Cloning, Characterization, and Expression of the Nitric Oxide-Generating
Nitrite Reductase and of the Blue Copper Protein Genes of
Achromobacter cycloclastes
Jang-Yi Chen,* Wei-Chao Chang,* Tschining Chang,† Wen-Chang Chang,*
,† Ming-Yih Liu,‡
William J. Payne,§ and Jean LeGall‡
,1*Institute of Biochemical Sciences, National Taiwan University, Taipei, Taiwan; †Institute of Biological Chemistry, Academia Sinica, Taipei, Taiwan; and ‡Departments of Biochemistry and Molecular Biology and §Microbiology,
University of Georgia, Athens, Georgia
Received January 11, 1996
The nitrite reductase (NIR) and blue copper protein (BCP) genes have been cloned from Achromobacter
cycloclastes and characterized. NIR gene encodes a protein of 378 amino acid residues including a putative
signal peptide of 37 residues. BCP gene encodes a protein of 148 residues with a 24-residue signal peptide. The DNA-derived amino acid sequence of NIR is in complete agreement with that from Edman degradation and the DNA coding sequence of BCP is also consistent with its partial N-terminal amino acid sequence. Both genes contain their own FNR box in the 59 upstream region and a TA-rich region that could be the transcription start site. These two genes are separated by at least 10 kb. Based on these observations it is very likely that these two genes, although functionally related, are regulated independently. Both proteins could be expressed in E. coli, and both of the expressed proteins could be recognized by their respective antisera. The expressed NIR dem-onstrates full enzymatic activity. The similarity of both proteins to the counterparts from Alcaligenes faecalis S-6 is discussed. © 1996 Academic Press, Inc.
Nitrite reductase (NIR) is one of the enzymes involved in the alternative respiration system of
the denitrifying bacteria. This copper-containing enzyme transfers electrons in the denitrifying
pathway: NO
3→
NO
2→
NO
→
N
2O
→
N
2(1), in which it reduces nitrite ion to nitric oxide. NIR
receives electrons from the azurin-like blue copper protein (BCP) (2,3). Both proteins have been
purified from several organisms. Their crystal structures have also recently been determined (4,5).
The crystal structure of NIR is a trimer composed of three identical subunits. Each subunit contains
one type I copper atom, and there are three type II copper atoms trapped in the space between the
subunits. The BCP contains only one type I copper. In Alcaligenes faecalis, the electrons are
transferred from the type I copper of the pseudoazurin to the type I copper then to type II copper
within NIR (6). However, the exact protein-protein interaction between BCP and NIR, the
mecha-nism of the electron flow from the environment to BCP, and the details of the NO
2−reduction
within NIR still remain uncertain. As part of the continuing effort to gain a clearer understanding
of the components of these reactions, we describe here the cloning and characterization of the
nucleotide sequences of the NIR and the BCP genes from Achromobacter cycloclastes and their
expression in Escherichia coli.
MATERIALS AND METHODS
Bacterial strains and plasmids. Achromobacter cycloclastes IAM1013 was used to construct a genomic library for the
screening of NIR and BCP genes. E. coli LE392 was used for genomic DNA cloning. E. coli JM109 used for plasmid cloning and sequencing was from Promega Inc. E. coli. strain M15 (PREP4), which derived from strain K12 was purchased from Qiagen Inc. and used for gene expression. The plasmids, pGemT, pGem3zf(+) (Promega) and pUC19, were used as cloning vector, and plasmid pQE30 (Qiagen Inc.) was used as expression vector.
Media. A. cycloclastes IAM1013 was grown anaerobically at 30°C in the following medium (per liter): nutrient broth,
1To whom correspondence should be addressed.
BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS219, 423–428 (1996) ARTICLE NO. 0249
423
0006-291X/96 $18.00
JOBNAME: BBRC 218#3 PAGE: 2 SESS: 18 OUTPUT: Fri Apr 12 01:53:54 1996
/xypage/worksmart/tsp000/68480f/57
2.3g; yeast extract, 1.2g; glycerol, 5ml; NaCl, 3g; and potassium nitrate, 0.5g. E. coli. strains were cultured in L-broth medium aerobically at 37°C. The concentration of ampicillin used for selection was 50mg/ml; for blue/white colony selection. IPTG, 0.5mM, and X-Gal, 40mg/ml (final concentration), were added to the medium. For expression of the cloned NIR protein and BCP under control of E. coli, T5 promotor, two lac operator sequences and 2mM IPTG were added.
Chemicals and Enzymes. All of the DNA modification enzymes and restriction endonucleases used were purchased from
Boehringer Mannheim GmbH or Promega Inc. [a-32P]dATP and [a-35S]dATP were from Amersham Inc. The nucleotide sequencing kits were the products of USB Inc. The recombinant gene expression and protein purification kits were purchased from Qiagen Inc.
Genomic library construction. All the methods used were as described (7). In general, genomic DNA of A. cycloclastes
was partially digested with Sau3AI restriction endonuclease, size-fractionated and then ligated with EMBL III vector. After
in vitro packaging, it was used to transfect E. coli strain LE 392.
Cloning of NIR and BCP genes. Two primers were designed based on the amino acid sequence of NIR: forward primer
corresponding to amino acids at positions 73 to 81: 59ATGGTNGTNCAYGARAAYGAYTAYG39, reverse primer corre-sponding to amino acids at positions 329 to 322: 59TCRTCRTTCCAYTCNCCNGTNAC39. A polymerase chain reaction (PCR) was performed using the above primers and genomic DNA of A. cycloclastes as template to produce a DNA fragment of 750 bp. After subcloning and sequencing to confirm its identity, this fragment was used to screen the genomic library. For the cloning of BCP gene, the same procedure was repeated, except the primers were designed based on their partial amino acid sequence as follows: forward primer corresponding to amino acids at positions 1 to 7: 5 9GCNGAYTTC-GARGTBCAYATG39, reverse primer corresponding to amino acids 43 to 37: 59YTCVACGTTCTTNCCCTTRTC39. The product of 129 bp fragment from a PCR was subcloned, sequenced and used as probe for screening of BCP gene.
Expression of BCP and NIR gene in E. coli. For NIR gene, a pair of synthetic oligonucleotides were synthesized as
primer: forward primer (NP1) beginning with the enterokinase cutting site (DDDDK) coding sequence followed by the codons of mature NIR protein; the reverse primer (NP2) containing the sequence of 39end and stop codon of NIR gene. After PCR amplification, the products were phosphorylated in order to ligate with pUC19 polylinker region on HincII site, then cleaved by BamHI and Hind III and ligated with pQE30 to produce NIR gene expression vector pQNIR. For BCP gene: forward primer (BP1) contained not only enterokinase cutting site coding sequence but also a Bam HI recognition sequence before the mature BCP gene. The reverse primer (BP2) contained a Hind III recognition sequence after 39 end coding sequence. The constructed vectors were checked by sequencing of the junction regions and transformed to E. coli strain M15. Cells carrying pQBC3 and pQNIR were precultured in LB medium, then IPTG was added to a final concentration of 2mM and incubated for 5 hrs. The cells were harvested and lysed. The cell extract was passed through Ni-NTA(nitrilo-tri-acetic acid) resin to the fused recombinant proteins which contain 6 × His tag. The fused protein purified could be cut by enterokinase after the sequence DDDDK amino acid sequence to release native products of NIR or BCP.
Immunological techniques. Both anti-NIR and anti-BCP sera were obtained by immunizing New Zealand White rabbits
with native NIR or BCP by surgical intrasplenic immunization according to the method of Hong et al. (8). Blood samples were taken between the fourth week and the tenth week. The antisera were used without further purification. Western blotting was carried out after SDS PAGE according to Bjerrum et al. (9). The membrane was first treated with rabbit anti-NIR or anti-BCP serum (1:5000) and followed by alkaline phosphatase conjugated goat anti-rabbit total Ig or IgG, then developed by incubation with nitroblue-tetrazolium and 5-bromo-4-chloro-3-indoyl-phosphate (Bcip).
RESULTS AND DISCUSSIONS
A genomic library of A. cycloclastes has been constructed with an efficiency of 7.5 × 10
5pfu/
m
g DNA. Both NIR and BCP genes are found inside this library and the nucleotide sequences for
NIR and BCP genes are shown in Fig. 1,A and Fig. 1,B, respectively. There are three open reading
frames containing the NIR structure sequence (Fig. 1,A). The protein deduced from the longest
reading frame is composed of 378 amino acid residues with a putative signal peptide of 37 residues.
The deduced amino acid sequence following the putative signal peptide is in complete agreement
with that described from Edman degradation. Comparison with the NIR from A. faecalis reveals
that the major difference is around this leader peptide. The mature proteins of these two NIR share
a sequence homology of 81%.
The longest open reading frame in BCP gene (Fig. 1,B) encodes a protein of 152 amino acid
residues. The mature protein starting from Ala29 to the C-terminal has 99.2% homology to that of
pseudoazurin (10) with only one difference at Gln89 which is a Glu in pseudoazurin. There are
three methionine codons before Ala29. The Met5 is preceded by a ribosome binding sequence (SD
sequence). Besides, the peptide from Met5 to Ala28 is a typical signal peptide as reported by
Duffaud et al. (11): Positively charged amino acid (Lys10) at the N-terminal, hydrophobic core to
form a helix (Gly12 to Ala24) and small amino acid (Ala) around the processing site at the
FIG. 1. Nucleotide sequence of NIR(A) and BCP(B) genes. Nucleotides and amino acids are numbered at the right and
left margin, respectively. The asterisk indicates the N-terminal of the mature protein. The putative gene regulatory elements: FNR box; SD sequence and TA-rich region (see text) are underlined.
JOBNAME: BBRC 218#3 PAGE: 4 SESS: 19 OUTPUT: Fri Apr 12 01:53:54 1996
/xypage/worksmart/tsp000/68480f/57
C-terminal. Therefore, the entire coding sequence in the BCP gene should begin from the second
ATG, and have 148 amino acid residues including the signal peptide of 24 residues. The partial
N-terminal sequence also matches completely with the DNA-derived sequence. When the sequence
was compared with its counterpart in A. faecalis S-6, the homology was only 66%, and even lower,
53.5%, with that of Pseudomonas ami (12). This is quite different from the case of NIRs where a
high degree of homology is found. Since BCPs are proposed to have similar function as electron
donor for NIR, it might mean that the structure is more flexible for the pseudoazurin/BCP than that
of NIR. In fact, those essential residues for maintaining the function of the BCP, such as copper
binding residues and hydrophobic face surrounded with lysine-residue ring, are conserved (13).
Both BCP and NIR genes contain the FNR box in their 5
9 upstream region (Fig. 1,A and B), a
structure that could be regulated, as it has been suggested, by the aerobic-anaerobic shift (14). On
the other hand, we could not find the typical Pribnow box in either gene. Instead, a TA-rich region
around base 110, TTAAT (Fig. 1,B), of the BCP and around base 412, TTGAATA (Fig. 1,A), of
the NIR gene were found. Since each of the two genes contains its own FNR box and the restriction
mapping shows that they are separated by at least 10 kb (data not shown), it is suggested that those
TA-rich regions could be the transcription start sites. If so, then these two genes would not form
an operon such as that found in E. coli (15).
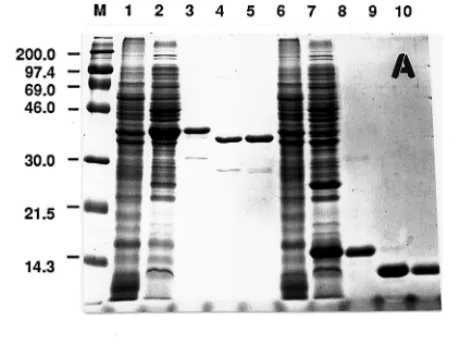
Both genes could be expressed in E. coli after IPTG induction (Fig. 2,A). Most of the proteins
were present in soluble fraction during the purification process (data not shown). The expressed
proteins were easily purified with a Ni
+2-NTA resin affinity column because of the 6× His tag
present at the N-terminal of the fusion protein. The 6× His tag could also be removed by
entero-kinase to release the native protein. The N-terminal sequencing confirmed that the expressed NIR
and BCP are identical to the native proteins. The absorption spectrum of recombinant BCP in the
oxidized state is the same as that of the native BCP, which could also be easily reduced by an agent
such as dithionite. The recombinant NIR shows the same optical absorption spectrum as the native
protein and has fully enzyme activity (data not shown). The expressed proteins were recognized by
the respective antisera as shown in Fig. 2, B.
FIG. 1—Continued.
Note: Both the NIR and BCP gene nucleotide sequences have been submitted to EMBL, the
accession numbers are Z48635 (NIR gene) and Z48669 (BCP gene).
ACKNOWLEDGMENTS
This study was supported by grants from National Science Council of the Republic of China to Wen-Chang Chang (NSC83-0418-B-001-016-BA and NSC84-2311-B-001-051-BA) and by National Science Foundation of the U.S.A. (MCB 9105711) to W.J.P. and J.L.
REFERENCES
1. Payne, W. J. (1985) in Denitrification in the Nitrogen Cycle (Golterman, H. Ly, Ed.), pp. 47–65, Plenum, New York. 2. Kakutani, T., Watanabe, H., Arima, K., and Beppu, T. (1981) J. Biochem. 89, 463–472.
3. Liu, M. Y., Liu, M. C., Payne, W. J., and LeGall, J. (1986) J. Bacteriology 166, 604–608.
4. Godden, J. W., Turley, S., Teller, D. C., Adman, E. T., Liu, M. Y., Payne, W. J., and LeGall, J. (1991) Science 253, 438–442.
5. Inoue, T., Nishio, N., Kai, Y., Harada, S., Ohshiro, Y., Suzuki, S., Kohzuma, T., Shidara, S., and Iwasaki, H. (1993)
J. Biochem. 114, 761–762.
FIG. 2. The protein profiles of expressed NIR and BCP analyzed in SDS–PAGE (A) and Western blotting (B). M,
protein markers with molecular weight in kDa. Lanes 1 and 6, cell extract of the host E. coli M15. Lane 2, cell extract of
E. coli M15 transformed with pQNIR and induced with IPTG. Lane 3, Ni-NTA resin purified recombinant NIR protein.
Lane 4, enterokinase released NIR. Lane 5, native NIR protein as marker. Lane 7, cell extract of E. coli M15 transformed with pQBC3 and induced with IPTG. Lane 8, Ni-NTA resin purified recombinant BCP protein. Lane 9, enterokinase released BCP protein. Lane 10, native BCP protein.
JOBNAME: BBRC 218#3 PAGE: 6 SESS: 19 OUTPUT: Fri Apr 12 01:53:54 1996
/xypage/worksmart/tsp000/68480f/57
6. Kukimoto, M., Nishiyama, M., Murphy, M. E. P., Turley, S., Adman, E. T., Horinouchi, S., and Beppu, T. (1994)
Biochemistry 33, 5246–5252.
7. Quertermous, T. (1990) in Current Protocols in Molecular Biology (Ausubel, F. M. et al., Eds.), pp. 5.8.1–5.8.5, Wiley, New York.
8. Hong, T. H., Chen, S. T., Tang, T. K., Wang, S. C., and Chang, T. H. (1989) J. of Immunological Method 120, 151–157. 9. Bjerrum, O. J., and Schafer-Nielsen, C. (1986) in Electrophoresis (Dunn, , Ed.), pp. 315–327.
10. Ambler, R. P. (1977) in The Evolution of Metalloenzymes, Metalloproteins and Related Materials (Leigh, G. J., Ed.), pp. 100–118, Symposium Press, London.
11. Duffaud, G. D., Lehnhardt, S. K., March, P. E., and Inouye, M. (1985) Curr. Top. Membr. Transp. 24, 65–104. 12. Petratos, K., Banner, D. V., Beppu, T., Wilkinson, K. S., and Tsernoglou, D. (1987) FEBS Lett. 218, 209–214. 13. Kukimoto, M., Nishiyama, M., Ohnuki, T., Turley, S., Adman, E. T., Horinouchi, S., and Beppu, T. (1995) Pro. Engng.
8, 153–158.
14. Shaw, D. J., and Guest, J. R. (1982) Nucleic Acid Res. 10, 6119–6130.
15. Hussain, H., Grove, J., Griffiths, L., Busby, S., and Cole, J. (1994) Mol. Microbio. 12, 153–163.