binding energy \vl~ich has a magnit.ude on the order of 1 e\‘. The distance from the surface is estimated from Ref. [37] for Eact.
VOL. 33
DER-RUENN SU
185
whole H-atom is desorbed. The charge transfer must then be involved in the process [3,15,36]. Of course. for far-surface distances, it is understood that the final state of nearby atoms is exerted by the van der Waals forces, of the same nature as those for intermolecular interactions. Therefore the initial and final momentum and energy considerations are not able to give us the physical picture of the adsorption and desorption processes. Thermal activation has been studied in superconducting junctions [36]. Abstracting, all of the above results lead us to the conclusion that AV is a potential barrier in front of the surface.
Recent developments regarding molecules on surfaces include many resonance tun-nelling processes. For electrons, there are rather complete theoretical results and for atomic or molecular tunnellings, we can also find some reports. Here we assume that we have a set of oscillator states in which the ad-atoms or molecules occupy. This consideration is equivalent to approsimatin g the potential well right in front of the surface by a harmonic potential as shown in Fig. 2. To find the dynamical configuration we count the number of tunnelling states with energies below the AV. !Ve also find the AV as a discrete function of temperatures similar to Ya. Azbel’s results [2]. Further \ve conclude that the magnitude of the AV is determined by the vibrntiorz frequerzcy. Starting with the Arrhenius-type law for the rate constant in chemical rextions
where A is the preesponential factor and Eact is the XV. For our system of ad-hydrogcn-atoms, a chemical rextion helps us to understa.nd
11 + JI,(Tl> -+
WTT) + 11,
(1)
which is modelled in one-dimension as is done in chemical physics. On the left hand side, we have the para-hydrogen molecule while on the right hand side we have the ortho-hydrogen molecule. In this reaction we ca.n see that the system conserves its energy. If we disregard the nuclear spins, no reaction has a.ctually occurred. Furthermore it is kinetically possible, a.nalogous to the well-known three equal ma.ss ball collision in elementary physics. But we need to add an AV of only 3.9 x lob2 eV per molecule to initiate the reaction. The dissociation energy of II2 is 44.75 x 10m2 eV. It seems that the formation of a neutral IIs cluster as the first step is not feasible for this reaction. We hestitate to apply the collapse of clusters mentioned above to this case [11,23]. Furthermore, the AV is less t1la.n l/10 of the dissociation energy of 112. Thus we a.re sure that the reaction occurs before or more often sta.tistically than the dissocia.tion and recombination processes take place, but because of the conservation of energy, Eq. (1) is in equilibrium so that no extra energies are necessary. All the energies are in balance. The necessity of the addition of this extra “involvment energy” for init.iating the react.ion is lvhat we need to investiga.te. F u r t h e r m o r e (I) occurs inside the equilibrium gas in a non-controlled condition; all the
---atoms and molecules behave as particles in the kinetic theory of gases. The kinetic energy
of the incident atoms, such as the H-atom in (l), is actually in the form of equipartition
energy. Referred to an equilibrium condition with energy conservation, this energy is the
only extra energy available to initiate the reaction. Therefore we must conclude that the
extra available energy during this reaction is the equipartition energy, The equipartition
energy for each degree of freedom is the main subject. Furthermore the degrees of freedom
are closely related to the dimensionality. In surface processes, here in particular the atomic
desorptions, the dimensionality is a subject of investigations. The results of the degrees of
freedom investigation are used to study the dimensionality of the process in modelling.
The Arrhenius-type law is given in the form of a Boltzmann factor with mono-energy,
AV,
Eact. Wecan easily see that
Eactis a
modelled valuewith the conjectured form of an
exponential function among the bulk energy distributions. This form of the law has been
applied to many quantities or observables. In this paper, we simplify the Arrhenius-type
law as a statistical average by modelling
and study the influences of tunnelling. We shall show that without tunnellings, there are no
activations. For the reactive collision
we propose that
e-P.Lr = J
eepEP(E)dEJP(E)dE ’
of hydrogen atoms above, for the energy concerned,
(2)
for a certain statistical distribution
P(E).As pointed out in a paper of Su [38], we are
really not able to have one single distinct atom incident in our experiments; we must treat
the incident atom as an atom inside an atomic (or molecular) beam with a planar incidence,
when an atomic adsorption process occurs in front of a surface. Similar to a plane-wave
incident on a surface, we can decompose the space into (2+1)-dimensions. The plane of
the “wave-front” has certain arbitrariness. We may think that the dynamics is “essentially
one-dimensional”, particularly with or without the charge and dipole inductions inside the
surfaces. We resolve this puzzle, i.e. the mathematical dimensionality of the system, by
using Boltzmann statistics as follows. The Boltzmann distribution can be expressed as
P(E)dE = eP(p-E)D(E)dE,
epp = X3N/V ; D(E) = density of stu2e.s ,
(3)
x = (2&2 ’
for X the thermal de Broglie wavelength of a molecule.
Here the chemical potential I_L is
calculated from the three-dimensional case. As is well-known in the statistical theory
of
._-_-VOL. 33
.
semiconductor heterostructures
results of the density of states.
effects
4
2mD(
END) a
2rk, =-dE2D h2 ’
DER-RUENNSU 187
etc., the dimensionality of the phase space is relevant to
For instance, for the two-dimentional case without edge
D(E~D) = constant C/Area x Energy Range.
For an atom incident on a surface, we use this result. Since we obtain black-body radiation
as our final result, this choice does have a certain physical meaning. Here to verify our
conjecture above, we calculate
Eact = p-’
In 2 =
FkT.We see that
Eactis the equipartition energy of approximately one degree of freedom which
confirms our one-dimensional conceptions.
Here we have self-consistently demonstrated
that our conjecture is verified by calculations. Of course, we need to adjust it as follows.
From the concept of AV, it is a barrier in the potential-energy-surface. The particle energy
in the reaction path must be larger than the AV so that the formation of products is
possible. Consequently, we can picture that only part of the energy
E > Eactis able to
pass over the activation complex and make measurable or effective collisions or proceed in
a reaction. From the probability idea in (3) we must have a cutoff at
Eactif there are no
tunnellings. This restriction is a classical condition with which a particle with energy less
than the barrier is not able to pass the barrier. To generalize our calculations, we use the
quantum theory which includes tunnelling. We propose to use Boltzmann statistics with
a cutoff to calculate the AV together with some tunnellings. Similar to (3) we use the
distribution
Pl(E)dE = Cep(p- E)O( E - E,,JdE
.
Then the AV becomes
&3Eac, = %kr e -@+,(E)dE + tu nnelling (Et < E,,i)
Jgcr f’~ (E)dE t t unnelling (Et < Eact) ’
(6)
with both sides functions of the AV. For tunnellings, we assume that we have a set of
discrete quantized tunnelling states with a set of eigen-energies
{El}.Therefore the AV
can be obtained from
S”
e-P&,, = E,ct eePEPl (E)dE + CePgT2
J& Pl( E)dE
t CePpTl
'
(7)
which leads to
e-P.% = e-PE~~~ ‘I2 + PTzeW.Lt 1 $
PTlePEacr
’For the one-dimensional case, we must obtain from (8)
(~1
Eact =
+kT;
T2=$kT+A
fi
for e = 2.718281.. ., the natural base number. IIere the functional relation between Tl, T2 and the temperature is given; the definition of the
T’ s
are given in (7). It is noted that for exactly one degree of freedom, this condition is extra so that no parameter can be used as an unknown to be determined. The proper solution for (8) isSeveral direct evidences follow from (8) and (9). (i) Tllere is no possibility to have AV without tunnelling states because if both
Tl
and T2 are zero, (8) is not able to be satisfied. (ii) From the solution (9), we see that the AV is determined by the tunnelling energies. (iii) There is no possibility to have an infinite number of sta.tes for the tunnellings such as hydrogen-like states. O t h e r w i s e b o t h T1 and T2 a.re infinities. From (9) the AV is again undetermined. On the other hand, if the system is modelled with N degrees of freedom, then for a generally continuous NEact = $kT ,
N=2[1+$) -ln2tln(,,J~)], 1
which gives a solution of N as a function of the temperature. l3ecause the modeling of a reaction is current interest [39], and since we may ha.ve the reduction of dimensionalities, the dimensionality of modeling the system is a.ctua.lly very relevant. This kind of modeling has alrea.dy been done in the above rea,ction (1). It is ma.nifest that (1) is one-dimensional and interpreted a.s “most stable”.
For the declined well right in front of the surface a.s shown in Fig. 2, we approximate it as a harmonic well since we alread- have had the ha.rmonic vibrations of the adsorba.tes there. Thus a.datoms start their vibrations and detach from the surface for la.rge vibra.tions after energy gains. Therefore these adatoms are able to be described by simple harmonic oscillators with energy eigenvalues (n + 1/2)L. The number of possible oscillation modes below the activation masiniunl, i.e. E,,*, is our number of tunnelling states. First, we consider only one tunnelling level,
VOL. 33 DER-RUENX ST; 189
+hw >
Eact > +huso that Tr = exp(-@hti/2); T2 =
esp(-dhw). IVeobtain in the final solution from (9)
Eact = +hu + kT
ln[( 1 + d-)/2],
(11)for
hw > kTIn
1+/m
2 .
(12)
From (11) we see that it is not exactly any number of degrees of freedom. The varia.tion
of the AV with
kTis shown in Fig. 3. Essentially there is no problem for this AV. To
understand the result, we study the exact one degree of freedom case
(12a)
in which the frequency w becomes negative if
kTis larger than 2.1353. For these high
temperatures, one degree of oscillation is not possible. Whereas from (11) we can obtain
more realistic results and find the thermodynamic determination of the AV for certain
temperatures. Recently Peksa et nl. tried to determine the ,4V of desorption for a metal
surface [JO]. For chemical reactions, we may have a negative .4V as reported by Hernandez et
nl. [39] with a reduced-dimensionality model. For such possibilities, we need further study.
It is interesting that here the first term of the AV is the energy eigenvalue of the oscillator
which is determined by the oscillation mechanism. The second term is determined totally
by the temperature.
Therefore
for a different system or mechanism, we have diflerent.4V’s.
In other words, we have shown that AV is not a quantity determined by dynamical
configurations.
The tunnelling character, here only the one tunnelling state, determines
the whole statistical nature, the activation. In other words,
the vilrrtltion frequency, suchas w a6ove determines the
AV. Condition (12) gives us the determination of the tunnelling
character. This equation determines the inclusion of the number of tunnelling states, i.e.
for higher temperatures we have to include two or more tunnelling states. This uncertainty
can make the definition of AV ambiguous. In general, if we get
ntunnelling states, we have
the condition
We must have the restriction
hw > kT
In
otherwise we must jump from n to n +
1for T,(“) and TJn) as the temperature is increased.
The results are obtained as
We obtain the quantities
T,(n) = e-phw/2
l -
e-
nphw1 _ e_Dhw
= ,-$3hwsinh(npLJ/2)
sinh(,DfLw/2) ’
(144
,pd”) 1OCI = -cash ( ; Bhu)2 ,$- :
phm
cosh( ;/3hw) 1+ ,/l+;tanh(+) coti, ),(14b)
EL:,) =
((n-
l)+++kT
x Ini
cosh( +/3hw)(
J
1 + 1 + g tanh( iphw) coth( zbhw) ) Pw
,wfihw
cosh( ;phw) 2
1
--+iIiuas
T --+ 0 for all 72
. W)Intuitively the a.bove limit is a simple one-dimensional harmonic oscillator at zero
tempera-ture physically, and the AV coincides with the oscillator ground state energy. The limiting
result may lead us to the wrong conclusion, that we have only one degree of freedom.
Because of the condition (13)
cosh( $?Fm) ( J1 + tw > kT x 111
1 + g tanh( :Bhw) coth( T/?hw) )
,-3hw
COSll( ;ptlLr’)
21
(15)
we have a maximum temperature restriction on the n-states configuration. We list this
maximum temperature for a given n in Table I for n = 1 - 6. Further we have plotted the
AV for various tempera.tures for R =
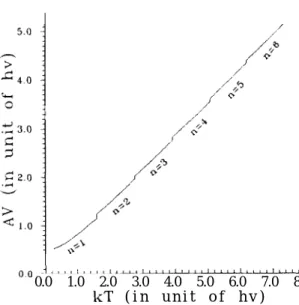
1 - Gin Fig. 3. In Fig. 3, the unit of hv is hv E hw
of the oscillator. Values of the AV show discreteness. Every jump from n to R. -I- 1 shows a
discontinuity and includes one more state with energy below the AV.
For the case where temperatures are estremly high, as a consequence of the above
discussion, we must have an AV out of the range of consideration although the activation
VOL. 33 DER-RUENN SU 191
00
i,,‘,I,“,l,‘,,I,*“l,,“l,,‘,I,,~,I,,,’l
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0kT ( i n u n i t o f h v )
FIG. 3. The activation energy plot for various temperatures: it increases discretely at certain given maximum temperatures which are tabulated in Table I. Labels of n are the number of tunneling states. It is clear that as the temperature increases over each maximum temperature, it tends to include one more tunneling state. The scale of abscissa and ordinate are both in the unit of the vibration mode, i.e. hv = hw.
TABLE I. For the number n of states below the activation energy, the value
T,,,
gives the maximum temperature. Above this temperature the number of states is increased by one.n 1 2 3 4 5 6
ICT,,,,,,
(in fiw) 1.607 2.780 3.930 5.071 6.216 7.355Pmin (in (~W>-') 0.622 0.360 0.254 0.197 0.161 0.136
mechanisms, such as the activation complex etc., are still there. The adatom diffusion on a surface has been investigated by Chen and Ying [41]. It indicates that the quantum tunnelling regime at low temperatures may pass to the classical regime at high temperatures, for a H/Ni( 100) system. We may assume that the motion along the perpendicular direction behaves with a similar mechanism. The statistical nature becomes that of black-body
radiation except that the chemical potential is not zero a.nd the zero point energy exits. We
ha.ve to neglect the statistical integral pa.rt and the dist,ribution function becomes
Mathema.tically we ha.ve an infinite AV. The expectation energy contributed from
eigcnfre-quency w is
where the ergodic distribution over w is assumed.
If the classical regime is a.dopted, we neglect the zero-point ground state
(17)
which is exactly the same as the black-body radiation ca.se. Black-body radiation has a
well-established theory. No further discussion is given here.
PI
PI
PI
PI
PI
[Gl
PI
PI
PI
WI
R E F E R E N C E S
R. E. Palmer a.nd I’. J. Rous, Rev. Mod. Phys.
64,390 (1992);
61,669 (1989).
M. Ya. Azbel, Phys. Rev. Lett. 68, 9s (1992).
A. G. Borisov, D. Teillet-Billy, and J. P. Gauyacq, Phys. Rev. Lett. 68, 2842
(1992).
C. Astaldi, A. Bianco, S. h[odesti, and E. Tosatti, Phys. Rev. Lett. 68, 90 (1994).
K.-D. Tsuei, Chemisqtion on A9etal Svrfclces, a talk presented at the National
Tai-wan University, Taipei (1992).
C.-H. Hsu, B. E. La.rson, h/l. El-Batanouny, and C. R. Willis, Phys. Rev. Lett. 66,
3164 (1991).
C. J. Karlsson, F. Owma.n, E. La.ndemark, Y.-C. Cha.o, P. hIHrtensson, and R. I. G.
Uhrberg, Phys. Rev. Lett. 72, 4145 (1994).
A.-S. hkktensson, C. Xyberg, and S. Andersson, Phys. Rev. Lett. 57, 2045 (1986).
D. M. Riffe, G. K. Wertheim, and P. II. Citrin, Phys. Rev. Lett. 65, 219 (1990).
K. A. II. German, C. B. Weare, and J. A. Yarmoff, Phys. Rev. Lett.
72, 3899
(1994).
VOL.33
DER-RUENN SI;
193
[ll]
K. Sato, H. II onna, S. Iwahuchi, T. Hirano, and H. Koinuma, Phys. Rev. B50, 2675
(1994).[la] J. P. Eisenstein, L. N. Pfeiffer, and K. VV. VVest, Phys. Rev. B50, 1760 (1994). [13] B. Hammer, K. W. Jacobsen, and J. K. Norskov: Phys. Rev. Lett. 69, 1971 (1992). [14] H. Liith, Szlrfaces and Interfaces of Solids (Springer-Verla.g, Berlin, 1993) p.436. [15] H. T. Liu and 2. Wu, Phys. Rev. Lett. 72, 3891 (1994).
[16] 0. Grizzi, M. Shi, H. Bu, and J. W. Rabalais, Phys. Rev. Lett. 63, 1408 (1989). [17] M. R. Sardela, H. H. Radamson, J. D. Ekberg, J. E. Sundgren, and G. V. Hansson,
Semicond. Sci. Technol. 9, 1272 (1994).
[la] P. J. Eenshuistra, J. II. M. Bonnie, J. Los, and II. J. Hopman, Phys. Rev. Lett. 60, 341 (19SS).
[19] R. I. Hall, I. C a dei, M. Landau, F. Pichou, and C. Schermann, Phys. Rev. Lett. 6 0 , 337 (1988).
[20] J. E. Miiller, Phys. Rev. Lett. 59, 2943 (1987).
[al] A. E. Ore1 and K. C. Kulander, Phys. Rev. Lett. 71, 4315 (1993). [22] W. D. Luedtke and U. Landman, Phys. Rev. Lett. 73, 569 (1994).
[23] M. Saeed, B. Yang, X. Tang, and L. F. DiMauro, Phys. Rev. Lett. 68, 3519 (1992). [24] F. K. Le G oues, P. hf. Mooney, and J. Tersoff, Phys. Rev. Lett. 71, 396 (1993). [25] J. E. Ayers and L. J. Schowalter, Phys. Rev. Lett. 72, 4055C (1992).
[26] B. Schmidt and R. Benny Gerber, Phys. Rev. Lett. 72, 2490 (1994). [27] E. T. Denisov, Kinet. Catal.-Engl. tr. 35, 293 (1994).
[28] J. W. Evans, Rev. Mod. Phys. 65, 1281 (1993). [29] T. T. Tsong, Phys. Rev. B44, 13703 (1991).
[30] H. Suhl, in The Physical Basis for Heterogeneous Catalysis, eds. E. Drauglis and R. I. Jaffee (Plenum, N. Y., 1975) p. 427.
[31] R. Bruckmeier, Ch. Wunderlich, and H. Figger, Phys. Rev. Lett. 72, 2550 (1994). [32] M. Watanabe and T. Iwata, Phys. Rev. Lett. 72, 3429 (1994).
[33] P. Guyot-S ionnest, Phys. Rev. Lett. 66, 1489 (1991).
[34] P. Dumas, Y. J. Chabal, and G. S. Higashi, Phys. Rev. Lett. 65, 1124 (1990). [35] D.-R. Su, Ch in. J. Phys. (Taipei) 32, 433 (1994).