Joao H.F. Pedra, David A. Johnson and John Y-J. Shyy
Huang,
Tzu-pin Shentu, Liang Wen, Brendan Gongol, Wei Sun, Xiao Liang, Ju Chen, Hsien-Da
Han Xiao, Min Lu, Ting Yang Lin, Zhen Chen, Gang Chen, Wei-Chi Wang, Traci Marin,
Endothelium Mediates Hemodynamic-Induced Atherosclerosis Susceptibility
Sterol Regulatory Element Binding Protein 2 Activation of NLRP3 Inflammasome in
Print ISSN: 0009-7322. Online ISSN: 1524-4539
Copyright © 2013 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Circulation
doi: 10.1161/CIRCULATIONAHA.113.002714
2013;128:632-642; originally published online July 9, 2013;
Circulation.
http://circ.ahajournals.org/content/128/6/632
World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/suppl/2013/07/09/CIRCULATIONAHA.113.002714.DC1.html
Data Supplement (unedited) at:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation
Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document.
Permissions and Rights Question and Answer
this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information about Office. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Circulation
in
Requests for permissions to reproduce figures, tables, or portions of articles originally published Permissions:
by guest on April 24, 2014 http://circ.ahajournals.org/
Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from http://circ.ahajournals.org/ by guest on April 24, 2014 Downloaded from
632
A
therosclerosis preferentially develops at branches and curvatures in the arterial tree. Cardiovascular risk factors such as hyperlipidemia, smoking, and hypertension increase the prevalence and severity of lesions in these atheroprone regions.1 At the cellular and molecular levels, disturbed flowpatterns with low shear stress such as those found at vascu-lar branches and curvatures increase the expression of genes such as interleukin 1β (IL-1β) and NADPH oxidase (NOX) to promote inflammatory and oxidative stresses in vascular endothelial cells (ECs).2,3 Such hemodynamic-induced
func-tionally disturbed endothelium predisposes localized areas to become atherogenic, with ensuing monocyte recruitment and foam cell formation. Although extensive studies have revealed EC gene expression profiles associated with the atheroprone flow patterns, the key molecular events link-ing mechanical stimuli to atherogenic responses remain undetermined.
Editorial see p 579
Clinical Perspective on p 642
Sterol regulatory element binding (SRE) protein 2 (SREBP2) plays a canonical role in cholesterol homeostasis by its transcrip-tional regulation of molecules involved in cholesterol biosyn-thesis and low-density lipoprotein (LDL) uptake.4 Cholesterol
depletion leads to increased expression of SREBP2, along with its intronic microRNA-33 (miR-33), which replenishes cellu-lar cholesterol by inducing genes that encode proteins such as HMG-CoA reductase and LDL receptor and suppressing the ATP-binding cassette transporter 1 (ABCA1).5,6 Conversely,
elevated levels of sterols suppress SREBP expression, thereby lowering cellular cholesterol levels. This homeostasis might be disrupted by atheroprone flow patterns, which induce sustained activation of SREBP1 and perturbation of EC function.7 Background—The molecular basis for the focal nature of atherosclerotic lesions is poorly understood. Here, we explored
whether disturbed flow patterns activate an innate immune response to form the NLRP3 inflammasome scaffold in vascular endothelial cells via sterol regulatory element binding protein 2 (SREBP2).
Methods and Results—Oscillatory flow activates SREBP2 and induces NLRP3 inflammasome in endothelial cells. The
underlying mechanisms involve SREBP2 transactivating NADPH oxidase 2 and NLRP3. Consistently, SREBP2, NADPH oxidase 2, and NLRP3 levels were elevated in atheroprone areas of mouse aortas, suggesting that the SREBP2-activated NLRP3 inflammasome causes functionally disturbed endothelium with increased inflammation. Mimicking the effect of atheroprone flow, endothelial cell–specific overexpression of the activated form of SREBP2 synergized with hyperlipidemia to increase atherosclerosis in the atheroresistant areas of mouse aortas.
Conclusions—Atheroprone flow induces NLRP3 inflammasome in endothelium through SREBP2 activation. This increased
innate immunity in endothelium synergizes with hyperlipidemia to cause topographical distribution of atherosclerotic lesions. (Circulation. 2013;128:632-642.)
Key Words: atherosclerosis ◼ endothelial cell ◼ NLRP3 protein, human ◼ shear stress
◼ sterol regulatory element binding proteins
© 2013 American Heart Association, Inc.
Circulation is available at http://circ.ahajournals.org DOI: 10.1161/CIRCULATIONAHA.113.002714
Received November 8, 2012; accepted June 6, 2013.
From the Division of Biomedical Sciences (H.X., T.Y.L., Z.C., T.M., T.-p.S., L.W., B.G., W.S., D.A.J., J.Y.-J.S.) and Center for Disease Vector Research and Department of Entomology (G.C., J.H.F.P.), University of California, Riverside; Institute of Vascular Medicine of Peking University Third Hospital, Beijing, China (H.X.); Department of Medicine, University of California, San Diego, La Jolla (M.L., Z.C., J.C., J.Y.-J.S.); Department of Biological Science and Technology, Institute of Bioinformatics and Systems Biology, National Chiao Tung University, HsinChu, Taiwan (W.-C.W., H.-D.H.); and Cardiovascular Research Center, Medical School, Xi’an Jiaotong University, Xi’an, China (X.L., J.Y.-J.S.).
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA. 113.002714/-/DC1.
Correspondence to John Y-J. Shyy, PhD, Department of Medicine/Division of Cardiology, University of California, San Diego, 9500 Gilman Dr, La Jolla, CA 92093. E-mail [email protected]
Sterol Regulatory Element Binding Protein 2 Activation
of NLRP3 Inflammasome in Endothelium Mediates
Hemodynamic-Induced Atherosclerosis Susceptibility
Han Xiao, MD, PhD; Min Lu, MD, PhD; Ting Yang Lin, MS; Zhen Chen, PhD;
Gang Chen, MD; Wei-Chi Wang, PhD; Traci Marin, MS; Tzu-pin Shentu, PhD;
Liang Wen, PhD; Brendan Gongol, MS; Wei Sun, MD; Xiao Liang, MD, PhD;
Ju Chen, PhD; Hsien-Da Huang, PhD; Joao H.F. Pedra, PhD; David A. Johnson, PhD;
John Y-J. Shyy, PhD
by guest on April 24, 2014 http://circ.ahajournals.org/
Xiao et al Shear-Induced Inflammasome and Atherosclerosis 633 The connection between hemodynamic-induced endothelial
dysfunction and inflammatory and oxidative stresses is poorly understood. However, aberrant lipid metabolism, unbalanced redox states, and innate immunity in phagocytes are linked through NLRP3 inflammasome with subsequent cleavage and activation of interleukin (IL)-1 family proteins.8 Moreover,
cho-lesterol crystals activate the NLRP3 inflammasome and increase the secretion of mature IL-1β in monocyte/macrophages.9,10 LDL
receptor–deficient mice receiving bone marrow–derived cells lacking NLRP3, ASC, or IL-1α/β were resistant to the develop-ment of diet-induced atherosclerosis.9 Alone, systemic activation
of NLRP3 inflammasome in macrophages does not explain the preferential localization of atherosclerosis in the arterial tree.
Disturbed flow can activate NOX and induce reactive oxy-gen species (ROS).11 This raises the possibility that
inflam-masome is involved in oxidative stress in ECs. Additionally, a recent report of minute cholesterol crystals appearing early in atherosclerotic lesions9 suggests a linkage between
hemo-dynamic stimuli, SREBP2 activation, and NLRP3 inflamma-some in ECs, all linked to atherosclerosis. Using in vitro and in vivo approaches, we report here that the atheroprone flow– induced endothelial inflammation and oxidative stress are mediated through SREBP2-elicited NLRP3 inflammasome. Our findings reinforce a primary role of EC innate immunity in the origin of atherosclerosis.
Methods
Antibodies and Reagents
Anti-SREBP2 antibody was from BD Transduction Laboratory and Abcam; anti–IL-1β, anti-caspase-1, and anti–α-tubulin, horseradish peroxide–conjugated anti-rabbit and anti-mouse antibodies were from Cell Signaling Technology; ABCG1, NLRP3, NOX2 anti-bodies were from Abcam; anti-ABCA1 antibody was from Millipore; and anti-ASC antibody was from Enzo Life Science. The caspase-1 inhibitor Z-YVAD-FMK was from Biovision. 25-Hydroxycholesterol (25-HC) and methyl-β-cyclodextrin were from Sigma.
Cell Culture
Human umbilical vein ECs (HUVECs) were cultured in medium M199 (Gibco) supplemented with 15% FBS (Omega), 3 ng/mL β-EC growth factor, 4 U/mL heparin, and 100 U/mL penicillin-streptomy-cin. Total cholesterol was measured with the Infinity Total Cholesterol Kit (Thermo Scientific). Caspase-1 activity was measured with the use of the Caspase-1/ICE Colorimetric Assay Kit (R&D Systems). Primary mouse lung ECs were isolated as described.12
Shear Stress Experiments
A circulating flow system was used to impose shear stress on conflu-ent monolayers of cells seeded on glass slides as described.13 A
recip-rocating syringe pump connected to the circulating system introduced a sinusoidal (1 Hz) component onto the shear stress. The atheropro-tective pulsatile shear flow (PS) or atheroprone oscillatory shear flow (OS) generated shear stresses of 12±4 or 1±4 dynes/cm2, respectively.
The flow system was enclosed in a chamber held at 37°C and venti-lated with 95% humidified air plus 5% CO2.
siRNA Knockdown
HUVECs at 50% to 70% confluence were transfected with SREBP2 siRNA, NLRP3 siRNA, ASC siRNA, NOX2 siRNA, or control siRNA at 20 nmol/L with Lipofectamine 2000 RNAi Max (Invitrogen). Experiments were performed with these cells at 48 hours after transfection.
Adenovirus Construction and Infection
Recombinant adenovirus encoding the mature form of SREBP2, that is, Ad-HA-SREBP2(N), was created, amplified, and titrated as reported previously.14 For adenovirus infection, the virus mixture
was added to 70% confluent cultured HUVECs and incubated for 12 hours. Adenovirus (Ad)-null was an infection control. The infected cells were then incubated in fresh growth medium for 24 hours before RNA or protein extraction.
Binding Site Prediction
The potential SREBP2 binding sites on selected human and mouse genes were predicted by use of the position weight matrix algorithm from TRANSFAC15 to scan the promoter regions of the genes. The
promoter regions were defined as −3000 to 500 from the transcrip-tional start site of the gene.
Chromatin Immunoprecipitation Assay
Chromatin immunoprecipitation assays were performed with stan-dard protocols16 using antibodies for SREBP2 (BD Transduction
Laboratory) and mouse IgG (Cell Signaling).
EC-SREBP2(N) Transgenic Mice
Animal experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of California, Riverside. The creation of the EC-SREBP2(N)-Tg mouse model is described in the online-only Data Supplement. Littermates car-rying ApoE−/− or EC-SREBP2(N)+/+ApoE−/− genotypes were
gen-erated by crossing EC-SREBP2(N)-Tg mice and ApoE−/− mice.
All mice were housed in colony cages with a 14-hour light/10-hour dark cycle and fed Rodent Diet 5001 (PMI Nutrition International) ad libitum unless otherwise indicated. Eight-week-old male EC-SREBP2(N)+/+ApoE−/− mice and their male
EC-SREBP2(N)−/−ApoE−/− littermates were fed a fat,
high-cholesterol diet containing 15% fat, 1.25% high-cholesterol, and 0.5% sodium cholate (Harlan Teklad) ad libitum. Eight weeks after the diet treatment, all mice were euthanized. In addition, 24-week-old male EC-SREBP2(N)+/+ApoE−/− mice and their male EC-SREBP2
(N)−/−ApoE−/− littermates were fed normal chow. Mouse aortas were
isolated to assess the extent and distribution of lesions by Oil Red O staining.17 Lesion area was measured with Image Pro Plus 6.0
(Media Cybernetics) and expressed as a percentage of the total area of aorta. Plasma levels of total cholesterol, high-density lipoprotein cholesterol, and triglycerides were determined with assay kits from Wako Pure Chemicals (Tokyo, Japan).
Statistical Analysis
Data are expressed as mean±SEM (n=3 unless otherwise noted). In parametric data, the Student t test or ANOVA was used to analyze the differences among groups if data were determined to be normally distributed. For nonparametric data, the Mann-Whitney U test with the exact method was used to analyze differences between 2 groups. Values of P<0.05 were considered statistically significant.
Results
Coinduction of SREBP2 and miR-33 in ECs by OS Initially, we examined the effects of PS and OS on the expression of SREBP2 in ECs. Imposition of OS, but not PS, activated SREBP2, as evidenced by the increased level of the mature form of SREBP2, namely SREBP2(N), and SREBP2 mRNA (Figure 1A and 1B). Given that miR-33 is intronic with SREBP-2, OS also elevated the level of miR-33 (Figure 1B). Additionally, the expression of SREBP2-targeted genes, that is, HMG-CoA reductase, HMG-CoA synthase, squalene synthase, and LDL receptor, was higher with OS
by guest on April 24, 2014 http://circ.ahajournals.org/
than PS (Figure 1C). The miR-33–targeted ABCA1, but not ABCG1, was downregulated at both the transcriptional and translational levels (Figure 1D and 1E). Thus, OS increased the expression of genes involved in cholesterol synthesis and uptake while decreasing ABCA1, which is involved in choles-terol efflux. In line with these changes, the cholescholes-terol content of HUVECs was greater with OS than PS (Figure 1F). One possible mechanism for OS induction of SREBP2 is that the action of OS is secondary to sterol depletion. Consequently, we examined whether sterol replenishment with 25-HC blocks the effect of OS on SREBP2 activation. Although 25-HC and methyl-β-cyclodextrin reduced and increased SREBP acti-vation in HUVECs, respectively, incubation of 25-HC had little effect on OS-induced SREBP2 expression (Figure 1G). Interestingly, overexpression of the mature form of SREBP2, that is, SREBP2(N) or premiR33, increased the cholesterol content (Figure IA and IB in the online-only Data Supplement. However, siRNA knockdown of SREBP2, but not miR-33, abolished OS-induced cholesterol accumulation (Figure IC and ID in the online-only Data Supplement). Consequently, we focused on the role of SREBP2 in OS-disturbed endothe-lial functions.
OS Induces NLRP3 Inflammasome in ECs via SREBP2
The atheroprone nature of OS is due largely to its imposition of inflammatory and oxidative stresses on ECs.18 Because
SREBP1a induces caspase-1–activated NLRP3 inflamma-some in macrophages, resulting in cleavage and secretion of IL-1 family cytokines,19 we investigated whether OS induces
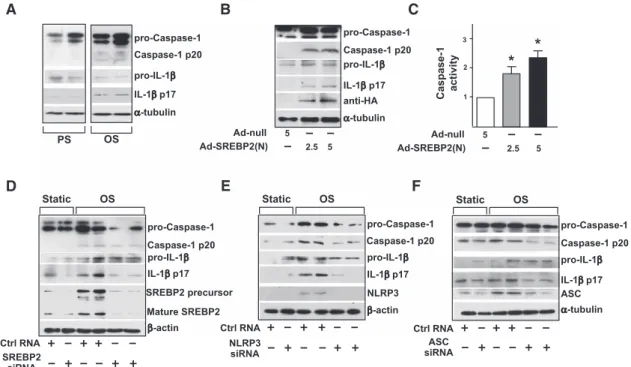
inflammasome in ECs. The levels of cleaved caspase-1 and IL-1β were greater in ECs with OS than PS (Figure 2A). Because OS induced SREBP2 maturation, we explored whether SREBP2(N) overexpression mimics OS to induce inflamma-some in ECs. As expected, increased cleavage of caspase-1 and IL-1β was found in ECs infected with Ad-SREBP2(N) encoding SREBP2(N) (Figure 2B). Consistent with this find-ing, ectopic expression of SREBP2(N) increased caspase-1 activity in ECs (Figure 2C). With siRNA knockdown of endogenous SREBP2, OS-induced cleavage of caspase-1 or IL-1β was reduced (Figure 2D). Similar results were found when the inflammasome components NLRP3 and ASC were knocked down (Figure 2E and 2F). Together, these results suggest that SREBP2 mediates OS-induced NLRP3 inflam-masome in ECs. 25-HC 25-HC C D E F
SREBP2 mRNA miR-33
PS OS 1 2 3 Relative Expression Relative Expression Squalene Synthase mRNA PS OS 0.5 1 1.5 2 Relative Expression PS OS LDLR mRNA Relative Expression0.5 1 1.5 2 HMGCoA Reductase mRNA 0.5 1.5 HMGCoA Synthase mRNA Relative Expression ABCA1 mRNA PS OS 0.5 1 ABCG1 mRNA PS OS 0.5 1 Relative
Expression Relative Expression
PS OS 1 2 PS OS 1 PS OS 1 2 3 B A G β-actin Mature Precursor ABCA1 ABCG1 β-actin PS OS β-actin SREBP2 Relative Expression Precursor Mature β-actin PS OS SREBP2 MβCD 2 4 6 SREBP2 precursor/a ctin (Relativ e Le ve l) PS OS 2 4 6 8 SREBP2 mature form/actin (Relativ e Le ve l) PS OS 0.5 1 ABCA1 0.5 1 ABCG1 PS OS Relative Level 130 70 55 MW Cholesterol (µg/mg) OS
*
*
*
*
*
*
*
*
*
*
PS OS 50 150*
250Figure 1. Oscillatory shear flow (OS)
induces sterol regulatory element binding protein 2 (SREBP2) and miR-33 in human umbilical vein endothelial cells (HUVECs). HUVECs were exposed to a pulsatile shear flow (PS; 12±4 dynes/cm2)
or OS (1±4 dynes/cm2) for 14 hours.
A, Representative immunoblot for
precursor SREBP2 and mature form of SREBP2. B, Analysis of levels of SREBP2
mRNA and miR-33 by quantitative real-time–polymerase chain reaction. C, The
mRNA levels of HMG-CoA synthase, HMG-CoA reductase, squalene synthase, and low-density lipoprotein receptor (LDLR). D, mRNA levels of ATP-binding
cassette transporter (ABC) A1 and ABCG1. E, Representative immunoblot
for ABCA1 and ABCG1. F, Total cellular
cholesterol level (n=8). G, Representative
immunoblot for precursor SREBP2 and mature form of SREBP2 in HUVECs treated with methyl-β-cyclodextrin (MβCD), 25-hydroxycholesterol (25-HC; 1 µg/mL), or OS plus 25-HC (1 µg/mL). Data are mean±SEM from at least 3 independent experiments. The Student
t test or Mann-Whitney U test with the
exact method was used. MW indicates molecular weight. *P<0.05.
Xiao et al Shear-Induced Inflammasome and Atherosclerosis 635
SREBP2 Upregulates NOX2 With Increased ROS Production
We then investigated the underlying mechanism by which SREBP2 regulates the OS-induced inflammasome in ECs. The ROS level is increased by NLRP3 activators such as asbes-tos and silica,20 and increased ROS are secondary messengers
essential for inducing NLRP3 inflammasome.20,21 Given that OS
is known to increase ROS production,22 we examined SREBP2
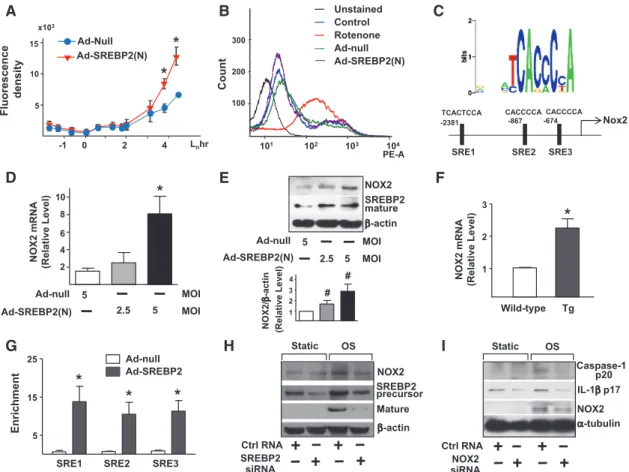
augmentation in relation to ROS production. The intracellular level of ROS increased in ECs infected with Ad-SREBP2(N) encoding the mature form of SREBP2 (Figure 3A). To assess the source of the increased level of ROS in ECs overexpress-ing SREBP2(N), we monitored ROS-generatoverexpress-ing mitochondria using MitoSOX, a selective mitochondrial superoxide indicator. Rotenone, the complex I inhibitor, greatly increased mitochon-drial ROS production, but Ad-SREBP2(N) overexpression was without effect (Figure 3B). Therefore, the increased ROS pro-duction caused by SREBP2 activation probably did not origi-nate from mitochondria. Bioinformatics predicted the presence of 3 SREs in the promoter region of the human NOX2 gene (−2381/−2367, −867/−861, and −674/668 bp; Figure 3C). Consistent with this prediction, the levels of NOX2 mRNA and protein increased in Ad-SREBP2(N)–infected HUVECs and in ECs isolated from transgenic mice overexpressing SREBP2(N) in endothelium [EC-SREBP2(N)-Tg] (Figure 3D–3F). Of note, the levels of other NADPH oxidase subunits (except
NOX1) were not significantly increased in these cells (Figure II in the online-only Data Supplement). Chromatin immuno-precipitation assay demonstrated an increase in the binding of SREBP2 to the 3 SREs containing the consensus sequence of CACC(T)CCA (Figure 3G). To investigate whether SREBP2 is required for OS-induced NOX2, we knocked down SREBP2 and found that the OS-induced NOX2 was partially suppressed (Figure 3H). Furthermore, NOX2 knockdown inhibited the OS-induced caspase-1 and IL-1β cleavage (Figure 3I). A simi-lar inhibitory effect was found in NOX2−/− mouse embryonic
fibroblasts (Figure III in the online-only Data Supplement). These results suggest that the SREBP2-transactivated NOX2 is required for OS-induced inflammasome in ECs.
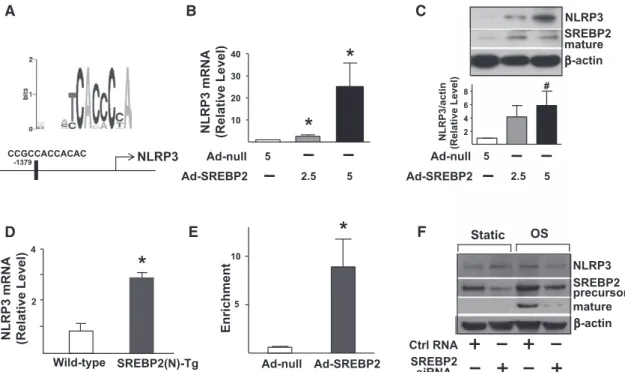
SREBP2 Upregulates NLRP3 Expression
Bioinformatics also predicted the presence of an SRE located at −1379/−1368 bp in the promoter region of the NLRP3 gene (Figure 4A). As expected, NLRP3 expression was increased in Ad-SREBP2(N)–infected HUVECs (Figure 4B and 4C) and ECs isolated from EC-SREBP2(N)-Tg mice (Figure 4D). Chromatin immunoprecipitation assays demonstrated an increase in the binding of SREBP2 to SRE containing the con-sensus sequence CCGCCACCACAC (Figure 4E). To inves-tigate whether SREBP2 is required for OS-induced NLRP3 expression, we knocked down SREBP2. Accordingly, the level of NLRP3 responding to OS was decreased (Figure 4F).
pro-Caspase-1 Caspase-1 p20 Caspase-1 p20 pro-Caspase-1 pro-IL-1β Caspase-1 p20 pro-Caspase-1 PS OS pro-IL-1β α-tubulin IL-1β p17 A B C D IL-1β p17 pro-IL-1β SREBP2 precursor Static OS Ctrl RNA β-actin Mature SREBP2 SREBP2 siRNA Caspase-1 p20 NLRP3 Static OS Ctrl RNA β-actin NLRP3 siRNA pro-IL-1β ASC Static OS Ctrl RNA α-tubulin ASC siRNA E F pro-Caspase-1 α-tubulin pro-IL-1β IL-1β p17 Caspase-1 p20 anti-HA pro-Caspase-1 Ad-null Ad-SREBP2(N) 5 2.5 5 Caspase-1 activity 1 2 3 * * Ad-null Ad-SREBP2(N) 5 2.5 5 IL-1β p17 IL-1β p17
Figure 2. Oscillatory shear flow (OS) induces NLRP3 inflammasomes in endothelial cells (ECs) via sterol regulatory element binding
protein 2 (SREBP2). A, Representative immunoblot for procaspase-1, caspase-1 p20, pro–interleukin (IL)-1β, and IL-1β p17 in human
umbilical vein ECs (HUVECs) exposed to pulsatile shear flow (PS) or OS for 14 hours and (B) in HUVECs treated with adenovirus-null
(Ad-null; 5 multiplicities of infection [MOI]) or adenovirus-SREBP2(N) tagged with HA [Ad-SREBP2(N), 2.5 or 5 MOI]. C, Caspase-1
activity of extracts from HUVECs treated with Ad-null 5 MOI or Ad-SREBP2(N) 2.5 or 5 MOI. Data are mean±SEM normalized to the Ad-null group for each independent experiment. The Mann-Whitney U test with the exact method was used. *P<0.05 vs Ad-null.
D, Representative immunoblot of procaspase-1, caspase-1 p20, pro-IL-1β, IL-1β p17, SREBP2 precursor, and mature form of SREBP2
in HUVECs transfected with 20 nmol/L control RNA or SREBP2 siRNA for 48 hours and then exposed to static conditions or OS for 14 hours. E, Representative immunoblot for procaspase-1, caspase-1 p20, pro-IL-1β, IL-1β p17, and NLPR3 in HUVECs transfected
with 20 nmol/L control RNA or NLRP3 siRNA for 48 hours and then exposed to static conditions or OS for 14 hours. F, Representative
immunoblot of procaspase-1, caspase-1 p20, pro-IL-1β, IL-1β p17, and ASC in HUVECs transfected with 20 nmol/L control RNA or ASC siRNA for 48 hours and then exposed to static conditions or OS for 14 hours.
by guest on April 24, 2014 http://circ.ahajournals.org/
Therefore, the OS-activated SREBP2 transactivates NLRP3 in ECs.
SREBP2(N) Overexpression Enhances Endothelial Inflammation
IL-1β stimulates the expression of chemokines, for example, monocyte chemoattractant protein 1, and adhesion molecules, for example, vascular cell adhesion molecule 1 and E-selectin, in ECs, which enhances leukocyte-endothelium interactions. In agreement with increased IL-1β secretion via SREBP2-augmented inflammasome, SREBP2(N) overexpression increased the mRNA levels of monocyte chemoattractant protein 1, vascular cell adhesion molecule 1, and E-selectin (Figure 5A). Increased expression of monocyte chemoat-tractant protein 1 and adhesion molecules was also found in ECs isolated from EC-SREBP2(N)-Tg mice compared with wild-type littermates (Figure 5B). The SREBP2(N)-induced inflammation in ECs was associated with increased monocyte
association, which was attenuated with caspase-1 inhibitor or NOX2 or NLPR3 siRNA knockdown (Figure 5C and 5D). Thus, NLRP3 inflammasome induced by OS via SREBP2 is functionally linked to endothelial proinflammatory responses. SREBP2-NOX2-Inflammasome Activation in Atheroprone Regions of the Mouse Aorta
We next investigated whether the differential regulation of SREBP2 by PS and OS in ECs cultured in the flow channel also translated to a functional or functionally disturbed endo-thelium in atheroprotective versus atheroprone regions in the mouse arterial tree. As shown in Figure 6A and 6B, the activation of NLRP3 inflammasome was evident in the aortic arch, where the endothelium is exposed predominantly to dis-turbed flow.23 As expected, the levels of the cleaved caspase-1
and IL-1β, that is, IL-1β p17, were higher in the aortic arch than in the thoracic aorta (Figure 6B). Consistent with this observation, the expression of IL-1β–regulated genes, that is,
NOX2 D Nox2 Enrichment H Fluorescence density G
SRE1 SRE2 SRE3
Ad-null Ad-SREBP2 Count 5 10 15
*
x103 Lnhr -1 0 2 4 Ad-SREBP2(N) Ad-Null SREBP2 precursor β-actin Mature Static OS C TCACTCCA CACCCCA -867 -674CACCCCA -2381SRE1 SRE2 SRE3
Ctrl RNA SREBP2 siRNA A B E F Tg Wild-type 5 NOX2 β-actin 2.5 5 SREBP2 mature MOI MOI Ad-null Ad-SREBP2(N) 1 2 3
*
*
*
*
NOX2 mRNA (Relative Level) Ad-null Ad-SREBP2(N) 5 2.5 5 MOI MOI
*
I 300 200 101 102 103 104 PE-A 100 Rotenone Ad-null Ad-SREBP2(N) Control Unstained 1 2 3 4 NOX2/ β-a ct in (Relati ve Le ve l) 2 4 6 8 10 5 15 25 Caspase-1 p20 IL-1β p17 α-tubulin Static OS NOX2 Ctrl RNA NOX2 siRNA NOX2 mRNA (Relative Level)*
# #
Figure 3. Sterol regulatory element binding protein 2 (SREBP2) upregulates NADPH oxidase 2 (NOX2) with an attendant increase in
reactive oxygen species (ROS) in endothelial cells (ECs). A, ROS production monitored by measuring H2DCFDA fluorescence in human
umbilical vein ECs (HUVECs) infected with adenovirus-null (Ad-null) or Ad-SREBP2(N) (5 multiplicities of infection [MOI]). B, Cytometric
analysis of HUVECs infected with Ad-null or Ad-SREBP2(N) (5 MOI) for 48 hours or treated with rotenone (40 μmol/L) for 3 hours and then stained with MitoSOX for 30 minutes. C, Depiction of the 3 putative SRE binding sites, at −2381/−2367 (SRE1), −867/−861 (SRE2), and
−674/668 (SRE3) bp upstream of the transcription initiation site in the human NOX2 promoter. NOX2 mRNA levels (D) and representative
immunoblot (and quantification) of NOX2 and the cleaved SREBP2 (E) in HUVECs treated with Ad-null (5 MOI) or Ad-SREBP2(N) (2.5 or
5 MOI). F, NOX2 mRNA levels in lung ECs from wild-type or EC-SREBP2(N)-Tg mice (n=8). G, Chromatin immunoprecipitation analysis
with antibodies against SREBP2 or IgG, soluble chromatin (≈500 bp in length) from HUVECs infected with Ad-null or Ad-SREBP2(N), and primers targeting the region spanning the 3 SRE binding sites in the NOX2 promoter. H, Representative immunoblot of NOX2, SREBP2
precursor, and mature form of SREBP2 in HUVECs transfected with 20 nmol/L control RNA or SREBP2 siRNA for 48 hours and then exposed to static or oscillatory shear flow (OS) for 14 hours. I, Representative immunoblot of caspase-1 p20, interleukin (IL)-1β p17, and
NOX2 in HUVECs transfected with 20 nmol/L control RNA or NOX2 siRNA for 48 hours and then exposed to static or OS for 14 hours. At least 3 independent experiments were performed; results are mean±SEM. The Mann-Whitney U test with the exact method was used. *P<0.05 vs Ad-null or wild-type; #P=0.05.
Xiao et al Shear-Induced Inflammasome and Atherosclerosis 637
monocyte chemoattractant protein 1, vascular cell adhesion molecule 1, intercellular adhesion molecule 1, and E-selectin, was increased in the aortic arch (Figure 6A). In addition, we separated intima (endothelium) from media and adventitia of thoracic aorta and aortic arch of C57BL/6 mice. As illustrated in Figure 6C, the expression levels of SREBP2, NOX2, and NLRP3 in the isolated endothelium in aortic arch were higher than those in thoracic aorta. Importantly, no differences were found between thoracic aorta and aortic arch in tissues con-taining media and adventitia. Higher levels of NLRP3, NOX2, and SREBP2 in the aortic arch were also verified by in situ hybridization (Figure IV in the online-only Data Supplement). Thus, the NLPR3 inflammasome is activated in the endothe-lium of atheroprone region of the arterial tree in vivo.
EC-specific SREBP2(N) overexpression mimicking OS induction of SREBP2 should render the thoracic aorta of EC-SREBP2(N)-Tg mice atheroprone. As expected, activation of NLRP3 inflammasome and induction of chemoattractants and adhesion molecules were seen in these arterial segments compared with corresponding areas in wild-type littermates (Figure 6D). SREBP2(N) overexpression also caused a functionally disturbed endothelium, as evidenced by impaired vasodilation responding to flow (Figure V in the online-only Data Supplement).
SREBP2(N) Overexpression in ECs Predisposes Atherosclerosis
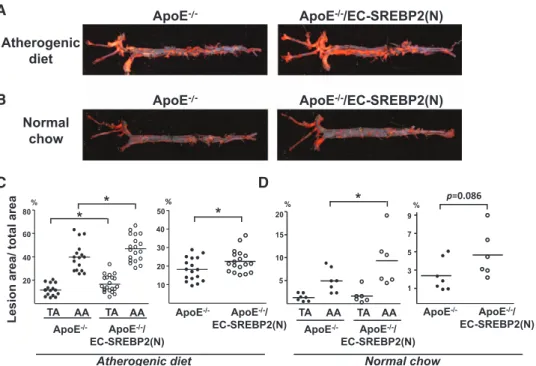
We introduced the ApoE-null background into EC- SREBP2(N)-Tg mice to investigate whether EC-specific
overexpression of SREBP2(N) leads to atherogenesis in ath-eroprotective regions in ApoE−/−/EC-SREBP2(N) mice. After
8 weeks of an atherogenic diet, the levels of total cholesterol and LDL were comparable between ApoE−/−/EC-SREBP2(N)
mice and their ApoE−/− littermates (Table 1). However, the
mean lesion area in thoracic aortas was ≈1.5-fold larger for ApoE−/−/EC-SREBP2(N)-Tg than ApoE−/− mice (17.4±1.7%
versus 11.5±1.3%; Figure 7A and 7C). The mean lesion area in the aortic arch was also larger for ApoE−/−/EC-SREBP2(N)
than ApoE−/− mice (47.7±2.5% versus 39.7±3.1%). The
order of lesion size was as follows: aortic arch of ApoE−/−/
EC-SREBP2(N) mice>aortic arch of ApoE−/− mice>thoracic
aorta of ApoE−/−/EC-SREBP2(N) mice>thoracic aorta of
ApoE−/− mice. The total lesion area as a sum of aortic arch,
thoracic aorta, plus abdominal aorta remained greater for ApoE−/−/EC-SREBP2(N) mice than control littermates
(22.5±1.4% versus 18.2±1.5%). These results demonstrate that local flow patterns synergize with other atherogenic fac-tors, for example, hyperlipidemia, in the formation of athero-sclerotic lesions. We also compared the lesion development in the 2 groups of animals fed normal chow for 24 weeks. As expected, the serum levels of total cholesterol and LDL were significantly lower with normal chow than with an athero-genic diet (Table 1). Compared with the ApoE−/− littermates,
ApoE−/−/EC-SREBP2(N) mice showed more lesions in aortic
arches, particularly the inner curvature of the arch and the ori-fices of the arch vessels (Figure 7B and 7D). Previous stud-ies by others showed that these areas have elevated nuclear
CCGCCACCACAC -1379 NLRP3 SREBP2 mature β-actin Ad-null Ad-SREBP2 5 2.5 5 NLRP3 C A D E B F Ad-null Ad-SREBP2 5 2.5 5 NLRP3 mRNA (Relative Level) Wild-type SREBP2(N)-Tg 2 4 Ad-null Ad-SREBP2 Enrichment Ctrl RNA β-actin mature NLRP3 SREBP2 precursor SREBP2 siRNA Static OS
*
*
*
10 20 30 40 2 4 6 8 NLRP3/actin (Relative Level ) 5 10 NLRP3 mRNA (Relative Level)*
#Figure 4. Sterol regulatory element binding protein 2 (SREBP2) overexpression upregulates NLRP3 inflammasome. A, Depiction of the
putative SRE binding site located at −1379/−1368 bp upstream of the transcription initiation site in the human NLRP3 promoter. NLRP3 mRNA levels (B) and representative immunoblot (and quantification) of NLRP3 and the mature form of SREBP2 (C) in human umbilical
vein endothelial cells (HUVECs) treated with adenovirus-null (Ad-null; 5 multiplicities of infection [MOI]) or Ad-SREBP2(N) (2.5 or 5 MOI).
D, NLRP3 mRNA levels in lung ECs from wild-type or EC-SREBP2(N)-Tg mice (n=8). E, Chromatin immunoprecipitation analysis with
antibodies against SREBP2 or IgG, soluble chromatin from HUVECs infected with Ad-null or Ad-SREBP2(N), and primers targeting the region spanning the SRE binding site in the NLRP3 promoter. F, Representative immunoblot of NLRP3, precursor SREBP2, and
mature form of SREBP2 in HUVECs transfected with 20 nmol/L control RNA or SREBP2 siRNA for 48 hours and then exposed to static conditions or oscillatory shear flow (OS) for 14 hours. Bar graphs represent mean±SEM from at least 3 independent experiments. The Mann-Whitney U test with the exact method was used. *P<0.05 vs Ad-null or wild-type; #P=0.05 vs Ad-null.
by guest on April 24, 2014 http://circ.ahajournals.org/
factor-κB and vascular cell adhesion molecule 1 activa-tion.23,24 Of note, with normal chow, lesion areas were
mar-ginal in the thoracic aortas of both ApoE−/−/EC-SREBP2(N)
and ApoE−/− mice, which reiterates the notion that
hyperlip-idemia synergizes with hemodynamic forces in the origin of atherosclerosis.
Discussion
The “response-to-injury” hypothesis states that endothe-lial dysfunction precedes the development of atheroscle-rosis.25 Much evidence suggests that atheroprone flow
patterns in the conduit arteries are a determining factor of atherogenesis. Furthermore, the Pathological Determinants of Atherosclerosis in Youth (PDAY) study provides unequivo-cal evidence that cardiovascular risk factors (eg, hyperlipid-emia, smoking, and hypertension) exacerbate atherosclerosis in atheroprone areas in the human arterial tree.26,27 Here, we
report that disturbed flow applied to ECs induced NLRP3 inflammasome via SREBP2 activation. Such SREBP2 activa-tion of NLRP3 inflammasome is sufficient for funcactiva-tionally disturbed endothelium leading to atherogenesis as supported by mouse models harboring the EC-SREBP2(N) transgene. Differential development of atherosclerotic lesions in the
aortic arch compared with thoracic aorta in mice with or with-out EC-specific expression of SREBP2(N) and in the pres-ence or abspres-ence of an atherogenic diet (Figure 7) provides a molecular basis for the hemodynamic-induced atherosclerosis susceptibility seen in the human arterial tree. We reasoned that endothelial expression of SREBP2(N) mimicking the effect of disturbed flow synergizes with hyperlipidemia (caused by an ApoE−/− background together with an atherogenic diet) to
accelerate atherosclerosis. The thesis is further supported by experiments using EC-SREBP2(N)-Tg or wild-type C57BL6 mice fed an atherogenic diet. Early atherosclerotic plaques developed in the aortic root of EC-SREBP2(N)-Tg mice but not control littermate mice (Figure VI in the online-only Data Supplement and Table 2). Thus, the translational relevance of this study is that the spatial localization and severity of ath-erosclerosis can depend on atheroprone flow coupled with cardiovascular risk factors via SREBP2(N)-induced NLRP3 inflammasome.
Clearly, IL-1β is a major atheroprone factor.28
Atherosclerosis was decreased in several rodent models lacking IL-1β or type I IL-1 receptor.29,30 In contrast, mice
deficient in IL-1 receptor antagonist show increased athero-sclerosis.31 Canakinumab, an anti-human IL-1β
monoclo-nal antibody, is currently being used in the Canakinumab B 1 2 * * 1 2 E-selectin Expression level MCP-1 Expression level VCAM-1 Expression level E-selectin Expression level MCP-1 Expression level 4 8 * 1 2 3
*
5 10 15 * 1 2 * VCAM-1 Expression level C Caspase-1 inhibitor Ad-null Ctrl Ad-null Ad-SREBP2(N) DFluorescence (AU) Ctrl Caspase-1
inhibitor 4 8 103 Ad-null Ad-SREBP2(N)
*
*
0.5 1 Fluorescence (AU) 106 1.5 CtrlRNA siRNANOX2 NLRP3siRNA
*
* *
Ctrl RNA NLRP3siRNA Ad-SREBP2(N) Ad-null Ad-SREBP2(N) NOX2 siRNA AFigure 5. Increased level of sterol
regulatory element binding protein 2 (SREBP2) causes endothelial cell (EC) NLRP3 inflammation.
A, Quantification of mRNA levels of
monocyte chemoattractant protein 1 (MCP-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin in human umbilical vein endothelial cells (HUVECs) treated with adenovirus-null (Ad-null; 5 multiplicities of infection [MOI]) or Ad-SREBP2(N) (2.5 or 5 MOI)
or (B) in lung ECs from wild-type or
EC-SREBP2(N)-Tg mice (n=8). C and D, HUVECs were infected with Ad-null
or Ad-HA-SREBP2(N) for 24 hours and then treated with or without caspase-1 inhibitor Z-YVAD-FMK (2 µmol/L) for 24 hours. In separate experiments, HUVECs were transfected with control RNA, NADPH oxidase 2 (NOX2) siRNA, or NLRP3 siRNA and then infected with Ad-null or Ad-HA-SREBP2(N) for 24 hours. The cells were incubated for 30 minutes with LeukoTracker-labeled THP1 cells. For quantification, parallel batches of treated cells were lysed and the fluorescence was measured. Data are mean±SEM from 3 independent experiments performed in triplicate. The Mann-Whitney U test with the exact method was used. *P<0.05 for comparisons with Ad-null or wild-type ECs.
Xiao et al Shear-Induced Inflammasome and Atherosclerosis 639
Anti-inflammatory Thrombosis Outcomes Study (CANTOS) to assess the efficacy of anti-IL-1β in reducing cardiovascular events.32 The antiatherosclerosis effect resulting from IL-1β
antagonism should involve the inhibition of NLRP3 inflam-masome in the endothelium and should be experimentally verified. Undoubtedly, NLRP3 inflammasome in monocyte/ macrophage is important for atherosclerosis, as suggested by
transplantation experiments with bone marrow deficient in NLRP3, ASC, or IL-1α/β.9 However, this
macrophage-asso-ciated mechanism would not contribute to functionally dis-turbed endothelium and ensuing atherosclerosis in our mouse models because the SREBP2(N) transgene was expressed only in ECs, not in bone marrow–derived macrophages (Figure VII in the online-only Data Supplement).
*
Relative expression (fold of control) TA AA 1 2 3 TA AA 2 4 6 8 TA AA 1 3 5 TA AA TA AA 2 4 6 8*
1 2 3 4*
SREBP2 NOX2 NLRP3 MCP-1 VCAM-1 ICAM-1 E-selectin
*
TA AA 2 4 6 TA AA 1 2 3 4*
*
*
*
A IL-1β p17 α-tubulin pro-IL-1β TA AA pro-Caspase-1 Caspase-1 p20 Pro-IL-1β IL-1β p17 TA AA TA AA 0.5 1.5 1*
5 10 15 20 pro-Caspase-1 Caspase-1 p20 TA AA 0.5 1 1.5 TA AA*
5 10 B*
2 4 6*
2 4 6*
1 3 5 1 2 3 4 1 2 3 4*
2 4 6 4 8*
WT Tg WT Tg WT Tg WT Tg WT Tg WT Tg WT TgSREBP2 NOX2 NLRP3 MCP-1 VCAM-1 ICAM-1 E-selectin
Relative expression (fold of WT )
*
C DIntima Media & Adventitia 0.5 1.0 1.5 TA AA SREBP2
Relative Expression (fold of control)
NOX2
Intima Media & Adventitia 1
2
3 TAAA
NLRP3
Intima Media & Adventitia 1 2 3 4
*
TAAA*
*
Figure 6. Sterol regulatory element
binding protein 2 (SREBP2)–NADPH oxidase 2 (NOX2)–inflammasome activation in atheroprone regions of mouse aortas. A, Quantification of
mRNA levels of SREBP2, NOX2, NLRP3, monocyte chemoattractant protein 1 (MCP-1), vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and E-selectin in the thoracic aorta (TA) and aortic arch (AA) of C57BL/6 mice (n=7). B, Representative immunoblot
of procaspase-1, caspase-1 p20, pro– interleukin (IL)-1β, and IL-1β p17 in TA and AA from C57BL/6 mice. C, The
levels of SREBP2, NOX2, and NLRP3 mRNA in aortic intima or media and adventitia. Data are mean±SEM of the relative mRNA normalized to that of β-actin (n=18). D, Quantification of mRNA levels of SREBP2, NOX2, NLRP3, MCP-1, VCAM-1, ICAM-1, and E-selectin in TA from wild-type (n=7) and EC-SREBP2(N)-Tg mice (n=7). The Mann-Whitney U test with the exact method was used. *P<0.05 for comparisons with TA or WT.
Table 1. Serum Lipid Profile of ApoE−/−/EC-SREBP2(N) and Their ApoE−/− Littermates Fed a Normal Chow or an Atherogenic Diet
Serum Lipids
Normal Chow, mg/dL* Atherogenic Diet, mg/dL†
ApoE−/− (n=7) ApoE−/−/EC-SREBP2(N) (n=6) ApoE−/− (n=17) ApoE−/−/EC-SREBP2(N) (n=18)
Total cholesterol 1284±272‡ 1224±334 1806±298 1967±576
Triglycerides 169±49 197±112 111±57 125±89
LDL 490±54 478±83 1055±168 1035±235
VLDL 30±14 39±22 21±12 29±22
HDL 764±253 707±374 730±177 903±365
EC indicates endothelial cell; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SREBP2, sterol regulatory element binding protein 2; and VLDL, very-low-density lipoprotein.
*Normal chow group was 24-week-old mice fed a normal chow.
†Atherogenic diet group was 8-week-old mice fed an atherogenic diet for 8 weeks. ‡All values are expressed as mean±SD averaged from the number of animals as indicated.
by guest on April 24, 2014 http://circ.ahajournals.org/
Reconciling published literature with the present report, we propose that the disturbed flow–increased NLRP3 inflam-masome in ECs and consequent production and secretion of IL-1β create a focal gradient of inflammatory cytokines and chemoattractants. This flow-elicited proinflammatory milieu recruits sentinel cells (ie, monocytes) and facilitates the retention and differentiation of monocytes in the sub-endothelial space. Indeed, atherosclerosis was found to be substantially lower in ApoE−/−/IL-1β−/− than ApoE−/−/IL-1β+/+
thoracic aortas.30
In macrophages, the SREBP2-coinduced miR-33a tar-gets ABCA1, which impairs reverse cholesterol transport.5,6
An antigomir against miR-33 decreases atherosclerosis in LDL receptor–null mice, with a concomitant increase in the level of high-density lipoprotein.33 Inhibition of miR-33 also
increases the level of high-density lipoprotein and lowers
that of very LDL triglycerides in nonhuman primates.34 In
line with SREBP2 induction, OS-induced miR-33a targets ABCA1 in ECs (Figure 1). Given that cholesterol crystals are an inflammasome inducer,9 disturbed flow patterns should
increase the cholesterol level, which arguably synergizes with SREBP2 to activate NLPR3 inflammasome. Conversely, the atheroprotective flow patterns should induce liver X receptors and hence upregulate ABCA1 to facilitate reverse cholesterol transport.35 Civelek et al36 reported no site-specific
differ-ences in endothelial ABCA1 expression between susceptible and protected sites of swine arteries. However, our published work showed that ABCA1 level is lower in the mouse aor-tic arch compared with thoracic aorta.35 The possible reason
for the discrepancy is the different species and diets used in the 2 studies. Given that hypercholesterolemia significantly increased ABCA1 expression in swine endothelium, it might be difficult to detect a site-specific difference in endothelial ABCA1 expression.
The intracellular and extracellular levels of sterols intri-cately regulate SREBP2, which in turn modulates cellular cholesterol homeostasis.4 Significantly, excessive amounts of
25-HC do not prevent OS induction of SREBP2 (Figure 1G), suggesting that the mechanotransduction mechanism over-rides that of the cholesterol-sensing system, leading to sus-tained SREBP2 activation. Thus, the disturbed flow–activated SREBP2 appears to disrupt cholesterol homeostasis in its activation of inflammasome. This argument can explain the synergism between atheroprone flow and hyperlipidemia in inducing atherosclerosis.
The endoplasmic reticulum stress/unfolded protein response is activated in ECs by atheroprone flow in vitro37 and is upregulated
in endothelium of swine atheroprone sites in vivo.38 We have
ApoE-/- ApoE-/-/EC-SREBP2(N)
Atherogenic diet Normal chow 10 30 50 * ApoE-/- ApoE-/-/ EC-SREBP2(N) %
Lesion area/ total are
a 20 40 TA AA 20 40 60 80% * * TA AA ApoE-/- ApoE-/-/ EC-SREBP2(N) 1 3 5 7 9 p=0.086 % ApoE-/- ApoE-/-/ EC-SREBP2(N) 5 10 15 20 TA AA TA AA * % ApoE-/- ApoE-/-/ EC-SREBP2(N) w o h c l a m r o N t e i d c i n e g o r e h t A A B D C
ApoE-/- ApoE-/-/EC-SREBP2(N)
Figure 7. Overexpression of sterol regulatory element binding protein 2 (SREBP2) in endothelial cells (ECs) enhances atherosclerosis.
A, Representative Oil Red O staining of aortas from (A) 8-week-old ApoE−/− (n=17) and ApoE−/−/SREBP2(N) mice (n=18) fed a high-fat diet
for 8 weeks and (B) 24-week-old ApoE−/− (n=7) and ApoE−/−/SREBP2(N) mice (n=6) fed a normal chow. C and D, Quantification of percent
lesion areas in the thoracic aorta (TA), aortic arch (AA), and whole aorta from groups in A and B. The Student t test or Mann-Whitney U
test with the exact method was used. *P<0.05.
Table 2. Serum Lipid Profile of EC-SREBP2(N) and Their Wild-Type Littermates Fed an Atherogenic Diet
Serum Lipids Wild-Type, mg/dL (n=5) EC-SREBP2(N), mg/dL (n=5) Total cholesterol 161±6* 176±30 Triglyceride 56±13 59±11 LDL 16±4 12±2 VLDL 105±31 107±20 HDL 62±36 68±20
EC indicates endothelial cell; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SREBP2, sterol regulatory element binding protein 2; and VLDL, very-low-density lipoprotein.
*All values are expressed as mean±SD averaged from the number of animals as indicated.
Xiao et al Shear-Induced Inflammasome and Atherosclerosis 641
previously shown that the unfolded protein response chaperone ATF6 inhibits SREBP2 activity by binding to SREBP2 in liver cells under glucose deprivation.14 This inhibitory effect does not
involve ATF6 regulation of SREBP2 maturation, that is, cleavage. In contrast, OS affects SREBP2 cleavage (Figure 1A). Support for the 2 distinct mechanisms of SREBP2 regulation comes from experiments showing that ATF6 and SREBP2 were coinduced in ECs under OS and that ATF6 knockdown by siRNA did not affect SREBP2 induction by OS (data not shown).
Among mechanosensitive signaling molecules, Akt and ade-nosine monophosphate–activated protein kinase regulate the endothelial phenotypic changes responding to atheroprone and atheroprotective flow, respectively.39 Akt positively regulates
SREBP40 through direct phosphorylation and transcriptional
activation via mTORC1. On the other hand, adenosine mono-phosphate–activated protein kinase inhibits SREBP-1c and -2 activities through Ser372 phosphorylation, which inhibits SREBP cleavage, nuclear translocation, and transcriptional activ-ity.41 Thus, the disturbed flow–activated Akt is likely involved in
SREBP2 activation in atheroprone areas, whereas SREBP2 sup-pression in atheroresistant areas is mediated at least in part by adenosine monophosphate–activated protein kinase. Thus, ade-nosine monophosphate–activated protein kinase activators such as statin and metformin may play a beneficial role similar to that of atheroprotective flow in phosphorylating SREBP2.
SREBP-1a is involved in the lipopolysaccharide-stimulated IL-1β production through activation of NLRP1a inflam-masome in macrophages.19 The SREBP1 promoter region
also contains an SRE binding site, which can be regulated by SREBP2.42 The OS-induced SREBP2 should also
acti-vate SREBP1 because ECs transfected with Ad-SREBP2(N) showed an increased expression of SREBP1 (Figure VIII in the online-only Data Supplement). However, we did not find increased levels of NLRP1a in ECs from EC-SREBP2(N)-Tg mice (Figure VIII in the online-only Data Supplement). Most, if not all, NLRP3 activators upregulate NOXs, with concur-rent elevation of the short-lived ROS, so NOX-mediated redox signaling is involved in NLRP3 inflammasome activation.20,21
Nevertheless, NOXs are not necessary for inflammasome acti-vation in hematopoietic cells because immune cells deficient in NOXs show normal or hyperactive activation of inflamma-some.43,44 In contrast, our data in Figure 3 suggest that SREBP2
transactivation of NOX2 is necessary for inducing NLRP3
inflammasome in ECs. Although the molecular basis of the dis-crepancies between macrophages and ECs remains unknown, SREBP2 induction of NOX2 is implicated in endothelial biol-ogy in that the major enzymatic product of NOX2 is ROS.45
As summarized in Figure 8, we demonstrated that athero-prone flow, like endogenous damage- and pathogen-associated molecules such as cholesterol crystals and lipopolysaccharide, induces NLRP3 inflammasome in endothelium via SREBP2 activation. This increased innate immunity in endothelium synergizes with hyperlipidemia to result in the focal nature of atherosclerosis.
Sources of Funding
This work was supported in part by National Institutes of Health grants HL89940 and HL105318, National Natural Science Foundation of China grant 81270349, National Science Council of the Republic of China (NSC 101-2311-B-009-003-MY3 and NSC 100-2627-B-009-002) and UST-UCSD I-RiCE Program (NSC 101-2911-I-009-101).
Disclosures
None.
References
1. Ross R. Mechanisms of disease: atherosclerosis: an inflammatory disease.
N Engl J Med. 1999;340:115–126.
2. Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: patho-physiological basis and clinical perspectives. Physiol Rev. 2011;91:327–387. 3. Davies PF. Hemodynamic shear stress and the endothelium in cardiovas-cular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6:16–26. 4. Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes.
Annu Rev Genet. 2007;41:401–427.
5. Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Näär AM. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569.
6. Rayner KJ, Suárez Y, Dávalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernández-Hernando C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. 7. Liu Y, Chen BP, Lu M, Zhu Y, Stemerman MB, Chien S, Shyy JY. Shear
stress activation of SREBP1 in endothelial cells is mediated by integrins.
Arterioscler Thromb Vasc Biol. 2002;22:76–81.
8. Wen H, Ting JP, O’Neill LA. A role for the NLRP3 inflammasome in metabolic diseases: did Warburg miss inflammation? Nat Immunol. 2012;13:352–357. 9. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG,
Abela GS, Franchi L, Nuñez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflam-masomes are required for atherogenesis and activated by cholesterol crys-tals. Nature. 2010;464:1357–1361.
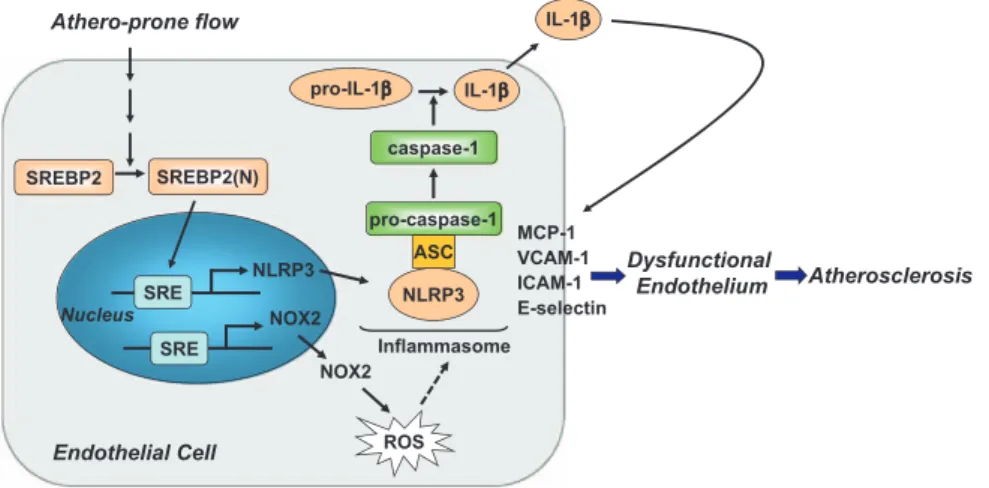
10. Rajamäki K, Lappalainen J, Oörni K, Välimäki E, Matikainen S, Kovanen PT, Eklund KK. Cholesterol crystals activate the NLRP3 inflammasome Athero-prone flow SREBP2(N) ROS pro-IL-1β IL-1β Nucleus IL-1β Dysfunctional Endothelium ASC pro-caspase-1 Inflammasome caspase-1 Endothelial Cell MCP-1 VCAM-1 ICAM-1 E-selectin SREBP2 NOX2 SRE NLRP3 SRE NOX2 Atherosclerosis NLRP3
Figure 8. Graphic summary of the
mechanism for sterol regulatory element binding protein 2 (SREBP2) activation of NLRP3 inflammasome in endothelium mediates atheroprone flow-induced atherosclerosis. ICAM-1, intercellular adhesion molecule 1; IL, interleukin; MCP-1, monocyte chemoattractant protein 1; NOX2, NADPH oxidase 2; ROS, reactive oxygen species; and VCAM-1. vascular cell adhesion molecule 1.
by guest on April 24, 2014 http://circ.ahajournals.org/
in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765.
11. Hwang J, Ing MH, Salazar A, Lassègue B, Griendling K, Navab M, Sevanian A, Hsiai TK. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: implication for native LDL oxida-tion. Circ Res. 2003;93:1225–1232.
12. Lim YC, Luscinskas FW. Isolation and culture of murine heart and lung endo-thelial cells for in vitro model systems. Methods Mol Biol. 2006;341:141–154. 13. Zhang Y, Lee TS, Kolb EM, Sun K, Lu X, Sladek FM, Kassab GS, Garland T Jr, Shyy JY. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler Thromb Vasc
Biol. 2006;26:1281–1287.
14. Zeng L, Lu M, Mori K, Luo S, Lee AS, Zhu Y, Shyy JY. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004;23:950–958.
15. Wingender E, Karas H, Knuppel R. TRANSFAC database as a bridge between sequence data libraries and biological function. Pac Symp
Biocomput. 1997;2:477–485.
16. Ni ZF, Ng DW, Liu JX, Chen ZJ. Chromatin immunoprecipitation (ChIP) assay. Protocol Exchange. 2009. doi:10.1038/nprot.2009.11. http://www. nature.com/protocolexchange/protocols/500. Accessed July 2, 2013. 17. Nakano K, Egashira K, Ohtani K, Gang Z, Iwata E, Miyagawa M, Sunagawa K.
Azelnidipine has anti-atherosclerotic effects independent of its blood pressure-lowering actions in monkeys and mice. Atherosclerosis. 2008;196:172–179. 18. Nigro P, Abe J, Berk BC. Flow shear stress and atherosclerosis: a matter of
site specificity. Antioxid Redox Signal. 2011;15:1405–1414.
19. Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, Raffatellu M, Osborne TF. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell
Metab. 2011;13:540–549.
20. Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbes-tos and silica. Science. 2008;320:674–677.
21. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140.
22. Matlung HL, Bakker EN, VanBavel E. Shear stress, reactive oxygen species, and arterial structure and function. Antioxid Redox Signal. 2009;11:1699–1709. 23. Suo J, Ferrara DE, Sorescu D, Guldberg RE, Taylor WR, Giddens DP.
Hemodynamic shear stresses in mouse aortas: implications for atherogen-esis. Arterioscler Thromb Vasc Biol. 2007;27:346–351.
24. Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion for-mation. Proc Natl Acad Sci U S A. 2000;97:9052–9057.
25. Landmesser U, Hornig B, Drexler H. Endothelial function: a critical deter-minant in atherosclerosis? Circulation. 2004;109(suppl 1):II27–II33. 26. McGill HC Jr, McMahan CA, Herderick EE, Tracy RE, Malcom GT,
Zieske AW, Strong JP. Effects of coronary heart disease risk factors on atherosclerosis of selected regions of the aorta and right coronary artery: PDAY Research Group: Pathobiological Determinants of Atherosclerosis in Youth. Arterioscler Thromb Vasc Biol. 2000;20:836–845.
27. McGill HC Jr, McMahan CA, Gidding SS. Preventing heart disease in the 21st century: implications of the Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study. Circulation. 2008;117:1216–1227. 28. Fearon WF, Fearon DT. Inflammation and cardiovascular disease: role of
the interleukin-1 receptor antagonist. Circulation. 2008;117:2577–2579. 29. Chamberlain J, Evans D, King A, Dewberry R, Dower S, Crossman D,
Francis S. Interleukin-1beta and signaling of interleukin-1 in vascular wall and circulating cells modulates the extent of neointima formation in mice.
Am J Pathol. 2006;168:1396–1403.
30. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M. Lack of interleukin-1beta decreases the sever-ity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc
Biol. 2003;23:656–660.
31. Devlin CM, Kuriakose G, Hirsch E, Tabas I. Genetic alterations of IL-1 receptor antagonist in mice affect plasma cholesterol level and foam cell lesion size. Proc Natl Acad Sci U S A. 2002;99:6280–6285.
32. Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J. 2011;162:597–605.
33. Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of miR-33 in mice promotes reverse cholesterol trans-port and regression of atherosclerosis. J Clin Invest. 2011;121:2921–2931. 34. Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils
JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, Khatsenko OG, Kaimal V, Lees CJ, Fernandez-Hernando C, Fisher EA, Temel RE, Moore KJ. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature. 2011;478:404–407.
35. Zhu M, Fu Y, Hou Y, Wang N, Guan Y, Tang C, Shyy JY, Zhu Y. Laminar shear stress regulates liver X receptor in vascular endothelial cells.
Arterioscler Thromb Vasc Biol. 2008;28:527–533.
36. Civelek M, Grant GR, Irolla CR, Shi C, Riley RJ, Chiesa OA, Stoeckert CJ Jr, Karanian JW, Pritchard WF, Davies PF. Prelesional arterial endothe-lial phenotypes in hypercholesterolemia: universal ABCA1 upregulation contrasts with region-specific gene expression in vivo. Am J Physiol Heart
Circ Physiol. 2010;298:H163–H170.
37. Feaver RE, Hastings NE, Pryor A, Blackman BR. GRP78 upregulation by atheroprone shear stress via p38-, alpha2beta1-dependent mechanism in endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:1534–1541. 38. Civelek M, Manduchi E, Riley RJ, Stoeckert CJ Jr, Davies PF. Chronic
endoplasmic reticulum stress activates unfolded protein response in arte-rial endothelium in regions of susceptibility to atherosclerosis. Circ Res. 2009;105:453–461.
39. Guo D, Chien S, Shyy JY. Regulation of endothelial cell cycle by laminar versus oscillatory flow: distinct modes of interactions of AMP-activated protein kinase and Akt pathways. Circ Res. 2007;100:564–571.
40. Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: cell signal-ing meets lipid metabolism. Trends Endocrinol Metab. 2010;21:268–276. 41. Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z,
Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M. AMPK phosphorylates and inhibits SREBP activity to atten-uate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388.
42. Amemiya-Kudo M, Shimano H, Yoshikawa T, Yahagi N, Hasty AH, Okazaki H, Tamura Y, Shionoiri F, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Sato R, Kimura S, Ishibashi S, Yamada N. Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. J Biol
Chem. 2000;275:31078–31085.
43. Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood. 2010;116:1570–1573.
44. van de Veerdonk FL, Smeekens SP, Joosten LA, Kullberg BJ, Dinarello CA, van der Meer JW, Netea MG. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci U S A. 2010;107:3030–3033. 45. Takac I, Schröder K, Brandes RP. The Nox family of NADPH oxidases:
friend or foe of the vascular system? Curr Hypertens Rep. 2012;14:70–78.
CLINICAL PERSPECTIVE
Atherosclerosis preferentially develops at branches and curvatures in the arterial tree, and the disturbed flow pattern imposed in the endothelium in these regions plays a major role in the preferentially localized atherosclerosis. In this study, we show that disturbed flow increases endothelial innate immunity via NLRP3 inflammasome in vitro and in vivo. The underlying mechanism involves the induction of sterol regulatory element binding protein 2 (SREBP2), which transactivates NADPH oxidase 2 and NLRP3. The increased innate immunity in endothelium predisposes hyperlipidemia to result in the focal nature of atherosclerosis. This newly defined SREBP2/NLRP3 inflammasome pathway suggests that SREBP2 could be a therapeutic target to prevent atherosclerosis initiation, which is in line with the antiatherosclerosis effect of interleukin-1β antagonism.
CIRCULATIONAHA/2013/002714R1