A Four-Gene Signature from NCI-60 Cell Line for Survival Prediction
in Non
–Small Cell Lung Cancer
Yi-Chiung Hsu,
1,3Shinsheng Yuan,
1Hsuan-Yu Chen,
1Sung-Liang Yu,
4Chia-Hsin Liu,
1Pin-Yen Hsu,
3Guani Wu,
1Chia-Hung Lin,
1Gee-Chen Chang,
6,7Ker-Chau Li,
1,8and Pan-Chyr Yang
2,3,5Abstract
Purpose: Metastasis is the main cause of mortality in non–small cell lung cancer (NSCLC) patients. Genes that can discriminate the invasion ability of cancer cells may become useful candidates for clinical outcome prediction. We identify inva-sion-associated genes through computational and laboratorial approach that sup-ported this idea in NSCLC.Experimental Design: We first conducted invasion assay to characterize the invasion abilities of NCI-60 lung cancer cell lines. We then systematically exploited NCI-60 microarray databases to identify invasion-associated genes that showed differential expression between the high and the low invasion cell line groups. Furthermore, using the microarray data of Duke lung cancer cohort (GSE 3141), invasion-associ-ated genes with good survival prediction potentials were obtained. Finally, we vali-dated the findings by conducting quantitative PCR assay on an in-house collected patient group (n = 69) and by using microarray data from two public western co-horts (n = 257 and 186).
Results: The invasion-associated four-gene signature (ANKRD49, LPHN1, RABAC1, and EGLN2) had significant prediction in three validation cohorts (P = 0.0184, 0.002, and 0.017, log-rank test). Moreover, we showed that four-gene signature was an indepen-dent prognostic factor (hazard ratio, 2.354, 1.480, and 1.670; P = 0.028, 0.014, and 0.033), independent of other clinical covariates, such as age, gender, and stage.
Conclusion: The invasion-associated four-gene signature derived from NCI-60 lung cancer cell lines had good survival prediction power for NSCLC patients. (Clin Cancer Res 2009;15(23):7309–15)
L
ung cancer is the most common cause of cancer deaths world-wide and non–small cell lung cancer (NSCLC) is the major dominant cell type (1, 2). For the early-stage lung cancer, sur-gery remains the treatment of choice. But the widespread metas-tasis of lung cancer often defeats this treatment and up to 40% of such patients may relapse within 5 years (3, 4). Therefore, identification of patients at the high risk of recurrence or metas-tasis after resection may help improve the management of NSCLC patients.Migration and invasion ability of cancer cells are the most crit-ical initial events of cancer metastasis (5). It is important to find gene signatures that may distinguish the invasion abilities of cancer cells to invade (6). Many studies on cancer metastasis have identified several molecules participating in tumor cell in-vasion and metastasis in different types of cancers (7). Microar-ray data have been widely used in discovering tumor subtypes, finding molecular characterization about tumor stage and meta-static status, and predicting patient survival (8–10). Various
Authors' Affiliations: Institutes of1
Statistical Science and2
Biomedical Sciences, Academia Sinica;3National Taiwan University Research Center for Medical Excellence-Division of Genomic Medicine; Departments of
4
Clinical Laboratory Sciences and Medical Biotechnology and5Internal Medicine, National Taiwan University College of Medicine, Taipei, Taiwan, Republic of China;6
Division of ChestMedicine, Departmentof Internal Medicine, Taichung Veterans General Hospital;7
Institutes of Biomedical Sciences and Molecular Biology, National Chung Hsing University, Taichung, Taiwan, Republic of China; and8
Department of Statistics, University of California, Los Angeles, Los Angeles, California Received 6/23/09; revised 8/26/09; accepted 8/29/09; published OnlineFirst 11/17/09.
Grant support: NSC97-3112-B-002-033; NSC97-3112-B-002-047-B4 MIB; Institute of Statistical Science, Academia Sinica, Taiwan; NSC97-2627-P-001-003; NSC98-2314-B-001-001-MY3; NSC95-3114-P-002-005-Y;
DOH96-TD-I-111-TM; and National Science Foundation grant DMS-0707160.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Ker-Chau Li, Pan-Chyr Yang and Shinsheng Yuan are co-corresponding authors. The wet lab done in the National Taiwan University and the dry lab done in Academia Sinica contributed equally.
Requests for reprints: Ker-Chau Li, Institute of Statistical Science Academia Sinica, Taipei 115, Taiwan, Republic of China. Phone: 886-2-2783-5611 ext. 207; Fax: 886-2-2783-1523; E-mail: kcli@stat.sinica.edu.tw.
F 2009 American Association for Cancer Research. doi:10.1158/1078-0432.CCR-09-1572
platforms of microarray have been applied to profile the gene expression patterns of the panel of NCI-60 cell lines (11).
We focused on the subset of the nine lung cancer cell lines in NCI-60 gene expression data set and used one set of public microarray data of western lung cancer patients to derive an invasion-associated gene signature for survival prediction. We validated the findings by three independent NSCLC cohorts. Our computational approach integrated both invasion profiles of lung cancer cell lines and gene expression profiles, and our results provided clues to interpret the mechanism of cancer metastasis.
Materials and Methods
Taiwanese cohort: patients and tissue specimen collections. A cohort that consisted of 69 patients from Taiwan was used for quantitative PCR (qPCR) validation conducted in our lab. Frozen specimens of lung cancer tissue came from 69 patients who underwent surgical resection of NSCLC at the Taichung Veterans General Hospital (VGHTC) between December 1999 and December 2003. The cohort included 27 adenocar-cinomas, 36 squamous cell caradenocar-cinomas, and 6 other types of cancer. The patients had not received adjuvant chemotherapy. The study was ap-proved by the institutional review board of the hospital. Written in-formed consent was obtained from all patients.
Cell culture. The nine NCI-60 lung cancer cell lines (A549/ATCC, NCI-H460, NCI-H23, NCI-H322M, EKVX, NCI-H226, NCI-H522, HOP-62, and HOP-92) were purchased from the National Cancer In-stitute. The cells were grown in tissue culture flasks at 37°C in 5% CO2in RPMI 1640 with 2 mmol/LL-glutamine and 10% fetal bovine
serum.
Matrigel invasion assay. The invasion capacities for cell lines were examined by using membrane invasion culture system to confirm their invasion ability, respectively. A HTS FluoroBlok insert contain-ing 8-μm pores (Falcon, Becton Dickinson) was coated with 30 μg Matrigel (BD). The cells were suspended in RPMI 1640 containing 10% fetal bovine serum and seeded into the upper wells of the chamber (2.5 × 104 per well). After incubating for 24 h at 37°C,
the membrane of the Transwell will be fixed for 10 min at room temperature with methanol and stained for 30 min with 50 μg/mL propidium iodide (Sigma) and the cell number in each blot was
counted under a microscope with Analytical Imaging Station system (Imaging Research, Inc.).
Cell line gene expression data. We used two microarray gene expres-sion profiling data sets for NCI-60 cell lines generated by Gene Logic using Affymetrix U95 and U133, respectively. The mapping of probes between U133 and U95 platforms was provided by Affymetrix.9The data sets were downloaded from the National Cancer Institute's Devel-opmental Therapeutics Program Web site.10
Public NSCLC cohorts. The NSCLC cohort we used to derive gene signature consisted of lung cancer patients from the Duke University. Gene expression of 111 lung tumor tissues was profiled by using HU133plus2 Affymetrix chip (12). We retrieved the data from the Gene Expression Omnibus (GSE 3141). Intensity values were log transformed to a base 2 scale.
The two western cohort data sets used for validation came from Shedden et al. (13). The patients were from four institutions, University of Michigan Cancer Center (UM), Moffitt Cancer Center (HLM), Me-morial Sloan-Kettering Cancer Center (MSK), and the Dana-Farber Can-cer Institute (CAN/DF), and divided into two cohorts, UM+HLM and MSK+CAN/DF, as in Shedden et al. We preprocessed the microarray data by normal score transformation.
The real-time reverse transcription-qPCR analysis. The expression of four signature genes and two control genes (TBP for cell lines and GAPDH for tissue specimens) was measured by reverse transcription-qPCR (RT-transcription-qPCR) with specific Taqman probes and primer sets; the transcripts were amplified with Taqman One-Step RT-PCR Master Mix Reagent (Applied Biosystems) and a detection system (ABI Prism 7900HT, Applied Biosystems). Gene expression was quanti-fied in relation to the expression of the respective control gene with the use of sequence detector software and the relative quantification method (Applied Biosystems). The choice of TBP and GAPDH as the internal control for real-time RT-PCR is based on their invari-ance of expression in cinvari-ancer cell lines and clinical cinvari-ancer specimens (14, 15).
Survival analysis. The association between survival and each gene expression profile from microarray is evaluated by univariate Cox pro-portional hazard regression analysis. A linear combination of the gene expression levels weighted by the regression coefficients was used to calculate the risk score for each patient.
More specifically, a patient's risk score was calculated as the sum of the levels of expression of each gene measured by microarray multiplied by the corresponding Cox regression coefficients. Patients were classified as having a high-risk signature or a low-risk gene signature, with the median of risk score as the threshold value. Sur-vival curves for both groups were obtained by the Kaplan-Meier method and compared using the log-rank test. Multivariate Cox pro-portional hazard regression analysis with stepwise selection was used to evaluate independent prognostic factors associated with pa-tient survivals, taking the gene signature, age, sex, stage, and histol-ogy as the covariates.
Except for the two places noted below, all tests were two sided, and a P value of <0.05 was considered statistically significant. One-sided tests were used in showing the consistency between qPCR data and the mi-croarray data (‡P values; Table 1), and in one-sided log-rank test (P va-lues of UM+HLM and MSK+CAN/DFcohorts) where the hypothesis of interest is the one-sided hypothesis that the survival time is longer for patients with the low-risk gene signature than that for patients with the high-risk signature, note the two-sided hypothesis that the survival time is different between the low-risk signature and the high-risk signature (16, 17).
Translational Relevance
Metastasis is the most life-threatening factor of malignantcancers. The migration and invasion abil-ity of cancer cells is the first critical factor of metas-tasis. Because most treatment failure and death in cancer patients are due to the highly metastatic po-tential of the neoplasm, finding biomarkers that may distinguish the invasive ability of different cancer cells can lead to the discovery of new gene signature to improve the prediction of patient survival. We identify novel invasion-associated genes from the nine lung cancer cell lines in the NCI-60 panel and validate the results thrice: twice with large-scale western lung cancer cohorts and once by our own oriental data. Our study may provide clues to the mechanism of cancer metastasis and pave ways for effective clinical outcome prediction of lung cancer. 9 http://www.affymetrix.com/support/technical/comparison_spreadsheets. affx 10 http://dtp.nci.nih.gov/mtargets/download.html
Results
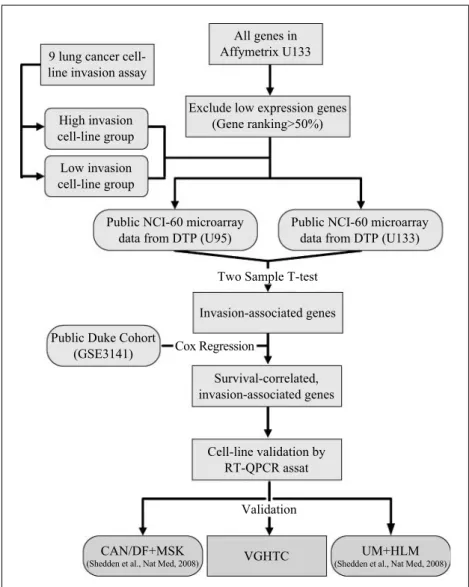
Strategy of in silico microarray approach to derive invasion-associated gene signature for outcome prediction of NSCLC patients. The strategy of our approach is illustrated in Fig. 1. Based on invasion ability of the nine lung cancer cell lines, we
divided them into two groups. We identified the invasion-associated genes from two Affymetrix NCI-60 microarray gene expression data sets by finding the genes with differential expression between the high and the low invasion groups. To reduce effect of microarray noises, we filtered out 50% of the probes with low average expression readings in data preprocessing.
Table 1. Four-gene signature from nine lung cancer cell lines predicts survival of 111 NSCLC patients
Gene symbol UniGene ID Probe ID (P)* Correlation coefficient (P)† HR‡(P)§ 95%CI of HR
ANKRD49 Hs.29052 219069_at (0.015) 0.571 (0.050) 0.578 (0.002) 0.410-0.818 RABAC1 Hs.11417 203136_at (0.005) 0.806 (0.004) 1.510 (0.020) 1.067-2.137 LPHN1 Hs.654658 219145_at (0.029) 0.728 (0.013) 0.775 (0.033) 0.614-0.980 EGLN2 Hs.515417 220956_s_at (0.048) 0.711 (0.016) 1.910 (0.041) 1.027-3.552 *P value was based on Student's t test for differential expression between the high and the low invasion cell lines at the given probe ID used in the gene; U133plus2 microarray data were used.
†P values for the correlation coefficients were given to test if qPCR and the microarray data for the lung cancer cell lines have positive correlation
or not.
‡The HRs are reported for the high-risk signature versus the low-risk signature, as determined by training Duke cohort. §Estimated by univariate Cox regression analysis in training Duke cohort.
Fig. 1. Schematic diagram for deriving the gene signature. For details, see Materials and Methods and Results. DTP, Developmental Therapeutics Program.
To examine which genes from our cell line–based search may lead to useful clinical biomarkers, we obtained a large-scale mi-croarray data set for tumor samples of western NSCLC patients together with patients' survival data from Gene Omnibus (GSE 3141), referred to as the Duke cohort (n = 111; ref. 12). We ap-plied the univariate Cox proportional hazard method to each of the invasion-associated genes obtained from cell line assay for finding genes whose expressions across patients correlate well with patient survival. Because genes with higher (lower) expres-sion levels for high invaexpres-sion cell lines are expected to be risk (protective) genes, we further inspect the direction of correla-tion with survival to identify genes consistent with their inva-sion abilities. For the genes that bypassed the consistency checking, we confirmed their expression pattern on nine cell lines by qPCR assay on the nine cell lines. Finally, we validate thisin silico–derived gene signature in three independent co-horts: two new U133plus2 microarray data of western NSCLC cohort (CAN/DF+MSK,n = 186; UM+HLM, n = 257; ref. 13) and a group of 69 Taiwanese NSCLC samples (VGHTC,n = 69) by qPCR assay.
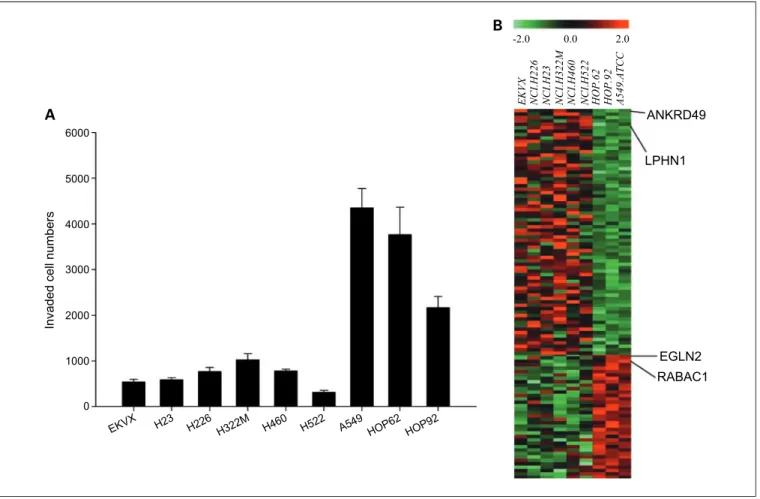
Invasion profile and in silico microarray analysis in lung cancer panel of NCI-60 cell lines. We determined the inva-sion ability of nine human NSCLC cell lines in NCI-60 (Fig. 2A) and found three cell lines, A549, HOP-62, and HOP-92, with much higher invasion ability than the other six cell lines (Fig. 2A). To find invasion-associated genes
from the U133plus2 NCI-60 microarray data set, gene ex-pression profiles for the high and the low invasion groups were compared by two-tailed t tests and confirmed with the U95 NCI-60 microarray data set. A total of 110 genes were found differentially expressed and their expression profiles formed two clusters (see Fig. 2B, wherein the top panel showed the expression profiles of 72 genes that correlated negatively with the invasive ability of lung cancer cell lines, whereas the bottom panel gave the expression profiles of 38 genes that correlated positively).
Survival-associated gene signatures. The 110 genes associat-ed with the invasion activity were identifiassociat-ed byin vitro invasion assay on NSCLC cell lines andin silico microarray analysis. How do they affect the clinical outcome of lung cancer patients? To investigate this issue, we take advantage of the availability of the Duke lung cancer cohort (12) and evaluated the correlation between the expression of each gene and the patient survival by univariate Cox proportional hazard regression. This analysis was done to determine which genes may be associated with pa-tient death. Protective genes have hazard ratio (HR) for death of <1, whereas risk genes have HR for death of >1. Of the 110 in-vasion-associated genes, 9 genes were found statistically signif-icant. We further checked if the protective/risk gene assignment was consistent with the direction of the invasion ability of the gene and found five genes consistent. One gene probe was lo-cated at a pseudogene site, which was not considered further.
Fig. 2. A, in vitro invasion activity profile of nine lung cancer cell lines in NCI-60. Columns, mean of the data (n = 3); bars, SD. B, heatmap of 110 gene expression profiles showing the differential expression between lowly invasive (group I) and highly invasive (group II) lung cancer cell lines.
For the remaining four genes,ANKRD49 and LPHN1 are protec-tive andRABAC1 and EGLN2 are risk genes (Table 1).
Risk scores were calculated by using the above four genes. Risk prediction was calculated as the sum of the levels of expres-sion of each gene measured by microarray, multiplied by the corresponding Cox regression coefficients. Patients in high- or low-risk group were classified by using the 50th percentile (me-dian) of the risk score distribution (median, 3.84; range, 2.5-6.68) as the cut point.
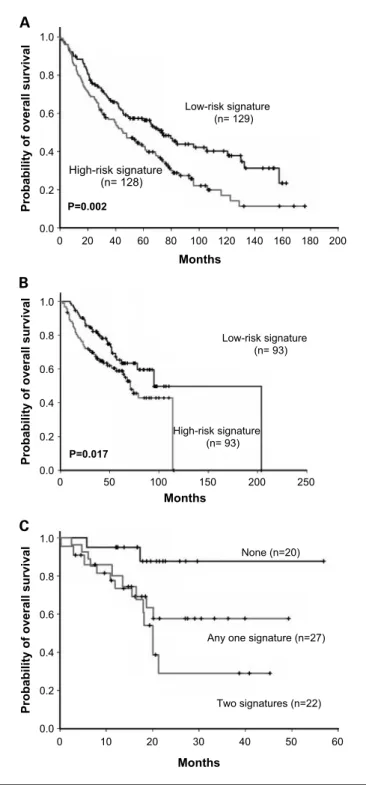
The Kaplan-Meier method was used to estimate overall surviv-al distribution for each group. Patients with high-risk gene sig-nature as predicted by the four-gene sigsig-nature had shortened median overall survival compared with patients with low-risk gene signature (24.33 versus 36.59 months;P = 0.001, log-rank test; Fig. 3A). The expression levels of these four candidate genes in the nine lung cancer cell lines were validated by doing the RT-qPCR assay and calculating the Pearson correlation between the qPCR data with the U133plus2 microarray data (Table 1).
Clinical characteristics of the NSCLC cohort are summarized according to their high or low risk of the four-gene signature (Supplementary Table S1). Multivariate Cox regression analysis with stepwise selection was used to search for independent prognostic factors associated with survival. The risk score by
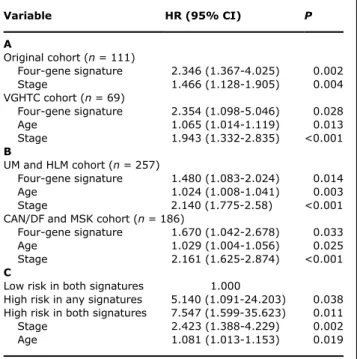
the four-gene signature was evaluated along with age, gender, tumor stage, and cell type. Patients with the high-risk signature had a higher risk for poor survival with adjusted HR of 2.35 [95% confidence interval (95% CI), 1.367-4.025;P = 0.002]. The four-gene signature was still a significant survival predictor after adjusting for tumor stage (Table 2A).
Independent validation of gene signature from Taiwanese data set by RT-qPCR and public western data set. To investigate to what extent the four genes were also valuable predictors for the Asian patients, we investigated the VGHTC cohort. We took lung cancer specimens from each patient and quantified the gene expression of four candidate genes by RT-qPCR to compute the patient risk scores for each patient and then di-vided patients into high- and low-risk groups in the same way as was done in the Duke cohort. We found that Kaplan-Meier survival curves for the high- and low-risk groups in the VGHTC cohort were separable and the result was statis-tically significant (P = 0.0184, log-rank test; F ig. 3B). To show that inclusion of the four-gene signature in the clinical model had useful survival prediction performance indepen-dent of cancer stage, we conducted a multivariate Cox regres-sion with backward deletion. From the initial set of five variables (age, gender, stage, cell type, and four-gene signa-ture), the final model contains only age, stage, and four-gene signature (Table 2A).
Moreover, we validated the four-gene signature in two new western cohorts (UM+HLM and CAN/DF+MSK). Kaplan-Meier survival curves were significantly separable (UM+HLM: P = 0.002, log-rank test; CAN/DF+MSK: P = 0.017, log-rank test; Fig. 4A and B), and four-gene signature was significant by multivariate Cox regression in both cohorts (Table 2B).
Fig. 3. Survival distributions of 111 patients in the Duke NSCLC cohort (A) and 69 patients in the VGHTC NSCLC cohort (B).
Table 2. Multivariate Cox regression analysis
Variable HR (95%CI) P A Original cohort (n = 111) Four-gene signature 2.346 (1.367-4.025) 0.002 Stage 1.466 (1.128-1.905) 0.004 VGHTC cohort (n = 69) Four-gene signature 2.354 (1.098-5.046) 0.028 Age 1.065 (1.014-1.119) 0.013 Stage 1.943 (1.332-2.835) <0.001 B UM and HLM cohort (n = 257) Four-gene signature 1.480 (1.083-2.024) 0.014 Age 1.024 (1.008-1.041) 0.003 Stage 2.140 (1.775-2.58) <0.001 CAN/DF and MSK cohort (n = 186)
Four-gene signature 1.670 (1.042-2.678) 0.033 Age 1.029 (1.004-1.056) 0.025 Stage 2.161 (1.625-2.874) <0.001 C
Low risk in both signatures 1.000
High risk in any signatures 5.140 (1.091-24.203) 0.038 High risk in both signatures 7.547 (1.599-35.623) 0.011 Stage 2.423 (1.388-4.229) 0.002 Age 1.081 (1.013-1.153) 0.019
NOTE: Four-gene signature for (A) Duke and VGHTC and (B) UM +HLM and CAN/DF+MSK NSCLC patients. (C) Multivariate Cox re-gression of concomitant four-gene and five-gene signatures as prognostic predictors in overall survival of VGHTC patients. Vari-ables were selected through stepwise selection method.
This reassures the clinical usefulness of our finding. The four invasion-associated genes might be potential molecular targets for lung cancer therapy.
Discussion
Many studies have developed useful gene signatures by ex-amining only the gene profiling data of the patient specimens
that they can access to (18–20). Differing from these works, our approach started with anin silico exploitation of the pub-lic domain microarray databases to derive the gene signature and concluded with the validation by our own clinical data as well as two public western clinical data. The public data we used in this report included two sets of NCI-60 cell line mi-croarray gene expression data and three mimi-croarray gene ex-pression data sets from a western patient cohort. We also conducted qPCR in our lab. As the genomic research gets dee-per and broader, it has become more and more difficult for any research team to generate all sorts of data on their own. Consequently, it is worth investigating methods of how to use the public data such as Gene Expression Omnibus, which is growing at an astonishingly fast pace. More and more such in silico microarray and bioinformatic approaches are antici-pated to foster the area of clinical genomics.
Previously, a five-gene signature (9), motivated by different cell line invasion assay and microarray platform, was also ob-tained for NSCLC. Although there is no overlap between the four genes identified in this article and the five-gene signa-ture, both achieve the same goal. Both signatures had signif-icant prediction power in multivariate Cox regression. Using both signatures, we stratified the VGHTC cohort into three groups and generated Kaplan-Meier plots to show their sur-vival differences (Fig. 4C). The combined signature is signif-icant under multivariate Cox regression with stage, gender, and age (Table 2B).
Among the four genes in our invasion-associated signature, the expression of RABAC1 and EGLN2 was found higher in high invasion lung cell lines, whereas that of LPHN1 and ANKRD49 was found higher in low invasion lung cell lines. RABAC1 (Rab acceptor 1) is a transmembrane protein, local-ized to the late Golgi, which also called PRA1 (21). It was involved with T-cell factor/β-catenin signaling and dephos-phorylation of extracellular signal-regulated kinase 1/2 (22). RABAC1 gene was highly expressed in breast, colon, lung, and ovary cancers (23). Interestingly, RABAC1 activated NF-κB signaling through latent membrane protein 1, which is an EBV-encoded oncoprotein (24). Moreover, infection of EBV was associated with several cancers (25), consistent with risk effect of RABAC1 in our signature. The EGLN2 was known as prolyl-hydroxylase-1, which acted like an oxygen sensor to inhibit hypoxia-induced activation of hypoxia-inducible factor-1α (26, 27). EGLN2 was also characterized as estrogen-induced tag-6, which has been reported to promote cell growth in breast cancer (28). It is likely that the function of EGLN2 in tumor tissues may be dependent on the oxygen level. LPHN1 encoded a member of the latrophilin 1, which was a member of G protein–coupled receptors associated with activation of neurons (29). Latrophilin 1 regulated cell adhesion and neurotoxin secretion (30) and brain-specific angiogenesis inhibitor 1. Brain-specific angiogenesis inhibitor 1 was widely expressed in both glial cells and neurons but has low expression in glioma cell lines (31). The function of ANKRD49 (ankyrin repeat domain 49) is little known. However, ankyrin repeat, a motif of 30– to 34–amino acid residues (32), was first identified in the sequence of yeast Swi6p, Cdc10p, and Notch (33), and Notch signaling is one important pathway in tumorigenic processes. Families of ankyrin repeat proteins mediating protein-protein interac-tion have been associated with development of human
Fig. 4. Survival distributions of 257 patients in the UM+HLM cohort (A) and 186 patients in the CAN/DF+MSK cohort (B). C, 69 patients in the VGHTC NSCLC cohort using the concomitant four-gene signature and five-gene signature.
cancer (34). The tumor suppressor genep16 (INK4A) was an ankyrin repeat protein that inhibited cell cycle progression to reduce proliferation of cancer cells (35).
We concluded that an invasion-associated gene signature de-rived from NCI-60 lung cancer cell lines by ourin silico micro-array approach had good survival prediction power in NSCLC patients. The signature requires only four genes. For clinical ap-plications, the smaller number of genes in the signature may have the greater potential of translating into practical methods
of lung cancer patient risk stratification and developing person-alized therapy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Hao Ho for contribution in data assembly.
References
1. Parkin DM. Global cancer statistics in the year 2000. LancetOncol 2001;2:533–43.
2. Jemal A, Siegel R, Ward E, et al. Cancer statis-tics, 2008. CA Cancer J Clin 2008;58:71–96. 3. Hoffman PC, Mauer AM, Vokes EE. Lung cancer.
Lancet2000;355:479–85.
4. Mountain CF. Revisions in the International Sys-tem for Staging Lung Cancer. Chest 1997;111: 1710–7.
5. Chambers AF, Groom AC, MacDonald IC. Dis-semination and growth of cancer cells in meta-static sites. Nat Rev Cancer 2002;2:563–72. 6. Steeg PS. Tumor metastasis: mechanistic
in-sights and clinical challenges. Nat Med 2006; 12:895–904.
7. Steeg PS, Bevilacqua G, Kopper L, et al. Evi-dence for a novel gene associated with low tu-mor metastatic potential. J Natl Cancer Inst 1988;80:200–4.
8. Potti A, Mukherjee S, Petersen R, et al. A ge-nomic strategy to refine prognosis in early-stage non-small-cell lung cancer. N Engl J Med 2006; 355:570–80.
9. Chen HY, Yu SL, Chen CH, etal. A five-gene sig-nature and clinical outcome in non-small-cell lung cancer. N Engl J Med 2007;356:11–20. 10. Roepman P, Jassem J, SmitEF, etal. An
im-mune response enriched 72-gene prognostic profile for early-stage non-small-cell lung can-cer. Clin Cancer Res 2009;15:284–90.
11. Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006;6:813–23.
12. Bild AH, Yao G, Chang JT, etal. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006;439:353–7. 13. Shedden K, Taylor JM, Enkemann SA, etal.
Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded vali-dation study. Nat Med 2008;14:822–7.
14. Shih JY, Yang SC, Hong TM, etal. Collapsin response mediator protein-1 and the invasion and metastasis of cancer cells. J Natl Cancer Inst 2001;93:1392–400.
15. Kurahashi T, Hara I, Oka N, Kamidono S, Eto H, Miyake H. Detection of micrometastases in pel-vic lymph nodes in patients undergoing radical cystectomy for locally invasive bladder cancer by real-time reverse transcriptase-PCR for cyto-keratin 19 and uroplakin II. Clin Cancer Res 2005;11:3773–7.
16. Green S, Benedetti J, Crowley J. Interdisciplin-ary statistics: clinical trials in oncology. 2nd ed. Boca Raton: Chapman & Hall/CRC; 2003. 17. Koch GG, Gillings DB. Encyclopedia of
statisti-cal sciences. New York: Wiley; 1988.
18. Raponi M, Zhang Y, Yu J, etal. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res 2006;66:7466–72.
19. Beer DG, Kardia SL, Huang CC, etal. Gene-expression profiles predictsurvival of patients with lung adenocarcinoma. NatMed 2002;8:816–24. 20. Lu Y, Lemon W, Liu PY, etal. A gene
expres-sion signature predicts survival of patients with stage I non-small cell lung cancer. PLoS Med 2006;3:e467.
21. Abdul-Ghani M, Gougeon PY, Prosser DC, Da-Silva LF, Ngsee JK. PRA isoforms are tar-geted to distinct membrane compartments. J Biol Chem 2001;276:6225–33.
22. Kim JT, Cho MY, Choi SC, etal. Prenylated Rab acceptor 1 (PRA1) inhibits TCF/β-catenin signaling by binding toβ-catenin. Biochem Biophys Res Commun 2006;349:200–8.
23. Fo CS, Coleman CS, Wallick CJ, Vine AL, Bachmann AS. Genomic organization, expres-sion profile, and characterization of the new pro-tein PRA1 domain family, member 2 (PRAF2). Gene 2006;371:154–65.
24. Liu HP, Wu CC, Chang YS. PRA1 promotes the intracellular trafficking and NF-κB signaling of EBV latent membrane protein 1. EMBO J 2006; 25:4120–30.
25. Thompson MP, Kurzrock R. Epstein-Barr virus and cancer. Clin Cancer Res 2004;10:803–21. 26. Kaelin WG. Proline hydroxylation and gene
ex-pression. Annu Rev Biochem 2005;74:115–28.
27. Erez N, Milyavsky M, Eilam R, Shats I, Goldfinger N, Rotter V. Expression of prolyl-hydroxylase-1 (PHD1/EGLN2) suppresses hypoxia inducible factor-1α activation and inhibits tumor growth. Cancer Res 2003;63:8777–83.
28. Seth P, Krop I, Porter D, Polyak K. Novel estro-gen and tamoxifen induced estro-genes identified by S A G E ( S e r i a l A n a l y s i s o f G e n e E x p r e s‐ sion). Oncogene 2002;21:836–43.
29. Krasnoperov V, Lu Y, Buryanovsky L, Neubert TA, Ichtchenko K, Petrenko AG. Post-translational proteolytic processing of the cal-cium-independentreceptor of α-latrotoxin (CIRL), a natural chimera of the cell adhesion protein and the G protein-coupled receptor. Role of the G protein-coupled receptor prote-olysis site (GPS) motif. J Biol Chem 2002;277: 46518–26.
30. Bjarnadottir TK, Fredriksson R, Schioth HB. The adhesion GPCRs: a unique family of G pro-tein-coupled receptors with important roles in both central and peripheral tissues. Cell Mol Life Sci 2007;64:2104–19.
31. Kaur B, BratDJ, Calkins CC, Van Meir EG. Brain angiogenesis inhibitor 1 is differentially expressed in normal brain and glioblastoma in-dependently of p53 expression. Am J Pathol 2003;162:19–27.
32. Sedgwick SG, Smerdon SJ. The ankyrin re-peat: a diversity of interactions on a common structural framework. Trends Biochem Sci 1999; 24:311–6.
33. Breeden L, Nasmyth K. Similarity between cell-cycle genes of budding yeastand fission yeast and the Notch gene of Drosophila. Nature 1987;329:651–4.
34. Li J, Mahajan A, Tsai MD. Ankyrin repeat : a unique motif mediating protein-protein interac-tions. Biochemistry 2006;45:15168–78. 35. Lin YC, Diccianni MB, Kim Y, etal. Human p16γ,
a novel transcriptional variant of p16(INK4A), co-expresses with p16(INK4A) in cancer cells and in-hibits cell-cycle progression. Oncogene 2007;26: 7017–27.