1
金屬陽離子於二氧化矽/水溶液之界面反應-分子層次之探討
計畫主持人:駱尚廉教授 計畫參與人員:官文惠博士 NSC 89-2211-E-002-077 執行機關:國立台灣大學環境工程研究所 執行期間:89.8.1~90.7.31
關鍵詞:金屬陽離子、二氧化矽、界面反應、
分子層次;metallic cations, SiO2, interfacereactions, molecular level
摘要
金屬陽離子之傳輸與流佈常受周遭固相介 質之影響,而在固/液界面發生吸附、水解、表 面沈澱等反應。而過去對界面現象之研究已證 實,處於兩相間的交界部份非常薄,最多不過 幾個分子厚;此一事實意味著分子層次的研究 方法,是探討界面反應所必須的工具。因此本 研究以二氧化矽為固相氧化物,探討 Al(III)離 子於不同系統 pH 值、不同 Al(III)濃度下之界 面吸附與水解現象。研究方法除傳統批式實驗 數據、表面錯合吸附模擬外,並加入量子方法 在分子層次對基本熱力學參數之計算,進而定 性並定量描述金屬陽離子在固相表面所表現的 吸附與水解行為。 經 CASTEP 量子理論計算後,擷取熱力學 上允許發生之反應型態,並合併這些吸附形式 模擬 Al(III)/SiO2(s)系統可得較佳之模擬結果。前言
在複雜且多元的自然環境中,幾乎沒有單 相(single phase)系統的存在,因此界面反應 一直是環境問題中最重要的課題之一。許多研 究者已經證實(Stumm, 1992; Lasaga and Gibbs, 1990; Adamson, 1990),處於兩相間的交界部 份,乃是一個具有一定厚度的界面層,這層的 結構和性質與相鄰兩相的性質都不一樣。按熱 力學的觀點,應將這一部份視為一個特殊的相。 根據量子力學的估算可知,這個界面相非常薄, 最多不過幾個分子厚;此一事實意味著分子層 次的研究方法,是探討界面反應所必須的工具。 金屬陽離子之傳輸與流佈均是環境工程所 關切之問題。惟其行為常受周遭固相介質之影 響,而在固/液界面發生吸附(adsorption)、水解 (hydrolysis)、表面沈澱(surface precipitation)等 反應。針對表面吸附水解反應,多數學者採用 表面錯合模式(surface complexation model)加以 描述與模擬,該模式將固相表面之官能基,當 作被吸附或形成表面沈澱金屬陽離子之配位基 (coordinating ligands) (Schindler, 1981; Schindlerand Stumm, 1987; Farley et al., 1985; Davis and
Kent, 1990)。研究者往往藉著實驗室批式吸附 實驗的數據用不同假設之模式,調整模式中幾 個特定的可調參數,使模式模擬結果盡可能符 合實驗所得之數據,並視最符合實驗數據之假 設模式為物種在固相表面之反應機制。上述之 模式均是根據表面物種之計量化學反應所推演 出來,常有同一物種,同一環境條件下,可能 同 時 符 合 兩 種 以 上 吸 著 機 制 之 情 況 出 現 (Goldberg, 1991)。 事實上,傳統的吸附實驗方法只能得到巨 觀的反應結果,而能符合實驗數據之假設模式 並不一定是真實的微觀反應機制。由量子力學 (quantum mechanics)發展出來的計算方法,已 被研究界面現象的科學家視為瞭解分子層次問 題的另一項有利工具。量子化學是以薛丁格方 程式(Schröedinger equation)來描述微小質點的 狀態,而計算化學之主要任務即在於運用各種 不同之數學方法解此方程式。所推演出來之方 法,在計算的過程中不使用任何經驗或半經驗 參數,僅利用宇宙常數,如蒲朗克常數(h)、電 子所帶電量(e)、光速(c)、質子與電子之質量等, 以計算分子層次之各項特性。 本研究將結合傳統吸附實驗數據與量子化 學對於分子層次之理論計算並配合吸附錯合-表 面沈澱模擬,進而完整描述 Al(III)在固相表面 所表現的吸附與水解行為。
材料與方法
研究首先針對 SiO2表面進行特性測定, 再依傳統批式實驗方法完成各種吸附實驗數 據;同時配合密度泛函法(DFT)計算金屬陽離子 在二氧化矽表面之反應熱力學參數、分子結構 等 ; 其 次 推 演 表 面 吸 附 錯 合 模 式 (surface complexation model),並以量子計算結果加以確 定表面之反應機制,修正模式之推求。 批式實驗 為求得 Al(III)在 SiO2(S)表面錯合吸附之平 衡常數值,先大致求得其在不同 pH 值下之飽 和吸附量,並設計從極低之表面覆蓋率(例如 幾個百分比之飽和吸附量)到百分之百覆蓋之 濃度。實驗進行前先配置 10g SiO2(s)/L 之貯備2
溶液,並先行曝 N2氣 2 hr 去除其中之 CO2。
所有的批式實驗均維持 1g SiO2(s)/L 之固液濃度
比。所有批式實驗均以不同濃度(0.01~1M) 之 KOH (Merck, >85% GR)、HNO3 (Merck, 65%
GR)溶液調整系統之 pH 值至所需之範圍。除 了背景離子強度對吸附影響試驗外,每個試驗 樣品均以 KNO3 溶液,維持背景離子強度為 0.1M。為比較不同初始 Al(III)濃度對邊緣曲線 之影響,改變實驗中 Al(III)離子初始濃度在 1×10-4~1×10-2 M,調整 pH 值範圍在 2~11,反 應時間為 48hr。實驗容器為 50mL 之有蓋 PE 瓶。反應係在恆溫振盪水槽中進行,調整轉速 150rpm;控溫在 25℃。實驗完畢後,先以 9500rpm 之轉速將溶液離心 15 min,再用 0.2μm 之濾膜 過濾離心後之上澄液,爾後將濾液保存於 4℃ 下,待累積一定數量之樣品後,採用原子吸收 光譜儀(Atomic Absorption Spectrophotometry, AA, Perkin-Elmer 5000)分析濾液中 Al(III)之含 量。
理論計算
本研究 採 用 國 科 會 國 家 高 速 電 腦 中 心 之 CASTEP (Cambridge Serial Total Energy Package) 計算軟體。是由劍橋大學卡文迪西實驗室的凝 體理論組所發展出來的一個解量子力學問題的 程式。凡給定一初始的原子排列,CASTEP 便 能解出諸如固體或表面等延伸系統最穩定時的 (基態)電子密度分佈、系統的總能量、以及 各原子的受力。CASTEP 用虛位勢來近似原子, 並用平面波作為基底來展開波函數。
結果與討論
針對氧化物固相表面反應,傳統方法均是 以表面錯合模式模擬實驗室之批式吸附實驗結 果,以最符合實驗數據之假設模式,作為研究 對象系統之唯一反應模式。但除非該系統能有 分子層次的光譜證據,否則所有的結果都僅限 於巨觀尺度之推論。既然表面反應發生在幾個 分子厚的範圍內,而光譜方法又有許多系統限 制,故以量子為出發點之理論計算,是研究固 相表面反應之最佳工具。 三層模式對 Al(III)在 SiO2表面吸附之模擬 表一為 SiO2 之表面各種可能之酸鹼及錯合反應 整理。表二為三層模式模擬所需之輸入參數。 由於針對此商品化之 SiO2,過去之文獻已有完整表面性質測定(Meng and Letterman, 1993a, b, 1996),故在本研究中不再重複估量。此外,本
研 究 以 3.3 版之 MINTEQA2 (Allison et al., 1991)做三層模式之模擬。圖 1 為各種表面錯 合反應對 0.1Mm 之 Al(III)/SiO2系統之表面吸 附反應模擬結果。由圖中可明顯看出,外層錯 合(outer-sphere complexes)吸附反應之假設於 實驗結果相差太多,故根據表面錯合模式之判 定標準,外層錯合反應發生之可能性可以完全 被 排 除 。 其 次 , 對 內 層 錯 合 ( inner-sphere complexes)反應之模擬結果顯示,內層單鉤水 解(SOAlOH+ type)與內層雙鉤((SO)2Al + type) 產物之模擬結果近乎一致,且與實驗點頗為符 合。但在 pH 值低於 3 的範圍,此兩種模擬結 果有低估的現象。而內層單鉤產物之模擬,在 pH<3 時高估實驗結果,而在 pH>3 之範圍內 又低估實驗結果。這種結果是可以理解的,因 為 Al(III) 在 較 低 的 pH 值 是 完 全 的 離 子 態 (Al(H2O)6 3+),而當 pH 值約高於 4 時,其會與 周圍水分子發生水解作用,而變成 AlOH(H2O)5 2+ 的型態,故以 SOAl2+為反應型態之假設則會低 估實驗結果。此時,根據傳統表面錯合模式之 處理原則,多半的研究者會下一個這樣的結論: Al(III)在水溶液中,會以內層單鉤吸附水解態, 或內層雙鉤錯合態兩種形式,吸附在 SiO2表面。 表一 三層模式模擬 SiO2表面反應之輸入參數
(Meng and Letterman, 1993a, b, 1996)
Site concentration (mmole/g) 1.67 Site density (sites/nm2
) 5.0 Surface area (m2 /g) 200 Capacitance,C1 (µF/cm 2 ) 125 Capacitance,C2 (µF/cm 2 ) 20 Log aint 1 K -1.5 Log aint 2 K -5.5 LogKintK -7.0 Log int N 3 K O
-1.5 圖 1 三層模式對 0.1mM Al(III)/SiO2系 統吸附反應之模擬結果 0 20 40 60 80 100 120 0 1 2 3 4 5 6 p H % Al(III) Removal exp. SOAl SOAlOH (SO)2Al SO-Al SO-AlOH
3
表二 SiO2之表面反應
surface reaction equilibrium expression surface ionization (inner-sphere)
SOH + H+ = SOH 2 + K a1 = exp(FΨ0/RT)[SOH2 +]/ [SOH][H+] [1] SOH = SO- + H+ Ka2 = exp(-FΨ0/RT)[SO -][H+]/[SOH] [2]
electrolyte adsorption (outer-sphere) SOH + K+ = SO-K + H+ K
K = exp(F(Ψβ-Ψ0)/RT)[SO-K][H
+]/[SOH][K+] [3]
SOH + NO3
- + H+ = SOH
2-NO3 KNO3 = exp(F(Ψ0 -Ψβ)/RT)[SOH2-NO3]/[SOH] [H +] [NO
3
-] [4]
inner-sphere surface complexes SOH + Al3+ = SOAl2+ + H+
K = exp(2FΨ0/RT)[SOAl2+][H+]/[SOH][Al3+] [5]
SOH + Al3+ + H
2O = SOAlOH
+ + 2H+
K = exp(FΨ0/RT)[SOAlOH+][H+]2/[SOH][Al3+] [6]
2SOH + Al3+ = (SO) 2Al
+ + 2H+ K = exp(FΨ0/RT)[(SO) 2Al
+][H+]2/[SOH]2[Al3+] [7]
outer-sphere surface complexes SOH + Al3+ = SO-−Al3+ + H+ K = exp(F(3Ψ
β-Ψ0)/RT)[SO--Al3+][H+]/[SOH][Al3+] [8]
SOH + Al3+ + H
2O = SO
-−AlOH2+ + 2H+
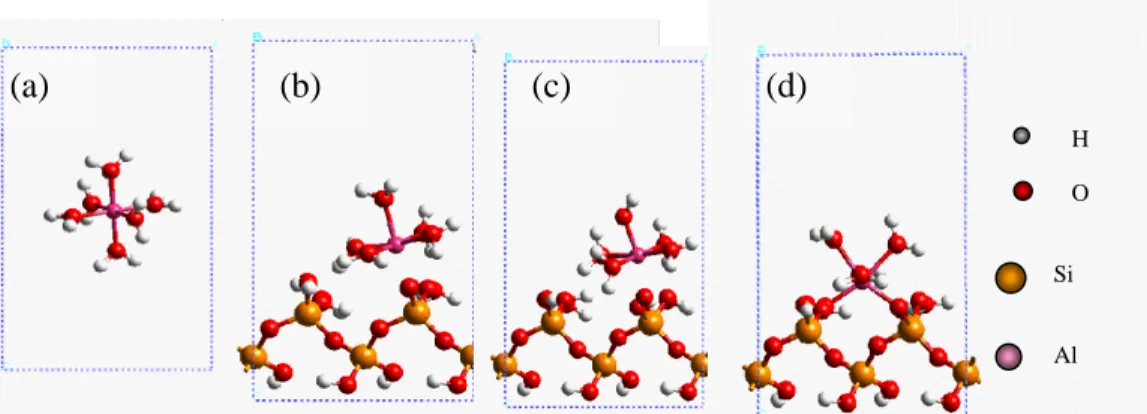
K = exp(F(2Ψβ-Ψ0)/RT)[SO--AlOH2+][H+]2/[SOH][Al3+] [9] 這樣的結論在巨觀研究上雖具相當之意義,但 其真實性卻值得商榷。有鑑於此,從量子觀點 出發的理論計算以驗證巨觀之模擬結果,實有 其必要性。 另外,在模擬過程發現,當液相中 Al(III) 初始濃度超過 1mM 時,無論改用何種反應假 設,都無法得到符合實驗點之模擬值;特別是 在 pH 值高於 5 的情況下,Al(III)移除率都只能 達 50%左右。由此可見,當液相中金屬陽離子 的濃度高至某一程度,其彼此間之交互作用相 較於表面吸附反應言,已不可將其忽略;即便 此時之表面覆蓋率仍相當的低。故此情況已違 反表面錯合反應之假設,必須將金屬陽離子間 之交互作用再考慮進去。 SiO2表面結構之建立 圖 2 為 SiO2塊材經[001]方向切割後之表面 結構。由於本研究之實驗系統係在水溶液環境 中反應,故 SiO2 表面呈現一完全水合狀態,亦 即表面之 O 原子全部以 H 原子作價鍵飽和處 理。此外,建構完成表面結構後,利用 CASTEP 程式將此結構作最佳化(optimized)計算。並 以此一表面與水合 Al(III)離子作其他反應之計 算。 Al(III)在 SiO2表面之反應計算 根 據 上 一 步 驟 之 計 算 結 果 , 將 以 SiO2(001)-(2×2)的最佳化後表面,作為往後所 要探討問題之表面。在圖 1 表面錯合模式模擬 中,針對表面吸附的型態總共做了兩大類共五 種的假設,而初步模擬之結果發現,外層吸附 之假設偏差太多,已被排除在具發生可能性之 外。故暫不考慮計算外層錯合之吸附型態。 根據熱力學之基本原理,當一個反應之Δ G 值必須小於 0,此反應屬自發反應,也就是 該系統在外界不對其作功的情況下,反應會自 然發生。本研究及利用此一基本觀念,利用 CASTEP 程式計算表二中各物種之總能量,再 用產物之總能減去反應物之總能,求得各假設 之吸附反應能量變化情形。另外,必須加以考 慮的是,離子態的 H+與 Al3+在水溶液中是不存 在的,真實的情況是 H+以 H 3O +的形式,Al3+以 配位 6 個水分子的形式 Al(H2O)6 3+存在。圖 3 分 別為水合 Al(III)、Al(III)內層單鉤吸附、Al(III) 內層單鉤吸附水解態、Al(III)內層雙鉤吸附態 之最佳化後結構圖。從此一過程中,反應之熱 力學本質始真實呈現,而不僅限於巨觀模擬實 驗結果好壞之人為判定。 表三為理論計算之能量變化值。由此計算 中,吾人可發現表二中之方程式(5)、(6)、(7), 其總能量變化均小於 0,表示其均為自發反應。 圖 2 SiO2塊材經[001]面切割並以 H 飽和後 之表面結構(2×2)
4
表三 各假設吸附型態之理論計算能量變化
inner-sphere surface complexes adsorption energy(ev)
Eq(5) SOH + Al3+ =SOAl2+ + H+ -7.29 Eq(6) SOH + Al3+ + H2O= -0.84 SOAlOH1+ + 2H+ Eq(7) 2SOH + Al3+ = (SO)2Al + + 2H+ -0.59 這個結果反映出在三層模式之模擬的確有其盲 點,在初始之表面錯合吸附模擬中,內層單鉤 能變化卻驅使反應往方程式之右邊走。故根據 此一結果,很顯然內層單鉤吸附的型態不應被 排除在可能發生之反應之外。 修正後之再模擬 前節顯示,表三中之 Eq(5)、(6)及(7)在熱 力學上都是可自發的,故吾人重新對 Al(III)/SiO2 系統作三層模式之模擬。這次,將內層吸附之 三種型態都一併放入模擬,結果示如圖 4,而 各產物之形成常數列如表四。很顯然,合併三 個方程式的模擬結果與實驗值之符合度,較任 單一反應模擬之結果都好。 表四 各假設吸附型態之形成常數 reaction formation constant logKSOAl Eq(5) 1.0
logKSOAlOH Eq(6) -3.0
logK(SO)2Al Eq(7) -0.2
結論
結合量子之分子層次計算評估表面吸附錯合 機制,較傳統巨觀實驗與模擬可得更精確之微 觀結果。
參考文獻
Admson, A. W., Physical Chemistry of Surfaces, John Wiley &
Sons, Canada, 1990.
Allison, J. D., D. S. Brown, K. J. Novo-gradac,
MINTEQA2/PRODEFA2, A Geochemical Assessment Model for Environmental Systems: Version 3.11 database and version 3.0 User’s Manual, Environmental Research Laboratory, U. S. EPA:
Athens, GA, 1991.
Davis, J. A., and D. B. Kent, in Mineral-Water Interface
Geochemistry (M. F. Hochella, Jr. and A. F. White, Eds.), pp.
177-260, Reviews in Mineralogy, No. 23, Mineral Soc. Am., Washinton, DC, 1990.
Farley, K. J., D. A. Dzombak, and F. M. M. Morel, “a Surface Precipitation Model for the Sorption of Cations on Metal Oxides,”J. Colloid Interface Sci. 106, 226-242, 1985.
Goldberg, S., ”Sensitivity of Surface Complexation Modeling to the Surface Site Density Parameter,” J. Colloid Interface Sci. 145,
1-9, 1991.
Lasaga, A. C. and G. V. Gibbs, “Ab Initio Quantum-Mechanical Calculations of Surface Reactions-A New Era?” in Aquatic Chemical Kinetics (Stumm W. Eds.) Wiley &Sons, Canada, 1990.
Meng, X. and R. D. Letterman, “Effect of Component Oxide Interaction on the Adsorption Properties of Mixed Oxides,”Environ Sci. Technol. 27, 970-975, 1993a.
Meng, X. and R. D. Letterman, “Modeling Ion Adsorption on Aluminum Hydroxide Modified Silica,”Environ Sci. Technol. 27,
1924-1929, 1993b.
Meng, X. and R. D. Letterman, “Modeling Cadmiunn and Sulfate Adsorption by Fe(OH)3/SiO2 Mixed Oxides,”Wat. Res. 30,
2148-2154, 1996.
Schindler, P. W. and W. Stumm, in Aquatic Surface Chemistry,(W.
Stumm, Ed.), pp.83-110. Wiley, New-York (1987).
Schindler, P. W., in Adsorption of Inorganics at Solid-Literquid interfaces (M. A. Anderson and A. J. Rubin, eds.), pp1-49. Ann
Arbor Science Pub., Ann Arbor. MI, 1981.
Stumm, W., Chemistry of the Solid-Water Interface: Processes at the Mineral-Water and Particle-Water Interface in Natural Systems,
John Wiley and Sons, New York, 1992.
致謝 感謝淡江大學化學系林志興教授在計算 上之討論;國科會高速電腦中心提供計算資源, 與中研院原分所提供之計算軟體界面使用。 0 2 0 4 0 6 0 8 0 1 0 0 1 2 3 4 5 p H % Al(III) Removal E q (5 )+ (6 )+ (7 ) e x p . (a) (b) (c) (d) H Si O Al
圖 3(a)水合 Al(III);(b)水合 Al(III)與 SiO2表面行內層單鉤吸附產物;(c)水合 Al(III)與 SiO2
表面行內層單鉤水解吸附;(d)水合 Al(III)與 SiO2表面行內層雙鉤吸附產物最佳化後結構