Editorial Manager(tm) for PLoS ONE Manuscript Draft
Manuscript Number: PONE-D-10-01146R1
Title: HDAC Inhibition Decreases the Expression of EGFR in Colorectal Cancer Cells Short Title: HDACs Are Implicated in EGFR Overexpression
Article Type: Research Article Section/Category: Clinical
Keywords: colorectal cancer; Epidermal growth factor receptor; Histone Deacetylase; Vorinostat; SP1 Corresponding Author: Ching-Chow Chen
Corresponding Author's Institution: National Taiwan University First Author: Chia-Wei Chou
Order of Authors: Chia-Wei Chou;Ming-Shiang Wu;Wei-Chien Huang;Ching-Chow Chen
Abstract: Epidermal growth factor receptor (EGFR), a receptor tyrosine kinase which promotes cell proliferation and survival, is abnormally overexpressed in numerous tumors of epithelial origin, including colorectal cancer (CRC). EGFR monoclonal antibodies have been shown to increase the median survival and are approved for the treatment of colorectal cancer. Histone deacetylases (HDACs), frequently overexpressed in colorectal cancer and several malignancies, are another attractive targets for cancer therapy. Several inhibitors of HDACs (HDACi) are developed and exhibit powerful antitumor abilities. In this study, human colorectal cancer cells treated with HDACi exhibited reduced EGFR expression, thereby disturbed EGF-induced ERK and Akt phosphorylation. HDACi also decreased the expression of SGLT1, an active glucose transporter found to be stabilized by EGFR, and suppressed the glucose uptake of cancer cells. HDACi suppressed the transcription of EGFR and class I HDACs were proved to be involved in this event. Chromatin immunoprecipitation analysis showed that HDACi caused the dissociation of SP1, HDAC3 and CBP from EGFR promoter. Our data suggested that HDACi could serve as a single agent to block both EGFR and HDAC, and may bring more benefits to the development of CRC therapy.
Suggested Reviewers: SHAO-CHUN Wang University of Cincinnati Medical Center [email protected]
Rakesh Kumar
The George Washington University [email protected]
Opposed Reviewers:
Thank you very much for your e-mail on Nov 17, 2010 that invites us to resubmit the manuscript (manuscript#: PONE-D-10-01146) entitled "HDAC Inhibition Decreases the Expression of EGFR in Colorectal Cancer Cells" We are now resubmitting our revised manuscript for consideration of publication in PLoS one.
We would like to express our sincere gratitude for the reviewer's effort and valuable suggestions for this manuscript. We have modified the manuscript accordingly and have addressed point by point how we modified the manuscript. We also have conform the journal guideline for manuscript length and tried to make the manuscript concise and comprehensive.
Thank you very much for your time in handling this manuscript. Sincerely yours,
Ching-Chow Chen, Ph.D. National Taiwan University
Department of Pharmacology, College of Medicine No.1, Jen-Ai Road, Section 1
Taipei 10018, Taiwan Phone: 886-2-23123456 ext. 88321 Fax: 886-2-23947833 E-Mail: [email protected] Editor PLoS one Manuscript #: PONE-D-10-01146 Dear Editor:
Thank you very much for your e-mail on Nov 17, 2010 that invites us to resubmit the manuscript (manuscript#: PONE-D-10-01146) entitled “HDAC Inhibition Decreases
the Expression of EGFR in Colorectal Cancer Cells” We are now resubmitting our
revised manuscript for consideration of publication in PLoS one.
We would like to express our sincere gratitude for the reviewer’s effort and valuable suggestions for this manuscript. We have modified the manuscript accordingly and have addressed point by point how we modified the manuscript. We also have conform the journal guideline for manuscript length and tried to make the manuscript concise and comprehensive.
Thank you very much for your time in handling this manuscript.
Sincerely yours, Ching-Chow Chen, Ph.D. Cover Letter 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
Point-by-point Responses to the Reviewers’ Comments Reply to Reviewer#1
Major issue
(a): The largest problem with this paper is a glaring lack of references to
publications directly related to these topics and over-interpretation of the
data. As one example, Zhou, Shaw and Davidson demonstrated in 2008
that SAHA was able to decrease levels of EGFR in ER-negative breast cancer cells through destabilization of mRNA levels, yet this paper is not cited. The presented ChIP data, while aptly performed, does not support the final model very conclusively. One would expect that for any gene where there is a decrease in transcription for there to be the exact changes observed by ChIP, but this does not support a direct mechanism as depicted in the final figure.
Our response:
1. As suggested, we have cited several important papers which are related to the HDAC-mediated regulation of EGFR in the section of Results (page 14, line 8-10) and Discussion (page 17, lines 5-10).
(Page 14, line 8-10)
Our result showed that TSA and SAHA significantly decreased the EGFR promoter activity (Fig.4B upper panel). It has been reported that HDACi decreased the EGFR mRNA stability in ER-negative human breast cancer cells [1]. Therefore, the stability of EGFR mRNA was examined.
(page 17, line 5-10)
Several studies show the inhibitory effect of HDACi on EGFR expression in human cancers. For example, FK-228, a depsipeptide HDAC inhibitor, is reported to decrease the expression of EGFR in lung cancer cells [2]. SAHA decreases the levels of EGFR in ER-negative breast cancer cells via mRNA destabilzaiton [1]. More recently, inhibition of HDAC6 is found to enhance the endocytosis of EGFR through increasing tubulin acetylation [3,4].
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
2. As suggested, we have modified the scheme (removing the acetyl- moiety of SP1) in the final figures to fit the results observed in ChIP experiments.
Fig.7. Schematic diagram of EGFR promoter in the basal state or treatement with HDACi. In the basal state, HDAC3, CBP and SP1 were both recruited to the promoter region and responsible for the transcription of EGFR (A). While treated with HDACi, the complex of HDAC3, CBP and SP1 were disrupted and dispersed from EGFR promoter, leading to the inactivation of EGFR transcription (B).
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
(b): As an added general point, the manuscript requires extensive grammatical
editing.
Our response:
As suggested, we had fixed several grammatical errors in manuscript.
Minor points
1. Fig.1- How many times have the western blot experiments been repeated and what is the statistical variation? This applies to essentially all of the western blot experiments in this paper. Fig 1A- The authors should state how much glucose was present during these measurements to allow for comparison with the data presented in Fig. 2.
Our response:
1. Each western blot experiment is performed three times and the statistical variation is in the range of ±15%.
2. As suggested, the glucose concentration used in Fig.1 is 1 mg/ml and has been stated in the figure legend.
2. Fig 3A--HDACi typically cause G1 arrest and only rarely G2 arrest, which the manuscript data demonstrates. The authors should explore this topic in the Discussion given that a functional G2 checkpoint typically protects cells from apoptosis induced by HDACi.
Our response:
As suggested, we had discussed this issue in the section of Discussion (page 19, lines 15-22).
(page 18, lines 14-20)
HDACi is reported to induce G2/M growth arrest as well as G0/G1 arrest in colorectal cancer cells, and the HDACi-mediated growth arrest consistently involves p21 induction [5-8]. In HCT116 cells, p21 is induced and the cell cycle is arrested in G2/M phase by silencing class I HDACs, especially HDAC3 [9]. Consistently, we found that SAHA induced p21 and G2/M arrest and re-expression of EGFR could alleviate these events.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
3. Fig 4C- I don't understand why knock-down of HDAC3 should have as much effect as either knockdown of HDAC1 or HDAC2 (which form a complex). To me this suggests that this is a very nonspecific effect. The authors should attempt to explain this perceived discrepancy.
Our response:
As suggested, we had discussed this issue in the section of Discussion (page 18, line 20 to page 19, line 3).
(page 18, line 17 to page 20, line 9)
HDAC3 has been reported to be maximally expressed in the proliferative compartment in mouse colon. Knockdown of HDAC3 induced a greater magnitude of G2/M and S phase arrest than that of HDAC1/2, suggesting that HDAC3 is more significant than HDAC1/2 in colon cell proliferation [10]. HDAC3 is a component of the NCoR-SMRT co-repressor complex, which is distinct from repressor complexes containing HDAC1 and HDAC2 (Sin3A and NuRD) [11], indicating the specific roles of HDAC isoform in gene repressing. In contrast, knockdown of HDAC1, 2 or 3 decreased the EGFR expression in varying degree, indicating that they share functional redundancy on promoting EGFR transcription. Ectopic express HDAC3 induced a greater magnitude of EGFR mRNA and a positive correlation between EGFR and HDAC3 expression in colon cancer patients. Therefore, HDAC3 may be most essential in EGFR transcription.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
4. Fig 5- The authors should quantify the amount of protein for Sp1 and EGFR.
Our response:
As suggested, we had quantified the amount of SP1 and EGFR in Fig.5.
Fig. 5. SP1 is essential for the EGFR transcription. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
5. In the discussion, the authors should attempt to reconcile their data with data from the literature that shows that treatment with both HDAC inhibitors and Akt/ERK signaling pathway inhibitors induces apoptosis more strongly than either alone. Some discussion of new clinical agents, such as CDUC-1017-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N- hydroxyheptanamide (CUDc-101) (Cai et al. and Lai et al.), that combined inhibition of both pathways in one compound as improvements of current generation HDACi would also potentially strengthen the manuscript.
Our response:
As suggested, we had cited the reference of CUDC-101 and a report which shows AEE788, a multiple receptor kinases inhibitor, synergize the HDACi-induced apoptosis, in the last paragraph of Discussion.
(page19, line 23 to page20, line 8)
It has been reported that HDAC inhibitors synergize with 5-FU in vitro and
in vivo to treat colon cancer through HDACi-induced downregulation of
thymidylate synthase, the 5-FU target enzyme [12]. Combination of 5-FU with SAHA has recently entered phase I/II trial to treat CRC [13,14]. Inhibition of MAPK and Akt signaling by AEE788, a multiple receptor tyrosine kinases inhibitor, synergistically potentiates HDAC-induced apoptosis in a broad spectrum of cancer cell lines [15]. Recently, a new compound, CUDC-101, which inhibit the activity of both EGFR and HDAC, is demonstrated to have powerful anticancer activity [16]. These reports strengthen the rationale of concurrent inhibition of EGFR and HDAC in cancer therapy. In this study, we showed that HDAC inhibitor alone is able to block EGFR transcription as well as HDAC, may provide a hint to superior strategy for colorectal cancer therapy.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
Reply to Reviewer#2
Major issue: Chen and colleagues demonstrated the HDACi , SAHA, can effectively
inhibit the suppression of transcription of EGFR by promoting dissociation of SP1, HDAC3 and CBP in the promoter. However, the authors didn't extensively mention the advantages of SAHA treatment in other cancers or cell lines. For examples, Fazzone published that HDACi and 5-FU can work synergistically to treat cancer. Should the authors further clarified whether it is a MSS dependent event? Since 3 of 4 cell lines are MSS and WiDR is actually derived from HT-29, it will be important to improve the quality of this manuscript.
Our response:
Thanks for this valuable suggestion. HCT116 cells (MSI-H), SW480 cells (MSS) and HT29 (MSS) and its derivates WiDr were used in this study. However, HCT116 is the only MSI cells we have and it’s not enough to tell whether this event is related to the microsatellite stability.
As suggested, we had made more discussion about the advantage of HDACi treatment in other cancers cell lines in the last paragraph of Discussion.
(page19, line 23 to page20, line 8)
It has been reported that HDAC inhibitors synergize with 5-FU in vitro and
in vivo to treat colon cancer through HDACi-induced downregulation of
thymidylate synthase, the 5-FU target enzyme [12]. Combination of 5-FU with SAHA has recently entered phase I/II trial to treat CRC [13,14]. Inhibition of MAPK and Akt signaling by AEE788, a multiple receptor tyrosine kinases inhibitor, synergistically potentiates HDAC-induced apoptosis in a broad spectrum of cancer cell lines [15]. Recently, a new compound, CUDC-101, which inhibit the activity of both EGFR and HDAC, is demonstrated to have powerful anticancer activity [16]. These reports strengthen the rationale of concurrent inhibition of EGFR and HDAC in cancer therapy. In this study, we showed that HDAC inhibitor alone is able to block EGFR transcription as well as HDAC, may provide a hint to superior strategy for colorectal cancer therapy.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
Minor points
1. Figure 1 A & B were never quoted in the manuscript.
Our response:
As suggested, we had quoted Fig.1A and 1B in the section of Results (page 12, line 7, 9 & 15)
2. Actin housekeeping gene was adopted in the normalization of real time PCR. Did the author exclude the possibility of simultaneously amplifying the psuedogene? In general, most investigators switch to use GAPDH for normalization.
Our response:
As suggested, we had performed a no-RT control to examine whether the pseudogene will be amplified. Both RT positive and negative were amplified by 35 cycles of PCR. There is no visible PCR amplicon in the RT negative control, indicating that the possibility of simultaneously amplifying the pseudogene is excluded.
Fig.1 Examination of pseudogene amplification. 2ug total RNA of
HCT116 cells was subjected to reverse transcription with or without reverse transcriptase. The PCR products were subjected to 2% agarose gel electrophoresis and visualized by ethidium bromide staining.
Suppl. Fig.1 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
References
1. Zhou Q, Shaw PG, Davidson NE (2009) Inhibition of histone deacetylase suppresses EGF signaling pathways by destabilizing EGFR mRNA in ER-negative human breast cancer cells. Breast Cancer Res Treat 117: 443-451. 2. Yu XD, Wang SY, Chen GA, Hou CM, Zhao M, et al. (2007) Apoptosis induced by depsipeptide FK228 coincides with inhibition of survival signaling in lung cancer cells. Cancer J 13: 105-113.
3. Gao YS, Hubbert CC, Yao TP (2010) The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J Biol Chem 285: 11219-11226.
4. Deribe YL, Wild P, Chandrashaker A, Curak J, Schmidt MH, et al. (2009) Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signal 2: ra84.
5. Heerdt BG, Houston MA, Augenlicht LH (1997) Short-chain fatty acid-initiated cell cycle arrest and apoptosis of colonic epithelial cells is linked to mitochondrial function. Cell Growth Differ 8: 523-532.
6. Schwartz B, Avivi-Green C, Polak-Charcon S (1998) Sodium butyrate induces retinoblastoma protein dephosphorylation, p16 expression and growth arrest of colon cancer cells. Mol Cell Biochem 188: 21-30.
7. Kobayashi H, Tan EM, Fleming SE (2003) Sodium butyrate inhibits cell growth and stimulates p21WAF1/CIP1 protein in human colonic adenocarcinoma cells independently of p53 status. Nutr Cancer 46: 202-211.
8. Xu WS, Perez G, Ngo L, Gui CY, Marks PA (2005) Induction of polyploidy by histone deacetylase inhibitor: a pathway for antitumor effects. Cancer Res 65: 7832-7839.
9. Wilson AJ, Byun DS, Popova N, Murray LB, L'Italien K, et al. (2006) Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem 281: 13548-13558.
10. Wilson AJ, Byun D-S, Popova N, Murray LB, L'Italien K, et al. (2006) Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. The Journal of biological chemistry 281: 13548-13558.
11. Jepsen K, Rosenfeld MG (2002) Biological roles and mechanistic actions of co-repressor complexes. J Cell Sci 115: 689-698.
12. Fazzone W, Wilson PM, Labonte MJ, Lenz HJ, Ladner RD (2009) Histone deacetylase inhibitors suppress thymidylate synthase gene expression and
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
synergize with the fluoropyrimidines in colon cancer cells. Int J Cancer 125: 463-473.
13. Wilson PM, El-Khoueiry A, Iqbal S, Fazzone W, Labonte MJ, et al. (2010) A phase I/II trial of vorinostat in combination with 5-fluorouracil in patients with metastatic colorectal cancer who previously failed 5-FU-based chemotherapy. Cancer chemotherapy and pharmacology: 979-988.
14. Fakih MG, Pendyala L, Fetterly G, Toth K, Zwiebel JA, et al. (2009) A phase I, pharmacokinetic and pharmacodynamic study on vorinostat in combination with 5-fluorouracil, leucovorin, and oxaliplatin in patients with refractory colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 15: 3189-3195.
15. Yu C, Friday BB, Lai JP, McCollum A, Atadja P, et al. (2007) Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin Cancer Res 13: 1140-1148.
16. Lai CJ, Bao R, Tao X, Wang J, Atoyan R, et al. (2010) CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res 70: 3647-3656. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
1
HDAC Inhibition Decreases the Expression of EGFR in Colorectal
Cancer Cells
Chia-Wei Chou, Ming-Shiang Wu, Wei-Chien Huang and Ching-Chow Chen*
Department of Pharmacology (C.W.C., C.C.C.) and Division of Gastroenterology,
Department of Internal Medicine and Primary Care Medicine (M.S.W.), College of
Medicine, National Taiwan University, and National Taiwan University Hospital,
Taipei, Taiwan; Center for Molecular Medicine and Graduate Institute of Cancer
Biology, China Medical University and Hospital (W.C.H.) *Manuscript
Click here to download Manuscript: revision-5.doc
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
2
Running title: HDACs are implicated in EGFR overexpression
*Corresponding author: Dr. Ching-Chow Chen, Department of Pharmacology
College of Medicine National Taiwan University, No.1, Jen-Ai Road, 1st Section
Taipei 10018, Taiwan; Phone: 886-2-23123456 ext. 88321. Fax: 886-2-23947833.
E-mail: [email protected] Text Pages: 27 Tables: 0 Figures: 7 References: 41 Abstract: 188 words Introduction: 673 words Results: 1263 words Discussion: 1059 words
Abbreviations: CRC, colorectal cancer; EGFR, epidermal growth factor receptor;
HDAC, histone deacetylase; HDACi, histone deacetylase inhibitor; TSA,
Trichostatin A; SAHA (Vorinostat), suberoylanilide hydroxamic acid; SGLT,
sodium-dependent glucose transporter; CBP, CREB binding protein; Ac-H3,
acetylated histone H3; Ac-H4, acetylated histone H4; H3K4me2, di-methylated
histone H3 at lysine 4; Act D, actinomycin D; MTM, mithramycin A; FCS, fetal calf
serum; Ab, antibody; PCR, polymerase chain reaction; ChIP, chromatin
immunoprecipitation. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
3
Abstract
Epidermal growth factor receptor (EGFR), a receptor tyrosine kinase which promotes
cell proliferation and survival, is abnormally overexpressed in numerous tumors of
epithelial origin, including colorectal cancer (CRC). EGFR monoclonal antibodies
have been shown to increase the median survival and are approved for the treatment
of colorectal cancer. Histone deacetylases (HDACs), frequently overexpressed in
colorectal cancer and several malignancies, are another attractive targets for cancer
therapy. Several inhibitors of HDACs (HDACi) are developed and exhibit powerful
antitumor abilities. In this study, human colorectal cancer cells treated with HDACi
exhibited reduced EGFR expression, thereby disturbed EGF-induced ERK and Akt
phosphorylation. HDACi also decreased the expression of SGLT1, an active glucose
transporter found to be stabilized by EGFR, and suppressed the glucose uptake of
cancer cells. HDACi suppressed the transcription of EGFR and class I HDACs were
proved to be involved in this event. Chromatin immunoprecipitation analysis showed
that HDACi caused the dissociation of SP1, HDAC3 and CBP from EGFR promoter.
Our data suggested that HDACi could serve as a single agent to block both EGFR and
HDAC, and may bring more benefits to the development of CRC therapy.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
4
Introduction
EGFR (also known as ErbB-1/HER1), which belongs to the ErbB family of receptor
tyrosine kinases, comprises an extracellular ligand-binding domain, a single
hydrophobic transmembrane domain and a cytoplasmic tyrosine kinase-containing
domain [1]. Ligand binding induces homo- or hetero-dimerization of receptor and
subsequent activation of the pathways including Ras/Raf/MEK/ERK and
PI3K/PDK1/Akt [1]. Most of colorectal cancer (CRC) is characterized with
overexpression of epidermal growth factor receptor (EGFR) and predicted with high
risk of metastasis and recurrence [2]. Targeting EGFR seems to be a promising
approach for the CRC treatment. Indeed, cetuximab, a human-mouse chimeric IgG1
antibody binds to the external domain of the EGFR, has been approved by FDA in
2004 for the treatment of metastatic colorectal cancer [3]. After that, a fully
humanized antibody, panitumumab, is also approved to treat CRC [4]. However,
accumulating evidences demonstrate that the effects of targeting EGFR in colorectal
cancer are largely limited due to the status of KRAS mutation [5]. The KRAS mutants
bypass EGFR to activate the Ras/Raf/MEK/ERK signals, and significantly weaken
the therapeutic effect of cetuximab [6]. Examination of KRAS status is now a
prerequisite for the use of cetuximab [7]. Although ~60% of CRC patients expressed
wild-type KRAS but only half of them benefits from cetuximab. Therefore, the KRAS
status is not the only determinant for the efficacy of EGFR target therapy [8].
Therefore, treatment with a broad spectrum of genetic backgrounds is urgently needed
and would benefit most patients irresponsive to cetuximab-based therapies.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
5
Although EGFR is a receptor tyrosine kinase and delivers signals after ligand
conjugation, its prosurvival effect can be independent to kinase activity. For example,
mice lacking EGFR are embryonic lethal but those harboring kinase-inactive mutants
only exhibit some epithelial defects [9,10]. In addition, loss of EGFR kinase activity
decelerates cell proliferaiton but loss of its expression ruins the glucose uptake and
leads to cell death [11-13]. Therefore, inhibition of EGFR expression may be a better
strategy for CRC therapy.
Histone deacetylases (HDACs) which removes the acetyl groups from histone to
silence the gene transcription are highly expressed in various tumors [14,15]. HDACs
have become one of the emerging targets for cancer therapy, and HDAC inhibitors
(HDACi) show promising anticancer activities [15]. Among various HDACi, SAHA
(Vorinostat) had been successfully approved for the treatment of cutaneous T cell
lymphoma (CTCL). HDAC family can be subdivided into four classes and the class I
HDACs, which includes HDAC1, HDAC2, HDAC3 and HDAC8, have been reported
to be highly expressed in colon cancer [16]. The pro-proliferative effects of HDACs
are connected to the transcriptional repression of cdk-inhibitor, p21, and knockdown
of HDAC 1, 2 and 3 reduced the growth of several colon cancer cells [17]. Therefore,
HDAC may serve as a potential target for CRC therapy, and SAHA had entered
clinical trials for the treatment of CRC [18].
In this study, we demonstrated that the EGF signaling in KRAS mutant cell lines,
HCT116 and SW480, was disrupted by HDACi through transcriptional repression of
EGFR expression, indicating that HDACi served as a single agent to block EGFR and
HDAC simultaneously. Loss of EGFR partially contributed to the cytotoxic effect of
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
6
HDAC inhibitors. In addition, the expression of SGLT1, an active glucose transporter
which is stabilized by EGFR, was also decreased by HDACi and led to the reduction
of glucose uptake in colon cancer cells. The mechanism underlying the transcriptional
repression of EGFR by HDACi was involved with the histones hypoacetylation and
the dissociation of SP1, HDAC3 and CBP from EGFR promoter. Our data suggested
that HDACi could serve as a single agent to concurrently block both EGFR and
HDAC, and may bring benefits to the CRC patients with a broader range of genetic
backgrounds. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
7
Materials and Methods
Ethics Statement
All patient-derived specimens were collected and archived under protocols approved
by Institutional Research Board of National Taiwan University Hospital and
supported by the National Science Council, Taiwan. A full verbal explanation of the
study was given to all participants. They consented to participate on a voluntary basis.
Materials
TSA was purchased from Sigma and SAHA were obtained from Merck. The
Myc-tagged HDAC1, 2 and 3 were provided by Dr. WM Yang (NCHU, Taiwan).
Antibodies specific for EGFR, p21, HDAC3, and actin were purchased from Santa
Cruz Biotechnology. Anti–Ac-histone H3, H4, and Sp1 antibodies were obtained
from Upstate. Anti-SGLT1 antibody was purchased from Abcam.
Cell culture
HCT-116 (from Van Dyke MW, M.D. Anderson) and SW480 (from TH Leu, NCKU)
human colon carcinoma cells were cultured in DMEM supplemented with 10% fetal
bovine serum; A431 (human epidermoid carcinoma cells) and MDA-MB-468 (human
breast adenocarcinoma cells) obtained from ATCC were maintained in RPMI
supplemented with 10% FCS.
RNA isolation, RT-PCR and real time PCR
Total RNA was isolated from HCT116 cell using Trizol reagent (Life Technology).
Reverse transcription reaction was performed using 2 μg of total RNA, reverse
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
8
transcribed into cDNA using oligo dT primer. cDNA was subjected to RT-PCR and
amplified 30 cycles using two oligonucleotide primers derived from published EGFR
or GAPDH sequence, including TGGAGCTACGGGGTGACCGT-3’ and
5’-GGTTCAGAGGCTGATTGTGAT-3’ (EGFR),
5’-AAGCCCATCACCATCTTC-CAG-3’ and 5’-AGGGGCCATCCACA-GTCTTCT-3’(GAPDH) and
5’-TGAC-GGGGTCACCCACACTGTGCCCATCTA-3’ and
5’-CTAGAAGCATTTGCG-GGGACGATGGAGGG-3’(Actin). The PCR products were subjected to 1.2%
agarose gel electrophoresis and visualized by ethidium bromide staining. Real time
PCR was performed with cDNA samples using the ABI Prism 7900 Sequence
Detection System (Applied Biosystems, Foster City, CA). Primers were as follows:
EGFR (forward primer, TTCCTCCCAGTGCCTGAAT-3’ reverse primer,
5’-GGTTCAGAGGCTGAT-TGTGAT-3’); Actin (forward primer,
5’-CCAACCG-CGAGAAGATGA-3’; reverse primer, 5’-TCCATCACGATGCCAGTG-3’). The
data were normalized by the Actin housekeeping gene detection.
Cell proliferation
For growth inhibition analysis, HCT116 cells were seeded at a density of 3 x 103 cells
per well in 96-well plates. After seeding, the growth medium was replaced with
medium containing indicated concentration of TSA. After 3 days, cell growth was
measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(Sigma, St. Louis, MO) colorimetric method. Cell cycle was determined by flow
cytometry using a propidium iodide stain buffer and analyzed on a BD FACS Calibur
cytometer with Cellquest software.
Measurement of Intracellular Glucose 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
9
Prior to harvesting, adherent cultures of control and TSA-treated cells in DMEM
containing 1 or 4.5 mg/ml glucose were washed twice with cold phosphate- buffered
saline (PBS) and then lysed with ion-freeH2O for 5min on ice. The glucose content
was measured with D-glucose measurement kit (GAHK-20, Sigma-Aldrich, St. Louis,
MO) according to the manufacturer’s protocol.
Transient transfection and luciferase activity assay
The EGFR promoter plasmid containing a firefly luciferase was transiently
transfected into HCT116 cells with Arrestin transfection reagent. Briefly, 0.9 μg of
plasmid DNA, 0.1 μg of Renilla luciferase, and 5 uL transfection reagents were
mixed, and the transfection protocol was carried out according to the manufacturer’s instructions (Promega). Six hours after transfection, the cells were cultured in the
normal complete medium for another 16 h. Then, the transfected cells were subjected
to luciferase assay. The firefly luciferaseactivity was normalized to that of the Renilla
luciferase.
Preparation and infection of shHDAC-expressing lentivirus
Briefly, 6 µg pCMV-dR8.91, 3 µg pMD2.G, and 9 µg shLuciferase,
pLKO-shHDAC1, pLKO-shHDAC2 or pLKO-shHDAC3 were cotransfected into HEK293T
cells using Lipofectamine 2000 (Invitrogen). The supernatants containing infectious
shLuciferase, shHDAC1, shHDAC2 or shHDAC3 lentivirus were collected on day 3
after transfection and stored at -80°C. For lentivirus infection, 2 x 105 HCT116 cells
were infected with shLuciferase, shHDAC1, shHDAC2 or shHDAC3 lentivirus at a
multiplicity of infection (MOI) of 1.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
10 Patients and specimen preparation
Specimens of tumor tissue and adjacent normal tissue of colon were obtained from 14
patients who have been pathologically diagnosed colon cancer and underwent surgical
resection at the National Taiwan University Hospital. Tissue specimens were ground,
then sonicated in the lysis buffer (50 mM Tris-HCl, pH 7.4, 1 mM EGTA, 150
mM NaCl, 5% Triton X-100) with protease inhibitors. The samples were
microcentrifuged to remove the larger debris and subjected to western analysis.
Chromatin immunoprecipitation assay
Cells were treated with 5 μM SAHA for 6 h and cross-linked with 1.42%
formaldehyde for 15 min. Cells in two 10-cm dishes were scraped in 1 ml of cold PBS,
centrifuged, and lysed in 1 mL of IP buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.5,
5 mM EDTA, 0.5% Nonidet P-40, and 1% Triton X-100) containing protease
inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 μM leupeptin and 1 μM aprotinin). The nuclear pellet was resuspended in IP buffer and sonicated to shear chromatin. The
sonicated lysates were immunoprecipited with antibodies against SP1, AcH3, AcH4,
H3K4Me2, CBP and HDAC3, respectively and the immune complexes were
recovered with protein A-Sepharose (Roche). The immunoprecipitated DNA and
input DNA were extracted by incubating with 100 μl of 10% Chelex (Bio-Rad), boiling to reverse the cross-link, and centrifuging to remove Chelex slurry. Real-time
PCR was performed with the purified DNA using the following primers: A: 5’- GTGAAAAACCCCACCGTTC-3’ and 5’- TCTGAAGGGGAGCAACCTTA-3’; B:
5’-AAGCTTCCGCGAGTTTCC-3’ and 5’- GAGGCTAAGTGTCCCACTGC-3’; C: 5’- ACCCTGGCACAGATTTGG-3’ and 5’- TGAGGAGTTAATTTCCGAGAGG-3’; D: 5’-CCAGTATTGATCGGGAGAGC-3’ and 5’- TTCCTCCAGAGCCCGACT-3’;
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
11
E: 5’-CTGAGGAAGGAACCCAAAAA-3’ and 5’-GGGAGGTCCTCTCAGAA AGC-3’.
Statistical analysis
Triplicate experiments were performed and results are presented as mean±SE. The
two- tailed Student’s t test was used to calculate the statistical significance between
group 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
12
Results
HDAC inhibitors disrupt the EGF signaling via silencing EGF receptor (EGFR) expression
To examine the antitumor effect of HDACi in colorectal cancer, KRAS wild type
(WiDR and HT29) and KRAS mutant cells (HCT116 and SW480) were treated with
SAHA or cetuximab for 48 hours, and cell viability was measured. SAHA reduced the
survival of these cells in a dose-dependent manner (Fig. 1A), suggesting the
independence of the KRAS status on the antitumor activity of HDACi. In contrast,
cetuximab had little effect on the cell viability (Fig. 1A). This result is consistent with
the previous study that colorectal cancer cells treated with cetuximab were killed
more efficiently by antibody-dependent cellular cytotoxicity (ADCC) which is absent
in in vitro system [19]. Since EGFR plays a significant role in CRC, the ability of its
ligand to trigger the downstream signal in KRAS mutant cells was examined. EGF
triggered both Akt and ERK phosphorylation in HCT116 cells and induced ERK
activation in SW480 cells (Fig. 1B), indicating that KRAS mutation doesn’t fully take
over the ligand-mediated ERK activation and also impling the significance of EGFR
in KRAS mutant cells. Moreover, pretreatment with HDAC inhibitors, TSA and
SAHA, disrupted the EGF-stimulated ERK and Akt phosphorylation in HCT116 cells
and ERK phosphorylation in SW480 cells (Fig. 1C). Since HDAC inhibitors blocked
both Akt and ERK phosphorylations, the very proximal component of EGF signaling
might be targeted by HDACi. Therefore, the expression of EGF receptor was firstly
examined. After treatment with TSA, the expression of EGFR was decreased in
HCT116, SW480, and HT29 cells. To identify whether this is a common phenomenon,
cells originated from different organs were used. After treatment with TSA, the
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
13
reduced EGFR expression was also seen in human skin (A431) and breast
(MDA-MB468) cancer cells (Fig.1D).
HDAC inhibitors reduce the expression of SGLT1 and decrease the intracellular glucose
In addition to EGF signaling, EGFR has been reported to be involved in the glucose
transport by associating and stabilizing the active glucose transporter, SGLT1 [13,20].
Since the expression of EGFR was reduced by HDACi in CRC cells, the levels of
SGLT1 expression and intracellular glucose in response to HDACi were also
examined. As expected, TSA reduced the SGLT1 expression (Fig.2A) and the
intracellular glucose concentration (Fig. 2B). Glucose replenishment retained the
intracellular glucose (Fig 2C) and rescued cells from the TSA-induced cell death (Fig
2D). These data suggested that the loss of EGFR and its partner, SGLT1, might be
involved in the cytotoxic effect of HDAC inhibitors.
Loss of EGFR is implicated in HDAC inhibitor-mediated cytotoxicity
HDAC inhibitors are shown to exert antitumor activity by arresting the cell cycle and
triggering apoptosis [15]. Consistently, SAHA increased sub-G1 population from
7.72% to 17.23% and G2/M population from 16.6% to 24.4% (Fig.3A). To elucidate
the role of EGFR in the antitumor activity of HDACi, cells were transfected with
myc-EGFR and then treated with SAHA for 24 hrs. Overexpression of myc-tagged
EGFR decreased the sub-G1 population and G2/M population (Fig.3A).
SAHA-induced p21 expression was also attenuated by the ectopic expression of EGFR
(Fig.3B). These data indicated that SAHA-reduced EGFR expression contributed to
the SAHA-induced apoptosis and cell cycle arrest.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
14
HDACs are implicated in the transcription of EGFR
Since the amount of EGFR protein is reduced after treatment with HDACi, the EGFR
gene transcription was examined. The mRNA level of EGFR was decreased
dramatically after treatment with TSA and SAHA (Fig.4A), suggesting HDACi
transcriptionally downregulate EGFR expression. This effect was further confirmed
by EGFR reporter assays. Our result showed that TSA and SAHA significantly
decreased the EGFR promoter activity (Fig.4B upper panel). It has been reported that
HDACi decreased the EGFR mRNA stability in ER-negative human breast cancer
cells [21]. Therefore, the stability of EGFR mRNA was examined. The de novo
transcription was stopped by actinomycin D and the EGFR mRNA was measured by
real-time PCR. The slope of EGFR mRNA degradation didn’t show a significant
difference between basal and TSA treatment (Fig. 4B lower panel), suggesting that
HDACi didn’t affect the degradation of EGFR mRNA in colorectal cancer cells. To further elucidate the involvement of HDACs in the transcription of EGFR,
myc-tagged HDAC1, HDAC2 or HDAC3 was ectopically expressed in HCT116 cells, and
EGFR mRNA was measured by RT-PCR. An increase of EGFR mRNA was found in
all these HDAC-expressing cells (Fig. 4C upper panel). Conversely, knockdown of
HDAC1, HDAC2 or HDAC3 by shRNA reduced the expression of EGFR protein
(Fig.4C lower panel). These data indicated that class I HDACs are crucial for EGFR
expression. The positive correlation between EGFR and HDAC3 expression was also
observed in fourteen pairs of human colon tumor and adjacent normal tissues (Fig.
4D). 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
15
SP1 is essential for EGFR transcription and HDAC inhibitor disturbs the binding of SP1 to EGFR promoter
There are several SP1 binding sites on the EGFR promoters and our previous studies
showed that HDACi affects the binding of SP1 to ADAMTS1or p21 promoters
[22,23]. Therefore, SP1 may participate in the HDACs-mediated EGFR expression.
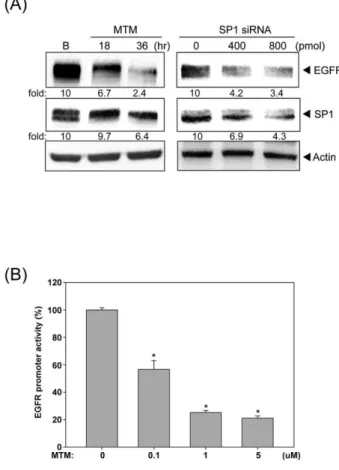
Indeed, inhibition of SP1 by mithramycin A (MTM) and siRNA significantly
decreased the EGFR expression (Fig.5A). Furthermore, MTM drastically reduced the
EGFR promoter activity (Fig.5B), indicating the critical role of SP1 in EGFR gene
transcription. The binding of SP1 to the EGFR promoter is further examined by
chromatin immunoprecipitation (ChIP). Five primer pairs (A, B, C, D and E) were
designed to evenly cover the regions (-1,200 to +1,000 bps) around transcription start
site (Fig.6A). Our data showed that the binding of SP1 to regions C and D was
significantly decreased after treatment with SAHA (Fig.6B). Furthermore, the
acetylation of Histone H3 and H4 on EGFR promoter was largely reduced, especially
in the regions nearby transcription start site (Fig.6B). The status of histone
methylation such as H3K4Me2, H3K9Me3 and H3K27Me3 was also examined.
SAHA didn’t change the residence of these methylation markers on EGFR promoter
despite of enriched H3K4Me2 was found (Fig.6B and data not shown). Since the
acetylation of histone H3 and H4 dropped dramatically after HDAC inhibition, the
occupancy of histone acetyltransferase (HAT) or HDAC on EGFR promoter was
examined. Our result showed that the recruitment of CBP to region D was
significantly decreased by SAHA (Fig.6B). Interestingly, the binding of HDAC3 to
the region D was attenuated, too (Fig.6B). These data showed the dissociation of SP1,
CBP and HDAC3 from EGFR promoter at the same time (Fig.7), implying that these
proteins may influence each other and affect their binding to the EGFR promoter.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
16
Discussion
EGFR and HDAC have been reported to be overexpressed in colorectal and various
cancers [1,15]. However, their relationship is not well-characterized. In this study, we
showed that HDAC inhibitors (HDACi) were able to disrupt the EGF-signaling in
colon cancer cells. EGFR expression in these cells as well as other origins such as
epidermoid (A431) and breast (MDA-MB468) was decreased by HDACi, suggesting
the potential of HDACi to treat EGFR overexpressing cancers. HDACi also reduced
the expression of an active glucose transporter, SGLT1, and thereby suppressed the
glucose uptake of colon cancer cells. More in-depth, we showed that SAHA induced
the dissociation of SP1/CBP/HDAC3 from the regions around EGFR transcription
start site where the histones became hypoacetylated. Our data indicated that the
HDAC inhibitors could serve as a single agent to block EGFR and HDAC, two
critical factors in CRC cells, and may provide a more effective therapy for a broader
range of indication.
Most solid tumors reside in a hypoxic environment and prefer the anaerobic
glycolysis rather than aerobic glycolysis, converting glucose to lactate and produce
fewer ATP with less oxygen consumption. Therefore, the glucose uptake is frequently
enhanced in tumors by overexpression of glucose transporters, such as GLUT1 and
SGLT1 [24]. Unlike GLUT1 that transports glucose passively, SGLT1 uses the
electro-chemical sodium gradient to transport glucose against the internal
concentration gradient. SGLT1 is expressed in human colon cancers, pancreatic
cancer, lung cancer and neoplastic lesions of head and neck [25-29]. It is found to be
stabilized by EGFR, and knockdown of EGFR decreases the SGLT1 expression and
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
17
glucose uptake [13]. Our data also showed that HDACi-mediated loss of EGFR, and
the concurrent reduction of SGLT1 expression and glucose uptake would eliminate
the overall pro-survival functions of EGFR.
Several studies show the inhibitory effect of HDACi on EGFR expression in
human cancers. For example, FK-228, a depsipeptide HDAC inhibitor, is reported to
decrease the expression of EGFR in lung cancer cells [30]. SAHA decreases the levels
of EGFR in ER-negative breast cancer cells via mRNA destabilzaiton [21]. More
recently, inhibition of HDAC6 is found to enhance the endocytosis of EGFR through
increasing tubulin acetylation [31,32]. In this study, we demonstrated that both EGFR
mRNA and its promoter activity were inhibited by HDAC inhibitors in colon cancer
cells, indicating that the de novo synthesis of EGFR was transcriptionally inhibited.
EGFR promoter is characterized with GC-rich, and TATA-less, and harbors multiple
specificity protein 1 (Sp1) binding sites [33]. In addition to SP1, several transcription
factors, such as AP-1, p53 and c-Jun, also participate in the EGFR transcription [34].
SP1 has been reported to regulate the basal EGFR promoter activity [35]. We showed
that inhibition or knockdown of SP1 could decrease the promoter activity and protein
expression of EGFR, emphasizing its crucial role in EGFR expression.
SP1 has been reported to be regulated by several post-translational modifications,
including phosphorylation, acetylation, ubiquitination and sumoylation [36]. It is
acetylated by p300 and deacetylated by HDAC [37]. Although acetylated SP1 could
increase the transcription of GC-box-dependent genes [37], accumulating data also
show that acetylation of SP1 decrease the its transcriptional activity. For example,
SP1 acetylation by HDACi reduces its ability to regulate 12(s)-lipooxygenase
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
18
LOX) expression. Ectopic expression of SP1 mutant, which cannot be acetylated at
lysine 703, increases 12S-LOX transcription, and deacetylation of SP1 is also
required for the transcription of COX-2 [38,39]. Our previous studies show that
HDACi affects the binding of SP1 to ADAMTS1 promoter and the association of SP1
and CBP on p21 promoter [22,23]. SP1 on EGFR promoter might be affected by
HDACi as well. Indeed, SP1 was dissociated from EGFR promoter after treatment
with HDACi, implying that acetylation may decrease the binding of SP1 to the EGFR
promoter. Surprisingly, the histones on EGFR promoter became hypoacetylated. This
could be explained by the concurrent dissociation of CBP, the histone
acetyltransferase (HAT).
HDACi is reported to induce G2/M growth arrest as well as G0/G1 arrest in colorectal
cancer cells, and the HDACi-mediated growth arrest consistently involves p21
induction [40-43]. In HCT116 cells, p21 is induced and the cell cycle is arrested in
G2/M phase by silencing class I HDACs, especially HDAC3 [17]. Consistently, we
found that SAHA induced p21 and G2/M arrest and re-expression of EGFR could
alleviate these events. HDAC3 has been reported to be maximally expressed in the
proliferative compartment in mouse colon. Knockdown of HDAC3 induced a greater
magnitude of G2/M and S phase arrest than that of HDAC1/2, suggesting that
HDAC3 is more significant than HDAC1/2 in colon cell proliferation [17]. HDAC3 is
a component of the NCoR-SMRT co-repressor complex, which is distinct from
repressor complexes containing HDAC1 and HDAC2 (Sin3A and NuRD) [44],
indicating the specific roles of HDAC isoform in gene repressing. In contrast,
knockdown of HDAC1, 2 or 3 decreased the EGFR expression in varying degree,
indicating that they share functional redundancy on promoting EGFR transcription.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
19
Ectopic express HDAC3 induced a greater magnitude of EGFR mRNA and a positive
correlation between EGFR and HDAC3 expression in colon cancer patients.
Therefore, HDAC3 may be most essential in EGFR transcription.
Association of HDACs with gene promoters are traditionally considered to repress
transcription and HDAC is thought to reactivate the silenced genes [45]. However,
HDACi is also reported to decrease the expression of thymidylate synthase, vascular
endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF) and
endothelial nitric oxide synthase (eNOS) [46-48]. It is suggested that gene
transcription primed by H3K4 methylation requires the dynamic cycle of histone
acetylation and deacetylation by transient HAT/HDAC binding [49]. In this study, we
found that EGFR promoter was enriched with H3K4 di-methylation, suggesting that
EGFR gene transcription may be primed by H3K4 methylation. HDAC3 and CBP
were both associated with EGFR promoter and concurrently dissociated after
treatment with HDACi, implying that dynamic HAT/HDAC binding is occurred.
Since CBP and HDAC3 are unable to directly bind gene promoter, SP1 may serve as
a bridge between CBP/HDAC3 and EGFR promoter (Fig. 6A). HDACi may induce
SP1 acetylation and leads to its dissociation from EGFR promoter, which disrupts the
dynamic binding of HDAC3 and CBP (Fig. 6B). Taken together, our results showed
that the SP1, HDAC3 and CBP were all dissociated from EGFR promoter after SAHA
treatment, suggesting their functional relevance on EGFR transcription.
It has been reported that HDAC inhibitors synergize with 5-FU in vitro and in vivo to
treat colon cancer through downregulation of thymidylate synthase, the 5-FU target
enzyme [46]. Combination of 5-FU with SAHA has recently entered phase I/II trial to
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
20
treat CRC [18,50]. Inhibition of MAPK and Akt signaling by AEE788, a multiple
receptor tyrosine kinases inhibitor, synergistically potentiates HDAC-induced
apoptosis in a broad spectrum of cancer cell lines [51]. Recently, a new compound,
CUDC-101, which inhibit the activity of both EGFR and HDAC, is demonstrated to
have powerful anticancer activity [52]. These reports strengthen the rationale of
concurrent inhibition of EGFR and HDAC in cancer therapy. In this study, we
showed that HDAC inhibitor alone is able to block EGFR transcription as well as
HDAC, and may provide a hint for superior strategy of colorectal cancer therapy.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
21
References
1. Normanno N, {De Luca} A, Bianco C, Strizzi L, Mancino M, et al. (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366: 2-16. 2. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, et al. (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. The New England journal of medicine 351: 337-345.
3. Rajpal S, Venook AP (2006) Targeted therapy in colorectal cancer. Clinical advances in hematology & oncology : H&O 4: 124-132.
4. Amado RG, Wolf M, Peeters M, {Van Cutsem} E, Siena S, et al. (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 26: 1626-1634.
5. Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, et al. (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360: 1408-1417.
6. Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, et al. (2009) Genetic prognostic and predictive markers in colorectal cancer. Nature reviews Cancer 9: 489-499.
7. Banck MS, Grothey A (2009) Biomarkers of Resistance to Epidermal Growth Factor Receptor Monoclonal Antibodies in Patients with Metastatic Colorectal Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 15: 7492-7501.
8. Linardou H, Papadimitriou CA, Dahabreh IJ, Kanaloupiti D, Siannis F, et al. (2008) Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. The Lancet Oncology 9: 962-972.
9. Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, et al. (1995) Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 376: 337-341.
10. Luetteke NC, Phillips HK, Qiu TH, Copeland NG, Earp HS, et al. (1994) The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes & Development 8: 399-413.
11. Ewald J (2003) Ligand- and kinase activity-independent cell survival mediated by the epidermal growth factor receptor expressed in 32D cells. Experimental Cell Research 282: 121-131.
12. Harari PM, Huang S-M (2004) Combining EGFR inhibitors with radiation or chemotherapy: will preclinical studies predict clinical results? International journal of radiation oncology, biology, physics 58: 976-983.
13. Fidler IJ, Weihua Z, Tsan R, Huang W-C, Wu Q, et al. (2008) Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer cell 13: 385-393.
14. Ritter CA, Arteaga CL (2003) The epidermal growth factor receptor-tyrosine kinase: a promising therapeutic target in solid tumors. Seminars in oncology 30: 3-11. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
22
15. Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nature reviews Drug discovery 5: 769-784.
16. Mariadason JM (2008) HDACs and HDAC inhibitors in colon cancer. Epigenetics : official journal of the DNA Methylation Society 3: 28-37.
17. Wilson AJ, Byun D-S, Popova N, Murray LB, L'Italien K, et al. (2006) Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. The Journal of biological chemistry 281: 13548-13558.
18. Wilson PM, El-Khoueiry A, Iqbal S, Fazzone W, Labonte MJ, et al. (2010) A phase I/II trial of vorinostat in combination with 5-fluorouracil in patients with metastatic colorectal cancer who previously failed 5-FU-based chemotherapy. Cancer chemotherapy and pharmacology: 979-988.
19. Levy EM, Sycz G, Arriaga JM, Barrio MM, von Euw EM, et al. (2009) Cetuximab-mediated cellular cytotoxicity is inhibited by HLA-E membrane expression in colon cancer cells. Innate Immun 15: 91-100.
20. Engelman JA, Cantley LC (2008) A sweet new role for EGFR in cancer. Cancer cell 13: 375-376.
21. Zhou Q, Shaw PG, Davidson NE (2009) Inhibition of histone deacetylase suppresses EGF signaling pathways by destabilizing EGFR mRNA in ER-negative human breast cancer cells. Breast Cancer Res Treat 117: 443-451. 22. Chou C-W, Chen C-C (2008) HDAC inhibition upregulates the expression of
angiostatic ADAMTS1. FEBS Letters 582: 4059-4065.
23. Lin Y-C, Lin J-H, Chou C-W, Chang Y-F, Yeh S-H, et al. (2008) Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer research 68: 2375-2383.
24. Ganapathy V, Thangaraju M, Prasad PD (2009) Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacology & therapeutics 121: 29-40.
25. Casneuf VF, Fonteyne P, {Van Damme} N, Demetter P, Pauwels P, et al. (2008) Expression of SGLT1, Bcl-2 and p53 in primary pancreatic cancer related to survival. Cancer investigation 26: 852-859.
26. Mahraoui L, Rodolosse A, Barbat A, Dussaulx E, Zweibaum A, et al. (1994) Presence and differential expression of SGLT1, GLUT1, GLUT2, GLUT3 and GLUT5 hexose-transporter mRNAs in Caco-2 cell clones in relation to cell growth and glucose consumption. Biochem J 298 Pt 3: 629-633.
27. Blais A (1991) Expression of Na(+)-coupled sugar transport in HT-29 cells: modulation by glucose. Am J Physiol 260: C1245-1252.
28. Ishikawa N, Oguri T, Isobe T, Fujitaka K, Kohno N (2001) SGLT gene expression in primary lung cancers and their metastatic lesions. Jpn J Cancer Res 92: 874-879.
29. Helmke BM, Reisser C, Idzko M, Dyckhoff G, Herold-Mende C (2004) Expression of SGLT-1 in preneoplastic and neoplastic lesions of the head and neck. Oral Oncol 40: 28-35.
30. Yu XD, Wang SY, Chen GA, Hou CM, Zhao M, et al. (2007) Apoptosis induced by depsipeptide FK228 coincides with inhibition of survival signaling in lung cancer cells. Cancer J 13: 105-113.
31. Gao YS, Hubbert CC, Yao TP (2010) The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J Biol Chem 285: 11219-11226.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
23
32. Deribe YL, Wild P, Chandrashaker A, Curak J, Schmidt MH, et al. (2009) Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signal 2: ra84.
33. Brandt B, Meyer-Staeckling S, Schmidt H, Agelopoulos K, Buerger H (2006) Mechanisms of egfr gene transcription modulation: relationship to cancer risk and therapy response. Clinical cancer research : an official journal of the American Association for Cancer Research 12: 7252-7260.
34. Johnson AC, Murphy BA, Matelis CM, Rubinstein Y, Piebenga EC, et al. (2000) Activator protein-1 mediates induced but not basal epidermal growth factor receptor gene expression. Molecular medicine (Cambridge, Mass) 6: 17-27. 35. Kageyama R, Merlino GT, Pastan I (1988) Epidermal growth factor (EGF)
receptor gene transcription. Requirement for Sp1 and an EGF receptor-specific factor. The Journal of biological chemistry 263: 6329-6336.
36. Waby JS, Bingle CD, Corfe BM (2008) Post-translational control of sp-family transcription factors. Current genomics 9: 301-311.
37. Koshiji M, To KKW, Hammer S, Kumamoto K, Harris AL, et al. (2005) HIF-1α Induces Genetic Instability by Transcriptionally Downregulating MutSα Expression. Molecular Cell 17: 793-803.
38. Chen C-J, Chang W-C, Chen B-K (2008) Attenuation of c-Jun and Sp1 expression and p300 recruitment to gene promoter confers the trichostatin A-induced inhibition of 12(S)-lipoxygenase expression in EGF-treated A431 cells. European journal of pharmacology 591: 36-42.
39. Tong X, Yin L, Giardina C (2004) Butyrate suppresses Cox-2 activation in colon cancer cells through HDAC inhibition. Biochemical and biophysical research communications 317: 463-471.
40. Kobayashi H, Tan EM, Fleming SE (2003) Sodium butyrate inhibits cell growth and stimulates p21WAF1/CIP1 protein in human colonic adenocarcinoma cells independently of p53 status. Nutr Cancer 46: 202-211.
41. Xu WS, Perez G, Ngo L, Gui CY, Marks PA (2005) Induction of polyploidy by histone deacetylase inhibitor: a pathway for antitumor effects. Cancer Res 65: 7832-7839.
42. Heerdt BG, Houston MA, Augenlicht LH (1997) Short-chain fatty acid-initiated cell cycle arrest and apoptosis of colonic epithelial cells is linked to mitochondrial function. Cell Growth Differ 8: 523-532.
43. Schwartz B, Avivi-Green C, Polak-Charcon S (1998) Sodium butyrate induces retinoblastoma protein dephosphorylation, p16 expression and growth arrest of colon cancer cells. Mol Cell Biochem 188: 21-30.
44. Jepsen K, Rosenfeld MG (2002) Biological roles and mechanistic actions of co-repressor complexes. J Cell Sci 115: 689-698.
45. Berger SL (2007) The complex language of chromatin regulation during transcription. Nature 447: 407-412.
46. Fazzone W, Wilson PM, Labonte MJ, Lenz HJ, Ladner RD (2009) Histone deacetylase inhibitors suppress thymidylate synthase gene expression and synergize with the fluoropyrimidines in colon cancer cells. Int J Cancer 125: 463-473.
47. Sasakawa Y, Naoe Y, Noto T, Inoue T, Sasakawa T, et al. (2003) Antitumor efficacy of FK228, a novel histone deacetylase inhibitor, depends on the effect on expression of angiogenesis factors. Biochem Pharmacol 66: 897-906.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63
24
48. Rossig L, Li H, Fisslthaler B, Urbich C, Fleming I, et al. (2002) Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res 91: 837-844.
49. Wang Z, Zang C, Cui K, Schones DE, Barski A, et al. (2009) Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138: 1019-1031.
50. Fakih MG, Pendyala L, Fetterly G, Toth K, Zwiebel JA, et al. (2009) A phase I, pharmacokinetic and pharmacodynamic study on vorinostat in combination with 5-fluorouracil, leucovorin, and oxaliplatin in patients with refractory colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 15: 3189-3195.
51. Yu C, Friday BB, Lai JP, McCollum A, Atadja P, et al. (2007) Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin Cancer Res 13: 1140-1148.
52. Lai CJ, Bao R, Tao X, Wang J, Atoyan R, et al. (2010) CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res 70: 3647-3656.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63