Synthesis and Reactivity of the Ruthenium
Cyclopropenyl Complex with a Tp Ligand
Yih-Hsing Lo, Ying-Chih Lin,* Gene-Hsiang Lee, and Yu Wang
Department of Chemistry, National Taiwan University, Taipei, Taiwan, 106 R.O.C.Received September 4, 1998
Treatment of Tp(PPh3)2Ru-CtC-Ph (1) with ICH2CN affords the cationic vinylidene complex [Tp(PPh3)2RudCdC(Ph)CH2CN]I (2). The neutral ruthenium cyclopropenyl complex Tp(PPh3)2RuCdC(Ph)CHCN (3) is then prepared by deprotonation of 2. Facile displacement of one phosphine ligand of 3 by CH3CN yields a diastereomeric mixture in a 4:1 ratio of the substitution product, Tp(PPh3)(CH3CN)RuCdC(Ph)CHCN (4). The cyclopropenyl ring in 3 and 4 is susceptible to ring opening by electrophiles such as CF3COOH, Ph3CPF6, and HgCl2. The substitution reaction of 3 with pyrazole is followed by an intramolecular nucleophilic addition of the nitrogen atom at the R-carbon atom to afford the metallacyclic complex, Tp-(PPh3)Ru(C3H3NN)CdC(Ph)CH2CN (8a). The reaction of 3 with CO in the presence of MeOH gives Tp(PPh3)(CO)RuC(OMe)dC(Ph)CH2CN (9). The reaction of CF3COOH with 9 yields Tp(PPh3)(CO)RuC(O)CH(Ph)CH2CN (11). The structures of 8a and 11 have been determined by X-ray diffraction analysis.
Introduction
Transition-metal cyclopropenyl and vinylidene com-plexes have been the focus of many recent investiga-tions. Organic cyclopropene is a highly strained cy-cloalkene with an estimated strain energy of more than 50 kcal/mol.1This molecule has played a crucial role in the development of an important concept of aromaticity, and its chemical reactivity has been extensively ex-plored.2 Interest in metal vinylidene complexes stems from their potential as reactive intermediates in organic and organometallic synthesis, as well as their applica-tion in catalytic processes.3 A key characteristic of vinylidene complexes appears to be the electrophilicity of the R-carbon, which adds, often easily, amines,4 alcohols,5-7phosphines,8and even fluoride.5We recently
reported facile synthesis of several ruthenium cyclopro-penyl complexes by deprotonation of cationic vinylidene complexes. The cationic nature of the vinylidene com-plex, along with the presence of an electron-withdrawing functionality, such as a CN group at Cγ of the vinylidene ligand, plays a role in enhancing the acidity of the proton next to the CN group. Thus the base successfully effects an intramolecular cycloaddition leading to for-mation of a neutral cyclopropenyl complex. We in fact found the facile cyclopropenation reaction of several ruthenium vinylidene complexes containing the cyclo-pentadienyl ligand. Therefore we thought a similar complex containing a tris(pyrazol-1-yl)borate ligand (Tp, B(C3H3N2)3) would be a suitable candidate for further investigating chemical reactivity of the ruthenium cy-clopropenyl complex. In the literature, research into the coordination chemistry of the Tp ligand has mostly focused on the first-row transition metals and group VI.9 The chemistry of Tp complexes of the second- and third-transition series and in particular that of group VIII10 remains less developed, although there is no obvious reason this should be the case. Herein, we report preparation of two neutral tris(pyrazol-1-yl)borato ruthenium cyclopropenyl complexes: Tp(PPh3)2 Ru-CdC(Ph)CHCN (3) and Tp(PPh3)(CH3 CN)-RuCdC(Ph)-CHCN (4). Electrophilic addition to these ruthenium cyclopropenyl complexes affords new vinylidene com-plexes. Reaction of 3 with pyrazole gives the metalla-cycle product, while in the presence of MeOH, reaction (1) (a) Special issue on strained organic compounds: Chem. Rev.
1989, 89. (b) Liebman, J. F.; Greenberg, A. Strained Organic Molelules;
Wiley: New York, 1978; p 91.
(2) Marier, G.; Periss, T.; Reisenauer, H. P.; Hess, B. A., Jr.; Schand, L. J. J. Am. Chem. Soc. 1994, 116, 2014. (b) Hopf, H.; Plagens, A.; Walsh, R. J. Chem. Soc., Chem. Commum. 1994, 1467.
(3) (a) Liebman, J. F.; Greenberg, A. Chem. Rev. 1976, 76, 311. (b) Halton, B.; Banwell, M. G. In The Chemistry of the Cyclopropnyl Group. Part 2; Patai, S., Rappoport, Z., Eds.; Wiley: Chichester, 1987; Chapter 21, p 1223.
(4) (a) Wang, Y.; Finn, M. G. J. Am. Chem. Soc. 1995, 117, 8045. (b) Bruneau, C.; Dixneuf, P. H. J. Chem. Soc., Chem. Commun. 1997, 507. (c) Bruce, M. I. Chem. Rev. 1991, 91, 197. (d) Trost, B. M. Angew. Chem. 1995, 107, 285. (e) Bianchini, C.; Peruzzini, M.; Romerosa, A.; Zanobini, F. Organometallics 1995, 14, 3152. (f) Bianchini, C.; Inno-centi, P.; Peruzzini, M.; Romerosa, A.; Zanobini, F. Organometallics
1996, 15, 272. (g) Werner, H.; Wiedmann, R.; Steinert, P.; Wolf, J.
Chem. Eur. J. 1997, 3, 127. (h) Trost, B. M.; Flygare, J. A. J. Am. Chem. Soc. 1992, 114, 5476. (i) Trost, B. M.; Dyker, G.; Kulawiec, R. J. Am. Chem. Soc. 1990, 112, 7809.
(5) Ting, P. C.; Lin, Y. C.; Lee, G. H.; Cheng, M. C.; Wang, Y. J. Am. Chem. Soc. 1996, 118, 6433.
(6) (a) Bruce, M. I.; Swincer, A. G.; Wallis, R. C. J. Organomet. Chem.
1979, 171, C5. (b) Bianchini, C.; Casares, J. A.; Peruzzini, M.;
Romerosa, A.; Zanobini, F. J. Am. Chem. Soc. 1996, 118, 4585. (7) (a) Barreett, A. G. M.; Carpenter, N. E. Organometallics 1987, 6, 2249. (b) Normbel, P.; Lugan, N.; Mathieu, R. J. Organomet.Chem.
1995, 503, C22.
(8) Senn, D. R.; Wong, A.; Patton, A. T.; Marsi, M.; Strouse, C. E.; Gladysz, J. A. J. Am. Chem. Soc. 1988, 110, 6096.
(9) Trofimenko, S. J. Prog. Inorg. Chem. 1986, 34, 115.
(10) (a) Albers, M. O.; Robinson, D. J.; Shaver, A.; Singleton, E. Organometallics 1986, 5, 2199. (b) Hiraki, K.; Ochi, N.; Kitamura, T.; Sasada, Y.; Shinoda, S. Bull. Chem. Soc. Jpn. 1982, 55, 2356. 10.1021/om980752q CCC: $18.00 © 1999 American Chemical Society
Publication on Web 02/24/1999
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
of 3 with CO gives the vinyl ether product. The mech-anism of these reactions is also reported.
Results and Discussion
Tp Metal Vinylidene Complex and Its Deproto-nation/Cyclopropenation. Treatment of the acetylide complex Tp(PPh3)2Ru-CtC-Ph (1) with ICH2CN af-fords the cationic vinylidene complex [Tp(PPh3)2 -RudCdC(Ph)CH2CN]I (2) with 90% yield. Complex 2 is soluble in CH2Cl2. Characteristic spectroscopic data of 2 consist of a strongly deshielded CR resonance as a triplet atδ 375.3 with JP-C) 16.5 Hz in the13C NMR spectrum and a singlet31P NMR resonance atδ 36.5 in CDCl3at room temperature, which is due to fluxional behavior of the vinylidene ligand.11For comparison, the Cp (Cp ) η5-C
5H5) analogue displayed a triplet CR resonance at δ 345.6 with JP-C ) 17.9 Hz in the 13C NMR spectrum and a singlet31P NMR resonance atδ 42.4.
The ruthenium cyclopropenyl complex Tp(PPh3)2 Ru-CdC(Ph)CHCN (3) can be readily prepared by depro-tonation of 2 with high yield. This reaction should be carried out at 0 °C in CH2Cl2 since complex 3 is thermally unstable. MeONa in MeOH is found to be a better deprotonation reagent (Scheme 1) even though reaction of 2 with n-Bu4NF (1 M in THF) or DBU (1,8-diazabicyclo[5.4.0]undecene) or KOH (dissolved in a minimum amount of H2O) also yields 3. Complex 3 is soluble in CH2Cl2, THF, and ether, but is insoluble in hexane. The diastereotopic center generated by this cyclopropenation reaction causes the two phosphine ligands to be nonequivalent. Thus, in the 31P NMR spectrum of 3, two doublet resonances atδ 48.8 and 48.1 with JP-P ) 29.9 Hz are observed. In the 1H NMR spectrum of 3, the singlet resonance atδ 1.18 is assigned to the CHCN group of the three-membered ring. The resonance of the same group in the Cp analogue appears at δ 1.40. Facile deprotonation indicates the acidic nature of the methylene protons of 2, which may be
ascribed to the combined effect of the cationic character and the presence of the electron-withdrawing CN sub-stituent of the vinylidene complex.5Complex 3 decom-poses in CDCl3 at room temperature, producing the vinylidene complex [Tp(PPh3)2RudCdC(Ph)CH2CN]Cl. The proton is believed to come from trace water in the solvent.
Facile Ligand Displacement of 3. We previously reported the analogous Cp complex of 3, which is stable with respect to the ligand substitution reaction; that is, the phosphine ligand bonds strongly to the ruthenium center, making the coordination site unavailable for an incoming substrate. In contrast, the Tp complex 3 is susceptible to ligand substitution reaction under rela-tively mild conditions. For example, treatment of 3 with CH3CN at room temperature affords the air-stable ruthenium cyclopropenyl complex Tp(PPh3)(CH3 CN)-RuCdC(Ph)CHCN (4) with high yield. Complex 4 is stable in ether, THF, and CH3CN, but decomposes in CDCl3 to produce a complicated mixture. Complex 4, prepared from 3, contains two diastereomers in a 4:1 ratio, and interestingly, the major one is stable and the minor one decomposes to some unidentifiable product. The1H NMR resonances attributed to the CHCN of the three-membered rings of the major and the minor products appear atδ 1.08 and 1.20, respectively. In the 13C{1H}NMR spectrum, the singlet resonance atδ 2.9 and the doublet resonance atδ 137.9 with2J
P-C) 11.4 Hz are assigned to the CHN and the ruthenium-bonded CR carbon of the major complex, respectively. The13C NMR spectrum of the minor product was not obtained because of its low stability.
Opening of the Three-Membered Ring by Elec-trophiles. Treatment of 3 with CF3COOH affords 2, indicating the basic character of the methyne carbon of the three-membered ring. No alkylation is observed when 3 is treated with CH3I, CH3CH2I, CH2dCHCH2 -Br, CHtC-CH2Br, and ICH2CN, but treatment of 3 with trityl hexafluorophosphate at 0 °C for 20 min affords the cationic vinylidene complex [Tp(PPh3)2 -RudCdC(Ph)CH(CPh3)CN]PF6 (5) with 69% yield (Scheme 1). There are a few examples in the literature in which electrophilic addition of CPh3+ resulted in (11) (a) Allen, D. L.; Gison, V. C.; Green, M. L.; Skinner, T. F.;
Bashikin, J.; Grebenik, P. D. J. Chem. Soc., Chem. Commum. 1985, 895. (b) Consiglio, G.; Morandini, F. Chem. Rev. 1987, 87, 761.
Scheme 1
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
formation of a C-C bond,15 even though CPh 3+ is usually considered a hydride abstractor. Treatment of 3 with HgCl2similarly affords the addition product, [Tp-(PPh3)2RudCdC(Ph)CH(HgCl)CN]Cl (6), while treat-ment of 4 with HgCl2 also gives [Tp(PPh3)(CH3 CN)-RudCdC(Ph)CH(HgCI)CN]Cl (7). Interestingly, only one diastereomer is observed for 7. Formation of these vinylidene complexes occurs by selective cleavage of the cyclopropenyl single bond near the metal center. This selectivity is similar to that reported for the asym-metrical cyclopropenes where the metal-substituted single bond was cleaved.16 Interestingly, reaction of MeONa with 6 gives the cyclopropenyl complex 3, and the reaction with 7 gives 4, but no reaction was observed in the reaction of 5 with MeONa, which may be attributed to the steric effect of the trityl cation.5
Reaction of 3 with Pyrazole and 3,5-Dimeth-ylpyrazole. The reaction of 3 with pyrazole in CH2Cl2 at room temperature did not yield the expected neutral substituted cyclopropenyl complex, but instead gave the metallacyclic complex Tp(PPh3)Ru-(C3H3 NN)CdC(Ph)-CH2CN (8a) (Scheme 2). An intermediate A was ob-served in the initial stage of the reaction monitored by spectroscopic methods. The 1H NMR spectrum of A shows two doublet resonances atδ 4.40 and 4.11 with JH-H) 18.4 Hz assignable to gem-protons of the CH2 -CN group. In the 31P NMR spectrum, the singlet resonance at δ 36.7 is assigned to the zwitterionic vinylidene complex A. We thus believe that the reaction first causes substitution of a phosphine ligand by a pyrazole molecule, followed by protonation to open the
three-membered ring to give the intermediate A. This is followed by nucleophilic addition of the neighboring nitrogen atom of the pyrazole group to CR of A to give the metallacyclic complex 8a (Scheme 2). Complex 8a is thermally robust and stable both in solution and in solid state. The1H NMR spectrum of 8a displays two doublet resonances centered atδ 3.35 and 3.18 with a coupling constant of JH-H) 17.3 Hz assignable to the CH2CN group. In the13C NMR spectrum, the doublet resonance atδ 163.3 with2J
P-C) 12.3 Hz is assigned to the vinyl CR. Reaction with 3,5-dimethylpyrazole gives a similar product, Tp(PPh3)Ru(Me2C3 HNN)-CdC(Ph)CH2CN (8b), in lower yield, which may be attributed to the steric effect. In the1H NMR spectrum of 8b, two doublet resonances atδ 3.25 and 3.17 with JH-H) 17.1 Hz are assigned to the CH2CN group. The doublet resonance atδ 162.2 with2J
P-C) 12.6 Hz in the13C NMR spectrum is assigned to the vinyl CR. A similar metallacyclic structure with a five-membered ring has also been reported recently.12
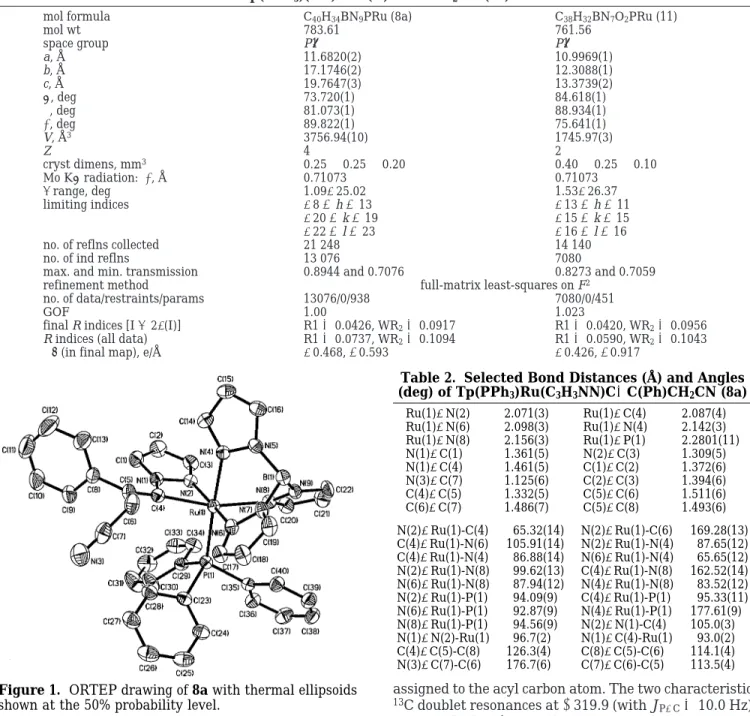
Structure of Metallacyclic Complex 8a. The mo-lecular structure of 8a has been determined by X-ray diffraction analysis (Table 1). An ORTEP diagram is shown in Figure 1, and Table 2 lists selected bond distances and angles. The structure 8a contains two crystallographically independent Ru complexes, al-though there is no essential structural difference be-tween them. The four-membered ring of 8a is puckered. The environment about the ruthenium metal center corresponds to a slightly distorted octahedron and it is obvious that the olefin in 8a is in an E configuration with the phenyl group trans to the Ru fragment, possibly because of steric reason. Three of the internal angles of the four-membered ring are 105.0(3)°, 96.7(2)°, and 93.0(2)°, while the fourth angle C(4)-Ru(1)-N(2) is 65.32(14)°. The three Ru-N(Tp) bond lengths (2.098(3), 2.142(3), and 2.156(3) Å) are slightly longer than the average distance of 2.038 Å in other chloro ruthenium Tp complexes.12,13 The Ru(1)-C(4) bond length of 2.087(4) Å is typical for a Ru-C single bond. The C(4)-C(5) bond length of 1.332(5) Å is typical for a CdC double bond, indicating the coordination of the sp2 carbon of the vinyl group. However, the bond angle Ru(1)-C(4)-N(1) of 93.0(2)° is much smaller than that of an idealized C (sp2) hybridization.
Reaction of 3 with CO in the Presence of MeOH. Reaction of 3 with CO in the presence of MeOH causes substitution of a phosphine ligand by CO followed by protonation of the three-membered ring by MeOH, possibly giving a vinylidene intermediate. This is then followed by addition of the MeO-to yield the vinyl ether complex Tp(PPh3)(CO)RuC(OMe)dC(Ph)CH2CN (9). In the1H NMR spectrum of 9, the two doublet resonances atδ 3.84 and 3.02 with JH-H) 16.8 Hz are assigned to the gem-protons. In the13C NMR spectrum, two doublet resonances attributed to the CO group and vinyl CR appear atδ 204.2 (with JP-C) 15.1 Hz) and 188.3 (with JP-C) 14.0 Hz), respectively. The νCOstretching in the IR spectrum of 9 appears at 1957 cm-1. In the absence of MeOH, the reaction gives an unstable complex, possibly a simple substitution product, Tp(PPh3 )(CO)-RuCdC(Ph)CHCN (B). We believe that the reaction may proceed through substitution as the first step, followed by opening of the cyclopropenyl ring by MeOH to give a (12) Christian, S.; Kurt M.; Roland S.; Kari K. Organometallics 1998,
17, 827.
(13) (a) Christian, S.; Kurt, M.; Roland, S.; Kari, K. J. Chem. Soc., Dalton Trans. 1997, 4209. (b) Gemel, C.; Trimmel, G.; Christian S.; Kurt M.; Roland S.; Kari K. Organometallics 1996, 15, 3998. (c) Gemel, C.; Trimmel, G.; Christian, S.; Kurt M.; Roland S.; Kari, K. Inorg. Chem. 1997, 36, 1076.
(14) Bell, R. A.; Chisholm, M. H.; Couch, D. A.; Rankel, L. A. Inorg, Chem. 1977, 16, 677.
(15) (a) Lews, J.; Parkins, A. W. J. Chem. Soc. A 1967, 1150. (b) Schrock. R. R.; Johnson, B. F. G.; Lewis, J. J. Chem, Soc., Dalton Trans.
1974, 951. (c) Harris, P. J.; Knox, S. A. R.; Mckinney, R. J.; Stone, F.
G. A. J. Chem. Soc., Dalton Trans. 1978, 1009.
(16) Padwa, A.; Blocklock, T. J.; Getman, N.; Hatanaka, N.; Loza, R. J. Org. Chem. 1978, 43, 1481.
Scheme 2
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
vinylidene ligand. Nucleophilic attack of MeO-at the R-carbon atom of the vinylidene ligand would then give the final product (Scheme 3). In our previous paper, we reported that MeOH is able to open certain three-membered rings.5The reaction of 9 with CF
3COOH in CH2Cl2 at room temperature ultimately leads to the formation of acylruthenium complex Tp(PPh3 )(CO)Ru-C(O)CH(Ph)CH2CN (11). When the reaction is followed by1H NMR spectroscopy, initial formation of the cat-ionic alkoxycarbene complex [Tp(PPh3 )(CO)RudC(OMe)-CH(Ph)CH2CN][CF3COO] (10) was apparent, then fol-lowed by a slow reaction to give complex 11. Chisholm and co-workers14 reported a similar reaction of Pt complexes.
When 9 was treated with HOAc in CH2Cl2, no reaction was observed. TheνCOstretching bands of 10 and 11 appear at 1998 and 1983 cm-1, respectively, in their IR spectra. The doublet resonance atδ 213.6 with 2J
P-C ) 13.4 Hz in the 13C NMR spectrum of 10 is
assigned to the acyl carbon atom. The two characteristic 13C doublet resonances atδ 319.9 (with J
P-C) 10.0 Hz) and 201.5 (JP-C) 12.8 Hz) are assigned to the carbene CR and the terminal CO group, respectively. In the1H NMR spectrum of 11, the two multiplet resonances at δ 3.75 and 3.21 are assigned to the CH2CN group. Orange-red crystals of 11 were obtained by slow diffu-sion of hexane into the ether solution of 11 at 0 °C for 3 days. The structure of 11 is determined by X-ray diffraction analysis. An ORTEP diagram is shown in Figure 2, and Table 3 lists selected bond distances and angles. The geometry around the metal center of 11 is approximately octahedral. The C(2)-O(2) bond length of 1.214(4) Å and the Ru-C(2)-C(3) bond angle of 124.7(2)° are characteristic for an acyl group. The Ru-C(2) bond length of 2.042(4) Å is typical of a Ru-C single bond. The C(1)-O(1) bond distance of 1.140(4) Å indicates terminal CO coordinate at the ruthenium center.
Concluding Remarks. The cyclopropenyl ruthe-nium complex 3 containing a Tp ligand was prepared from deprotonation of the vinylidene precursor. Unlike Table 1. Crystal and Intensity Collection Data for Tp(PPh3)Ru(C3H3NN)CdC(Ph)CH2CN (8a) and
Tp(PPh3)(CO)RuC(O)CHPhCH2CN (11)
mol formula C40H34BN9PRu (8a) C38H32BN7O2PRu (11)
mol wt 783.61 761.56 space group P1h P1h a, Å 11.6820(2) 10.9969(1) b, Å 17.1746(2) 12.3088(1) c, Å 19.7647(3) 13.3739(2) R, deg 73.720(1) 84.618(1) β, deg 81.073(1) 88.934(1) γ, deg 89.822(1) 75.641(1) V, Å3 3756.94(10) 1745.97(3) Z 4 2 cryst dimens, mm3 0.25× 0.25 × 0.20 0.40× 0.25 × 0.10 Mo KR radiation: γ, Å 0.71073 0.71073 θ range, deg 1.09-25.02 1.53-26.37 limiting indices -8 e h e 13 -13 e h e 11 -20 e k e 19 -15 e k e 15 -22 e l e 23 -16 e l e 16
no. of reflns collected 21 248 14 140
no. of ind reflns 13 076 7080
max. and min. transmission 0.8944 and 0.7076 0.8273 and 0.7059
refinement method full-matrix least-squares on F2
no. of data/restraints/params 13076/0/938 7080/0/451
GOF 1.00 1.023
final R indices [I > 2σ(I)] R1 ) 0.0426, WR2) 0.0917 R1 ) 0.0420, WR2) 0.0956
R indices (all data) R1 ) 0.0737, WR2) 0.1094 R1 ) 0.0590, WR2) 0.1043
∆F (in final map), e/Å -0.468, +0.593 -0.426, +0.917
Figure 1. ORTEP drawing of 8a with thermal ellipsoids
shown at the 50% probability level.
Table 2. Selected Bond Distances (Å) and Angles (deg) of Tp(PPh3)Ru(C3H3NN)CdC(Ph)CH2CN (8a)
Ru(1)-N(2) 2.071(3) Ru(1)-C(4) 2.087(4) Ru(1)-N(6) 2.098(3) Ru(1)-N(4) 2.142(3) Ru(1)-N(8) 2.156(3) Ru(1)-P(1) 2.2801(11) N(1)-C(1) 1.361(5) N(2)-C(3) 1.309(5) N(1)-C(4) 1.461(5) C(1)-C(2) 1.372(6) N(3)-C(7) 1.125(6) C(2)-C(3) 1.394(6) C(4)-C(5) 1.332(5) C(5)-C(6) 1.511(6) C(6)-C(7) 1.486(7) C(5)-C(8) 1.493(6) N(2)-Ru(1)-C(4) 65.32(14) N(2)-Ru(1)-C(6) 169.28(13) C(4)-Ru(1)-N(6) 105.91(14) N(2)-Ru(1)-N(4) 87.65(12) C(4)-Ru(1)-N(4) 86.88(14) N(6)-Ru(1)-N(4) 65.65(12) N(2)-Ru(1)-N(8) 99.62(13) C(4)-Ru(1)-N(8) 162.52(14) N(6)-Ru(1)-N(8) 87.94(12) N(4)-Ru(1)-N(8) 83.52(12) N(2)-Ru(1)-P(1) 94.09(9) C(4)-Ru(1)-P(1) 95.33(11) N(6)-Ru(1)-P(1) 92.87(9) N(4)-Ru(1)-P(1) 177.61(9) N(8)-Ru(1)-P(1) 94.56(9) N(2)-N(1)-C(4) 105.0(3) N(1)-N(2)-Ru(1) 96.7(2) N(1)-C(4)-Ru(1) 93.0(2) C(4)-C(5)-C(8) 126.3(4) C(8)-C(5)-C(6) 114.1(4) N(3)-C(7)-C(6) 176.7(6) C(7)-C(6)-C(5) 113.5(4)
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
its Cp analogue, the Tp cyclopropenyl complex 3 un-dergoes facile phosphine substitution reaction with several donor molecules. This property was employed to prepare novel complexes of Ru. For example, CH3
-CN readily displaces one of the phosphine ligands of 3 to give a mixture of diastereomers in a 4:1 ratio. Reaction of 3 with pyrazole gives the metallacyclic product 8a resulting from a displacement reaction followed by nucleophilic addition of the nitrogen atom to the CR. Reaction of 3 with CO in the presence of MeOH follows a similar pathway to yield the vinyl ether complex 9.
Experimental Section
General Procedure. All manipulations were performed
under nitrogen using vacuum-line, drybox, and standard Schlenk techniques. CH3CN and CH2Cl2were distilled from CaH2and diethyl ether, while THF was from Na/ketyl. All other solvents and reagents were of reagent grade and used without further purification. NMR spectra were recorded on Bruker AC-200 and AM-300WB FT-NMR spectrometers at room temperature (unless stated otherwise) and are reported inδ units with residual protons in the solvent as an internal
standard (CDCl3,δ 7.24; CD3CN,δ 1.93; C2D6CO,δ 2.04). FAB mass spectra were recorded on a JEOL SX-102A spectrometer. Tp(PPh3)2RuCl17 was prepared by following the method re-ported in the literature. Elemental analyses and X-ray dif-fraction studies were carried out at the Regional Center of Analytical Instruments at National Taiwan University.
Preparation of Tp(PPh3)2Ru-CtC-Ph (1). To a 50 mL MeOH solution of Tp(PPh3)2RuCl (3.93 g, 4.50 mmol) were added excess phenylacetylene (4.17 mL, 10 equiv) and excess Et3N, and the solution was heated to reflux for 90 min. The yellow precipitates formed and, after cooling, were filtered off and washed with MeOH and hexane. This yellow solid product was dried under vacuum to give compound 1 (3.34 g, 79% yield). Spectroscopic data for 1: 1H NMR (CDCl
3): δ 7.58 (d, JH-H) 2.1 Hz, 1H, Tp), 7.40 (d, JH-H) 2.1 Hz, 2H, Tp), 7.24-6.91 (m, PPh3, Tp), 5.54 (t, JH-H) 2.1 Hz, 2H, Tp), 5.31 (t, JH-H) 2.1 Hz, 1H, Tp), 5.20 (d, JH-H) 2.1 Hz, 1H, Tp).13C NMR (acetone): δ 135.8 (t, JP-C) 12.3 Hz, CR), 145.7-122.7 (Ph, Tp, PPh3,Cβ).31P NMR (CDCl3): δ 48.6. MS (FAB) m/z: 940.1 (M+), 678.1 (M+- PPh3), 577.1 (M+- PPh3, C2Ph), 363.0 (M+ - PPh3, C2Ph, Tp). Anal. Calcd for C53H44N6BP2Ru (938.75): C, 67.81; H, 4.72; N, 8.95. Found: C, 68.07; H, 4.54; N, 8.45.
Synthesis of the Vinylidene complex [Tp(PPh3)2
-RudCdC(Ph)CH2CN]I (2). To a Schlenk flask charged with (17) Nathaniel, W.; Alock, N. W.; Burns, I. D.; Claire, K. S.; Hill, A. F. Inorg. Chem. 1992, 31, 2906.
Scheme 3
Figure 2. ORTEP drawing of 11 with thermal ellipsoids
shown at the 50% probability level.
Table 3. Selected Bond Distances (Å) and Angles (deg) of Tp(PPh3)(CO)RuC(O)CH(Ph)CH2CN (11) Ru-C(1) 1.846(4) Ru-C(2) 2.042(3) Ru-N(1) 2.125(3) Ru-N(5) 2.162(3) Ru-N(3) 2.208(3) Ru(1)-P(1) 2.3599(9) C(1)-O(1) 1.140(5) N(7)-C(5) 1.130(4) C(2)-C(3) 1.571(5) C(3)-C(4) 1.543(5) C(4)-C(5) 1.456(5) C(2)-O(2) 1.214(4) C(1)-Ru-C(2) 94.42(13) C(1)-Ru-N(1) 89.36(13) C(2)-Ru-N(6) 90.38(11) C(1)-Ru-N(5) 171.93(13) C(2)-Ru-N(5) 88.83(12) N(1)-Ru-N(5) 83.22(11) C(1)-Ru-N(3) 89.20(12) C(2)-Ru-N(3) 172.25(12) N(1)-Ru-N(3) 82.80(10) N(5)-Ru-N(3) 86.72(10) C(1)-Ru-P(1) 92.69(11) C(2)-Ru-P(1) 95.22(9) N(1)-Ru-P(1) 173.87(8) N(5)-Ru-P(1) 94.37(8) N(3)-Ru-P(1) 91.45(7) O(2)-C(2)-C(3) 114.1(3) O(1)-C(1)-Ru 172.9(3) O(2)-C(2)-Ru 121.1(2) C(3)-C(2)-Ru 121.1(2) C(4)-C(3)-C(2) 107.8(3) N(7)-C(5)-C(4) 176.6(5)
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
complex 1 (3.10 g, 3.30 mmol) in 50 mL of CH2Cl2was added ICH2CN (0.46 mL, 3.5 mmol). The clear solution was stirred for 16 h, and then the solvent was reduced to about 5 mL. This mixture was slowly added to 90 mL of a vigorously stirred diethyl ether solution. The green precipitate thus formed was filtered off and washed with diethyl ether and hexane to give compound 2 (3.29 g, 90%). Spectroscopic data for 2: 1H NMR (CDCl3): δ 7.89 (br, 1H, Tp), 7.62 (br, 2H, Tp), 7.42-6.94 (m, PPh3, C2Ph, Ph), 6.78 (br, 1H, Tp), 6.66 (br, 1H, Tp), 5.73 (br, 2H, Tp), 5.60 (br, 1H, Tp), 5.47 (br, 1H, Tp), 3.08 (s, 2H, CH2). 13C NMR (CDCl 3): δ 375.3 (t, JP-C) 16.5 Hz, CR), 146.2-106.8 (m, Ph, Tp, PPh3,Cβ), 117.4 (CN), 11.4 (CH2).31P NMR (CDCl3): δ 36.5. MS (FAB) m/z: 980.5 (M+- I), 718.4 (M+ -I, PPh3), 577.2 (M+- I, PPh3, C2PhCH2CN). Anal. Calcd for C55H46N7BIP2Ru (1105.7): C, 59.74; H, 4.19; N, 8.87. Found: C, 59.67; H, 4.25; N, 8.74.
Synthesis of the Cyclopropenyl Complex Tp(PPh3)2
Ru-CdC(Ph)CHCN (3). To a solution of 2 (1.02 g, 0.92 mmol) in
20 mL of CH2Cl2at 0 °C was added a solution of MeONa (10 mL, 1 M in MeOH). The mixture was stirred for 10 min, and the color changed from green to yellow. The solvent was removed under vacuum at 0 °C, and the solid residue was extracted with cool ether. The extract was filtered through Celite. Solvent of the filtrate was removed under vacuum to give 3 (0.82 g, 91% yield). Spectroscopic data for 3: 1H NMR (0 °C, acetone): δ 7.81 (d, JH-H ) 2.1 Hz, 1H, Tp), 7.77 (d, JH-H) 2.3 Hz, 1H, Tp), 7.59 (d, JH-H) 2.3 Hz, 1H, Tp), 7.40-6.94 (m, 33H, Ph, PPh3), 6.89 (d, JH-H) 2.1 Hz, 1H, Tp), 6.58-6.54 (m, 2H, Ph PPh3), 5.97 (d, JH-H) 2.3 Hz, 1H, Tp), 5.83 (d, JH-H) 2.3 Hz, 1H,Tp), 5.62 (t, JH-H) 2.1 Hz, 1H, Tp), 5.44 (t, JH-H) 2.3 Hz, 1H, Tp), 5.39 (t, JH-H) 2.3 Hz, 1H, Tp), 1.18 (s, 1H, C2PhCHCN).31P NMR (0 °C, acetone): δ 48.8, 48.1 (AB, JP-P) 29.9 Hz). MS (FAB) m/z: 980.4 (M++ 1), 718.3 (M++ 1 - PPh3), 577.2 (M++ 1 - PPh3, C2PhCHCN).
Synthesis of the Cyclopropenyl Complex Tp(PPh3
)-(CH3CN)RuCdC(Ph)CHCN (4). To a solution of 3 (1.00 g, 1.02 mmol) in 20 mL of CH2Cl2was added 10 mL of CH3CN. The solution was stirred for 30 min, the color changed from yellow to brown, and then the solution was filtered through Celite. Solvent of the filtrate was removed under vacuum, and the residue was washed with hexane to give 4 (0.62 g, 80.0% yield). Two diastereomers of 4 in a 4:1 ratio are observed. The minor isomer in solution decomposed after 2 h at room temperature. Spectroscopic data for 4: 1H NMR (acetone) major compound: δ 7.85 (d, JH-H) 2.2 Hz, 1H, Tp), 7.81 (d, JH-H) 2.2 Hz, 1H, Tp), 7.69 (d, JH-H) 2.2 Hz, 1H, Tp), 7.40-7.07 (m, Ph), 6.86 (1H, Tp), 6.78 (1H, Tp), 6.76 (1H, Tp), 6.02 (t, JH-H) 2.2 Hz, 1H, Tp), 5.98 (t, JH-H )2.2 Hz, 1H, Tp), 5.85 (t, JH-H)2.2 Hz, 1H, Tp), 2.22 (s, 1H, CH3CN), 1.08 (s, 1H, C2PhCHCN); minor compound: δ 8.10 (1H, Tp), 7.40-7.07 (m, Ph, Tp, PPh3), 5.74 (1H, Tp), 5.61 (1H, Tp), 5.96 (1H, Tp), 2.29 (s, 1H, CH3CN), 1.20 (s, 1H, C2PhCHCN).13C NMR (CDCl3) major compound: δ 137.9 (d, JP-C) 11.4 Hz, CR), 146.9-105.6 (m, Ph, PPh3, Tp), 123.7 (CH3CN), 114.4 (CN), 3.6 (CH3CN), 2.9 (CH).31P NMR (acetone): δ 56.7, 56.0 (4:1). MS (FAB) m/z: 759.3 (M++ 1), 718.3 (M++ 1 - CH 3CN), 577.1 (M+ + 1 - CH
3CN, C2PhCHCN). Anal. Calcd for C39H33N8 -BPRu (756.56): C, 61.91; H, 4.40; N, 14.81. Found: C, 62.01; H, 4.46; N, 14.50.
Preparation of [Tp(PPh3)2RudCdC(Ph)CH(CPh3
)CN]-PF6(5). To a solid mixture of 3 (0.25 g, 0.26 mmol) and Ph3 -CPF6(0.12 g, 0.3 mmol, in drybox) at 0 °C, was added 30 mL of CH2Cl2. The mixture was stirred for 20 min, and the color changed from yellow to green. Then the solvent was removed under vacuum. The residual solid was washed with 2× 20 mL of benzene and was dried under vacuum to give complex
5 (0.25 g, 69% yield). Spectroscopic data for 5: 1H NMR (acetone): δ 7.92 (d, JH-H) 2.3 Hz, 1H, Tp), 7.83 (d, JH-H) 2.3 Hz, 1H, Tp), 7.71 (d, JH-H) 2.3 Hz, 1H, Tp), 7.4-6.9 (m, Tp, Ph), 6.83 (d, JH-H) 2.3 Hz, 1H, Tp), 5.81 (d, JH-H) 2.2 Hz, 1H, Tp), 5.66 (d, JH-H) 2.2 Hz, 1H, Tp), 5.63 (t, JH-H) 2.2 Hz, 1H, Tp), 5.54 (t, JH-H) 2.2 Hz, 1H, Tp), 4.91 (s, 1H, CH).13C NMR (acetone): δ 371.3 (t, J P-C ) 16.2 Hz, CR), 146.1-106.3 (m, Ph, Tp, CPh3, PPh3,Cβ), 122.3 (CN), 21.2 (CH). 31P NMR (acetone): δ 35.1, 33.6 (AB, J P-P) 26.7 Hz). MS (FAB) m/z: 1222.1 (M+- PF6), 978.8 (M+- PF6,CPh3), 838.8 (M+ - PF6, CPh3, C2PhCHCN), 577.2 (M+ - PF6, CPh3, C2PhCHCN, PPh3). Anal. Calcd for C74H60N7BP3F6Ru (1366.06): C, 65.06; H, 4.43; N, 7.18. Found: C, 65.34; H, 4.41; N, 7.29.
Synthesis of [Tp(PPh3)2RudCdC(Ph)CH(HgCl)CN]Cl
(6). To a solid mixture of 3 (0.51 g, 0.52 mmol) and HgCl2(0.15 g, 0.52 mmol) at 0 °C was added 30 mL of CH2Cl2. The mixture was stirred for 30 min, and the color changed from yellow to green. Then the solvent was removed under vacuum. The residual solid was extracted with 2× 20 mL of ether, and the solvent was removed under vacuum to give complex 6 (0.45 g, 69% yield). Spectroscopic data for 6: 1H NMR (acetone): δ 8.01 (d, JH-H) 2.2 Hz, 1H, Tp), 7.86 (d, JH-H) 2.3 Hz, 1H, Tp), 7.75 (d, JH-H) 2.3 Hz 1H, Tp), 7.49-7.07 (m, Tp, Ph), 6.85 (d, JH-H) 2.2 Hz 1H, Tp), 5.85 (d, JH-H) 2.2 Hz, 1H, Tp), 5.70 (d, JH-H) 2.2 Hz, 1H, Tp), 5.68 (t, JH-H) 2.2 Hz, 1H, Tp), 5.56 (t, JH-H ) 2.2 Hz, 1H, Tp), 3.19 (s, 1H, C2 -PhCHCNHg).13C NMR (CDCl 3): δ 375.1 (t, JP-C) 15.4 Hz, CR), 146.8-107.5 (m, Ph, Tp, PPh3,Cβ), 120.3 (CN), 23.1 (CH). 31P NMR (acetone): δ 35.5, 34.1 (AB, J P-P ) 26.9 Hz). MS (FAB) m/z: 1214.3 (M++ 1 - Cl), 978.8 (M+- Cl, HgCl), 838.8 (M+ - Cl, HgCl, C 2PhCHCN), 577 (M+ - Cl, HgCl, C2 -PhCHCN, PPh3). Anal. Calcd for C55H45N7BCl2HgP2Ru (1249.28): C, 52.87; H, 3.63; N, 7.85. Found: C, 52.42; H, 4.04; N, 8.07.
Synthesis of [Tp(PPh3)(CH3
CN)RudCdC(Ph)CH(HgCI)-CN]Cl (7). To a solid mixture of 4 (0.17 g, 0.17 mmol) and
HgCl2(0.046 g, 0.17 mmol) was added 30 mL of CH2Cl2. The mixture was stirred for 10 min, and then the solvent was removed under vacuum. The residual solid was extracted with 2 × 20 mL of ether, and after filtration, the solvent was removed under vacuum to give 7 (0.12 g, 73% yield). Spectro-scopic data for 7: 1H NMR (acetone): δ 7.78 (d, J
H-H) 2.2 Hz, 1H, Tp), 7.76 (d, JH-H) 2.3 Hz, 1H, Tp), 7.75 (d, JH-H) 2.3 Hz, 1H, Tp), 7.43-7.03 (m, Tp, Ph), 6.40 (d, JH-H) 2.2 Hz, 1H, Tp), 6.33 (br, 1H, Tp), 6.10 (t, JH-H) 2.2 Hz, 1H, Tp), 5.93 (t, JH-H) 2.2 Hz, 1H, Tp), 3.69 (s, 1H, C2PhCHCNHg), 2.45 (s, 3H, CH3CN).13C NMR (acetone): δ 369.3 (d, JP-C) 16.1 Hz, CR), 146.8-107.5 (m, Ph, Tp, PPh3,Cβ), 135.0 (CH3 -CN), 124.6 (-CN), 25.1 (CH), 4.6 (CH3CN).31P NMR (acetone): δ 45.9. MS (FAB) m/z: 994.8 (M+- Cl), 758.8 (M+- Cl, HgCl), 618.4 (M+ - Cl, HgCl, C2PhCHCN), 577.2 (M+- Cl, HgCl, C2PhCHCN, CH3CN).
Reaction of 6 with MeONa. To a solution of 6 (0.43 g,
0.34 mmol) in 20 mL of CH2Cl2at 0 °C was added a solution of MeONa (1 mL, 1 M in MeOH). The mixture was stirred for 10 min, and then the solvent was removed at 0 °C under vacuum, the solid residue was extracted with cool ether, and the extract was filtered through Celite. Solvent of the filtrate was removed under vacuum to give 3 (0.245 g, 71% yield).
Reaction of 7 with MeONa. To a solution of 7 (0.37 g,
0.35 mmol) in 20 mL of CH2Cl2at 0 °C was added a solution of MeONa (1 mL, 1 M in MeOH). The mixture was stirred for 25 min, and the color changed from green to yellow. The solvent was removed at 0 °C under vacuum, the solid residue was extracted with cool ether, and the extract was filtered through Celite. Solvent of the resulting solution was removed under vacuum to give 4 (0.17 g, 63% yield).
Preparation of Tp(PPh3)Ru(C3H3NN)CdC(Ph)CH2CN
(8a). To a solid mixture of 3 (1.02 g, 1.02 mmol) and pyrazole
(0.07 g, 1.02 mmol) was added 30 mL of CH2Cl2. The mixture was stirred for 60 min, and the color changed from yellow to bright yellow. Then the solvent was removed under vacuum. The residual solid was extracted with 2× 20 mL of ether, and the solvent was filtered through Celite. Solvent of the resulting
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
solution was removed under vacuum to give 8a (0.61 g, 79% yield). Spectroscopic data for 8a: 1H NMR (acetone): δ 7.84 (d, JH-H) 2.1 Hz, 1H, Tp), 7.83 (d, JH-H) 2.2 Hz, 1H, Tp), 7.82 (d, JH-H) 2.1 Hz, 1H, TP), 7.43-7.10 (m, Ph, Tp), 7.15 (d, JH-H) 2.2 Hz, 1H, Tp), 7.08 (d, JH-H) 2.2 Hz, 1H, Tp), 7.02 (d, JH-H) 2.1 Hz, 1H, Tp), 6.68 (d, JH-H) 2.1 Hz, 1H, Tp), 6.17 (t, JH-H) 2.2 Hz, 1H, Tp), 5.97 (t, JH-H) 2.1 Hz, 1H, Tp), 5.94 (d, JH-H) 2.2 Hz, 1H, Tp), 5.85 (t, JH-H) 2.2 Hz, 1H, Tp), 5.61 (t, JH-H) 2.1 Hz, 1H, Tp), 3.35 (d, JH-H) 17.3 Hz, 1H, C2PhCHHCN), 3.18 (d, JH-H)17.3 Hz, 1H, C2 -PhCHHCN).13C NMR (acetone): δ 163.3 (d, J P-C) 12.3 Hz), 147.4-127.1 (Ph, PPh3, Tp), 119.6 (CN), 14.1 (C2PhCH2CN). 31P NMR (acetone): δ 61.3. MS (FAB) m/z: 758.2 (M+ + 1), 718.1 (M+- PZ), 577.1 (M+- PZ, C2PhCHCN). Anal. Calcd for C40H34N9BPRu (938.75): C, 61.31; H, 4.37; N, 16.09. Found: C, 61.47; H, 4.44; N, 16.30. An intermediate was observed if the reaction was monitored by NMR spectroscopy within 20 min. Spectroscopic data for A: 1H NMR (acetone):
δ 8.03 (br, 1H, Tp), 7.96 (br, 1H, Tp), 7.83 (br, 1H, Tp), 7.43-7.10 (m, Ph, Tp), 7.65 (br, 1H, Tp), 6.53 (br, 1H, Tp), 6.24 (br, 1H, Tp), 6.03 (br, 1H, Tp), 5.83 (br, 1H, Tp), 5.40 (br, 1H, Tp), 4.40, 4.11 (two d, JH-H ) 18.4 Hz, 2H, CH2CN). 31P NMR (acetone): δ 36.7. Preparation of Tp(PPh3)Ru(Me2C3HNN)CdC(Ph)CH2
-CN (8b). To a solid mixture of 5b (0.40 g, 0.41 mmol) and
3,5-dimethylpyrazole (0.056 g, 0.82 mmol) was added 30 mL of CH2Cl2. The mixture was stirred for 20 min, and then the solvent was removed under vacuum. The residual solid was extracted with 2 × 20 mL of ether, and the solution was filtered through Celite. Solvent of the filtrate was removed under vacuum to give 8b (0.163 g, 49% yield). Spectroscopic data for 8b: 1H NMR (acetone): δ 7.81 (d, J
H-H) 2.0 Hz, 1H, Tp), 7.51 (d, JH-H) 2.0 Hz, 1H, Tp), 7.42 (d, JH-H) 2.0 Hz, 1H, Tp), 7.43-6.84 (m, Ph, Tp), 6.60 (d, JH-H) 2.1 Hz, 1H, Tp), 6.13 (t, JH-H) 2.0 Hz, 1H, Tp), 5.97 (t, JH-H) 2.1 Hz, 1H, Tp), 5.94 (d, JH-H) 2.1 Hz, 1H, Tp), 5.61 (t, JH-H) 2.0 Hz, 1H, Tp), 3.25, 3.17 (two d, JH-H) 17.1 Hz, 2H, CH2CN), 2.38, 2.13 (s, 3H, (CH3)2C3HNN).13C NMR (acetone): δ 162.2 (d, JP-C) 12.6 Hz), 146.1-127.1 (PPh3, Tp, Ph), 118.7 (CN), 15.3 (C2PhCH2CN), 16.8, 15.3 ((CH3)2C3HNN). 31P NMR (acetone): δ 61.0. MS (FAB) m/z: 814.2 (M+ ), 718.1(M+ -Me2C3HNN), 577.1 (M+- Me2C3HNN, C2PhCHCN). Synthesis of Tp(PPh3)(CO)RuC(OMe)dC(Ph)CH2CN
(9). A solution of 3 (1.5 g, 1.53 mmol) was dissolved in
methanol under CO atmosphere. After stirring for 50 min, the yellow solution became brown. The solution was filtered through Celite, and the solvent of the resulting solution was removed under vacuum to give the product 9 (1.01 g, 85% yield). Spectroscopic data for 9: IR (acetone): νCO1957 cm-1 (s).1H NMR (acetone): δ 8.00 (d, J H-H) 1.9 Hz, 1H, Tp), 7.82 (m, 2H, Tp), 7.78 (d, JH-H) 2.2 Hz, 1H, Tp), 7.52-7.03 (m, Ph, Tp), 6.34 (d, JH-H) 1.9 Hz, 1H, Tp), 6.27 (t, JH-H) 2.2 Hz, 1H, Tp), 5.92 (t, JH-H) 2.2 Hz, 1H, Tp), 5.82 (d, JH-H) 1.9 Hz, 1H, Tp), 3.84 (d, JH-H) 16.8 Hz, 1H, C2PhCHHCN), 3.02 (d, JH-H) 16.8 Hz, 1H, C2PhCHHCN), 2.85 (s, 3H, OMe). 13C NMR (acetone): δ 204.2 (d, J P-C) 15.1 Hz, CO), 188.3 (d, JP-C) 14.0 Hz, CR), 146.5-124.5 (Ph), 119.9 (CN), 56.1 (OMe), 22.4 (C2PhCH2CN). 31P NMR (acetone): δ 46.9. MS (FAB) m/z: 777.3 (M+), 605.2 (M+- (OMe)CdC(Ph)(CH 2CN)), 577.1 (M+- (OMe)CdC(Ph)(CH
2CN), -CO). Anal. Calcd for C39H34 -O2N7BPRu (775.56): C, 60.39; H, 4.42; N, 16.09. Found: C, 60.71; H, 4.51; N, 16.41.
Synthesis of [Tp(PPh3)(CO)RudC(OMe)CH(Ph)CH2
CN]-[CF3COO] (10). Complex 9 (0.072 g, 0.093 mmol) was dis-solved in 0.5 mL of CD3C(O)CD3at 0 °C, and CF3COOH (0.03
mL) was added. After 5 min, the solvent was removed under vacuum, and the product was washed with hexane and was identified as 10. Spectroscopic data for 10: IR (acetone): νCO 1998 cm-1(s). 1H NMR (acetone): δ 8.38 (br, 1H, Tp), 8.10 (br, 1H, Tp), 7.98-7.05 (m, Ph, Tp), 6.67 (br, 1H, Tp), 6.35 (br, 1H, Tp), 6.30 (br, 1H, Tp), 6.10 (br, 1H, Tp), 5.51 (br, 1H, Tp), 5.31 (dd, JH-H) 5.4 Hz, JH-H) 10.1 Hz, 1H, CCHPhCH2 -CN), 4.78 (s, 3H, OMe), 3.31 (m, 2H, C2PhCH2CN).13C NMR (acetone): δ 319.9 (d, JP-C) 10.0 Hz, CR), 201.5 (d, JP-C) 12.8 Hz, CO), 162.2 (q, JF-C) 43.1 Hz, CF3COO), 146.5-123.5 (Ph), 118.7 (q, JF-C) 282.0 Hz, CF3COO), 117.5 (CN), 70.4 (OMe), 64.2 (CCPhCH2CN), 29.0 (C2PhCH2CN). 31P NMR (acetone): δ 36.9. MS (FAB) m/z: 778.3 (M+), 605.2 (M+ -(OMe)CH(Ph)(CH2CN)), 577.1 (M+- (OMe)CH(Ph)(CH2CN), -CO). Synthesis of Tp(PPh3)(CO)RuC(O)CH(Ph)CH2CN (11). Complex 9 (0.072 g, 0.093 mmol) was dissolved in 0.5 mL of CD3C(O)CD3at 0 °C, and CF3COOH (0.03 mL) was added. After 36 h, the solvent was removed under vacuum, and the product was washed with hexane and was identified as 11. Spectroscopic data for 11: IR (acetone): νCO1983 cm-1(s).1H NMR (acetone): δ 7.98-7.11 (m, Ph, Tp), 6.49 (br, 1H, Tp), 6.24 (br, 1H, Tp), 6.04 (br, 1H, Tp), 5.99 (br, 1H, Tp), 3.75, 3.21 (m, 2H, CH2CN), 3.21 (m, 1H, C2PhCHHCN), 2.41 (m, 1H, C2HPhCH2CN). 13C NMR (acetone): δ 213.6 (d, JP-C) 13.4 Hz, CR), 207.5 (d, JP-C) 12.6 Hz, CO), 146.5-123.5 (Ph), 119.7 (CN), 54.6 (CCHPhCH2CN), 24.0 (C2PhCH2CN). 31P NMR (acetone): δ 41.3. MS (FAB) m/z: 761.2 (M+), 605.2 (M+ - C(O)CH(Ph)(CH2CN)), 577.1 (M+- C(O)CH(Ph)(CH2CN), -CO). Anal. Calcd for C38H31O2N7BPRu (760.53): C, 60.01; H, 4.11; N, 12.89. Found: C, 59.87; H, 3.95; N, 13.18.
X-ray Analysis. Dark red crystals of 8a suitable for X-ray
diffraction study were grown directly from CH2Cl2. A suitable single crystal of dimensions 0.25× 0.25 × 0.20 mm3was glued to a glass fiber and mounted on an SMART CCD diffractome-ter. The data were collected using 3 kW sealed-tube molyb-denum KR radiation (T ) 295 K). Exposure time was 5 s per frame. Sadabs (Siemens area detector absorption) absorption correction was applied, and decay was negligible. Data were processed and the structure was solved and refined by the SHELXTL program. The structure was solved using direct methods and confirmed by Patterson methods refining on intensities of all data (13 076 reflections) to give R1 ) 0.0426 and wR2 ) 0.0917 for 13076 unique observed reflections (I > 2σ(I)). Hydrogen atoms were placed geometrically using the riding model with thermal parameters set to 1.2 times that for the atom to which the hydrogen is attached and 1.5 times that for the methyl hydrogens.
The procedures for the structure determination of 11 were similar to that of 8a. The final residuals of the refinement R1 (wR2) were 0.042 (0.0956). Final values of all refined atomic positional parameters (with esd’s) and tables of thermal parameters are given in the Supporting Information.
Acknowledgment. We thank the National Science Council, Taiwan, the Republic of China, for support of this work.
Supporting Information Available: Tables of atomic
coordinates, bond lengths and angles, anisotropic thermal parameters, and hydrogen atom positions for 8a and 11. This material is available free of charge via the Internet at http://pubs.acs.org.
OM980752Q
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009