行政院國家科學委員會專題研究計畫 成果報告
應用寡核
酸微陣列的技術開發嚴重急性呼吸道症候群
(Severe acute respiratory syndrome,SARS)及相關非典
型病毒之快速檢測法
計畫類別: 個別型計畫 計畫編號: NSC92-2751-B-002-003-Y 執行期間: 92 年 07 月 01 日至 93 年 11 月 30 日 執行單位: 國立臺灣大學醫學院內科 計畫主持人: 楊泮池 共同主持人: 白果能 報告類型: 完整報告 處理方式: 本計畫可公開查詢中 華 民 國 94 年 3 月 17 日
應用寡核苷酸微陣列的技術開發嚴重急性呼吸道症候群
及相關非典型病毒之快速檢測法
The rapid and reliable method for Severe Acute Respiratory
Syndrome (SARS) and related viral atypical pathogen detection by
oligonucleotide microarray
楊泮池/白果能 台大醫學院/中央研究院
ABSTRACT
Most methods for viral disease diagnosis are geared towards specific detection of a single or several known targets. This project aims to develop viral conserved sequences at genus level and establish a library of viral probes for discriminating not only known vertebrate viruses but also emerging and uncharacterized vertebrate viruses based on the sequenced viral genomes and oligonucleotide microarray technology. Several viral pathogens were successfully identified by 70-mer virus oligonucleotide test array to demonstrate the feasibility of the methodology. The viral conserved and specific sequence database covering 30 vertebrate viral families, 97 genera and 624 species is constructed and available on line at the website of http://genestamp.sinica.edu.tw/virus/index.htm.
INTRODUCTION
Viral infections have remained as major health problems in the world in spite of all the medical advances in the past century. New viral infections, such as the SARS, continue to emerge. Most methods for viral disease diagnosis are geared towards specific detection of a single target. When presented with a symptomatic patient with unknown etiology, a number of assays are often executed in parallel to reach a final diagnosis, which is a time-consuming and labor-intensive procedure. Recently DNA microarray (chip) permits simultaneous monitoring and analysis of a large number of viral pathogens in one hybridization reaction (Wang et al., 2002). The method selected ten highly homologous 70mer sequences from each virus in a viral genus as microarray probes and therefore sacrifices the diagnostic capacity of specific viral targets. Here we present an alternative and improved methodology to simultaneously identify uncharacterized viruses at genus level and specific viruses at species or subtype level. Finally this project aims to establish a library of viral detection probes based on the sequenced viral genomes and oligonucleotide microarray technology for detection of a wide variety of viruses.
ALGORITHM
More than 3,000 completely sequenced viral genomes covering 30 vertebrate viral families, 97 genera and 624 species were obtained from NCBI RefSeq and GenBank databases. Viral genomes with sequence similarity more than 99.5% were merged together and the resulting 2,641 non-redundant genomes were used for the following analysis. The procedure for identifying conserved sequences of a viral genus to serve as microarray probes is depicted in Figure 1. For a given genus of viruses, each viral genome sequence was aligned with other genomes by the BLASTN program. Any segment of the viral genome having 75% local sequence similarity in a 50 base window with any other genome was considered significantly homologous (Kane et al., 2000). All the segments with significant homology to the maximum number of other genomes were extracted from each viral genome in the genus. Only three homologous segments at most were reserved for each virus according to the segment length ranking. Only three 70mer probes at most with 40-60% of GC content were selected from each reserved segment based on the following criterion: one 70mer probe selected for the segment length of 70-140 nt; two probes for 140-280 nt segments; and three probes for segments longer than 280 nt. We then merged all the 70mer candidate probes with sequence similarity larger than 75% if needed to limit the number of total 70mer probes within 30. The resulting 70mer probes are the conserved sequence set, which guaranties to identify each viral genome in the genus.
In order for in silico validation of the methodology, SARS Coronavirus (SARS-CoV) was removed from the genus of Coronavirus and treated as a prediction genome. Other viral genomes, the so-called calibration genome set, were served to choose the conserved sequence set based on the aforementioned algorithm. The resulting non-redundant conserved sequences were then aligned with SARS-CoV genome. One of the conserved sequence set had 80% sequence homology to SARS-CoV genome (Fig. 2), which demonstrates the feasibility of the conserved sequence searching approach.
EXPERIMENTAL VERIFICATION
To experimentally verify the approach of probe design, a test virus chip with 128 70mer oligonucleotides covering 26 specific viral species (Table 1) and 3 viral genera (Coronavirus, Flavivirus, and Enterovirus) were construct. The 70mer probes of specific viruses were designed using the algorithm as described in Chang and Peck (2003). One mock HeLa cell culture and six viral genomes (SARS-CoV, Japanese encephalitis virus (JEV), Dengue virus II/III/IV and Enterovirus 71) extracted from
infected cell cultures were individually labeled with Cy-3 fluorescent dye and hybridized to the oligonucleotide array. The viral sample treatment and microarray hybridization protocol were conducted as described (Wang et al., 2002). A procedure was established for automatic virus identification based on the microarray data without visualization. We first estimated the distribution of non-target signal (background) intensities from all 128 spots of the mock chip plus the intersection of non-conserved spots and non-specific spots of all the other seven chips. Totally we included 780 non-target signal spots from all eight chips. We then used the 99.5th percentile of the background signal distribution (that is 1768) as the cut-off signal intensity for later virus classification. The above 8 chip data were converted to a color visualization scheme for better explanation (Fig. 3). Any spot signal intensity larger than the cut-off value on a chip was considered non-background, caused by target
virus hybridization, and labelled with a solid-cycle spot on the chip. Given the hybridization result of one particular virus chip, the decision rule for viral identification is described as follows: (i) Normalize all the non-background conserved spots for each of the three genera: norm(Flavivirus), norm(Coronavirus), and
norm(Enterovirus), where norm(x) is the ratio of the number of non-background x-genus spots divided by total number of x-genus spot; (ii) The sample is classified to
be x-genus virus if norm(x) ≥ 0.1; (iii) The specific virus can be identified from what virus the non-background spot in the specific virus probe region belongs to; (iv) If there are multiple specific viruses identified, pick the one with highest average signal intensity. The decision rule resulted in perfect match of all the 8 chips in viral genus classification and of 7 chips in specific virus classification except for the JEV chip (Fig. 1E) which came with cross-hybridization to 3 non-target Flavivirus_viruses: Louping ill virus, West Nile virus and Murray Valley encephalitis virus. The signal ratios of JEV to the three viruses were 5.2, 30.6, and 30.3, respectively. Therefore the virus was correctly classified as JEV. Besides, only one of two SARS-CoV specific probes in the SARS-CoV chip produced detectable hybridization signal (Fig. 3F). To avoid the oligonucleotide sequence-dependent effect (Chou et al., 2004), we designed 5 specific probes for each virus in our database. It is noteworthy that two viral infections from different viral genera can be accurately detected at one hybridization reaction (Fig. 3H). Additionally, due to the lack of complete sequenced
Figure 2. In silico validation of the conserved sequence design methodology. (A)
Through the multiple sequence alignment by ClustalW, the genus of Coronavirus without SARS-CoV genome had four main non-redundant conserved sequence sets. (B) BLAST result displays that the 1st conserved sequence set has high sequence similarity with SARS-CoV genome.
genome, Enterovirus 71 was not contained in our viral database for the aforementioned analysis. However, the virus was still detected and identified as the Enterovirus genus (Fig 3G), which fully demonstrates the feasibility of the viral probe design algorithm and decision rule.
REFERENCES
Chang,P.-C. and Peck, K. (2003) Design and assessment of a fast algorithm for identifying specific probes for human and mouse genes. Bioinformatics, 19, 1311-1317.
Chou,C.-C., Chen,C.-H., Lee,T.-T. and Peck,K. (2004) Optimization of probe length and the number of probes per gene for optimal microarray analysis of gene expression. Nucleic Acids Res., 32, e99.
Kane,M.D., Jatkoe,T.A., Stumpf,C.R., Lu,J., Thomas,J.D. and Madore,S.J. (2000) Assessment of the sensitivity and specificity of oligonucleotide (50mer) microarrays. Nucleic Acids Res. 28, 4552-4557.
Figure 3. Test virus chip layout and multiple viral detection results. The digital
numbers represent different specific viruses. Please refer to Table 1 for detailed virus information.
Wang,D., Coscoy,L., Zylberberg,M., Avila,P.C., Boushey,H.A., Ganem,D. and DeRisi,J.L. (2002) Microarray-based detection and genotyping of viral pathogens.
Proc. Natl. Acad. Sci. USA, 99, 15687-15692
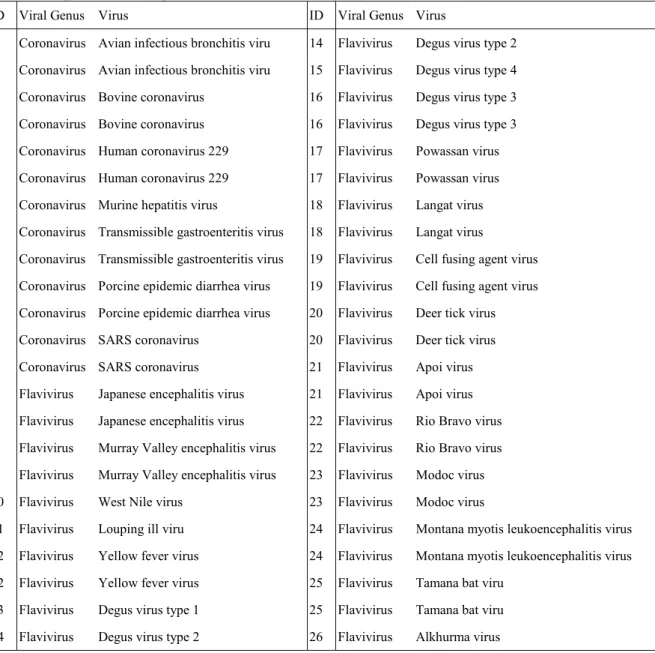
Table 1. Specific viral species in the 70-mer oligonucleotide test array. ID Viral Genus Virus ID Viral Genus Virus
1 Coronavirus Avian infectious bronchitis viru 14 Flavivirus Degus virus type 2 1 Coronavirus Avian infectious bronchitis viru 15 Flavivirus Degus virus type 4 2 Coronavirus Bovine coronavirus 16 Flavivirus Degus virus type 3 2 Coronavirus Bovine coronavirus 16 Flavivirus Degus virus type 3 3 Coronavirus Human coronavirus 229 17 Flavivirus Powassan virus 3 Coronavirus Human coronavirus 229 17 Flavivirus Powassan virus 4 Coronavirus Murine hepatitis virus 18 Flavivirus Langat virus 5 Coronavirus Transmissible gastroenteritis virus 18 Flavivirus Langat virus
5 Coronavirus Transmissible gastroenteritis virus 19 Flavivirus Cell fusing agent virus 6 Coronavirus Porcine epidemic diarrhea virus 19 Flavivirus Cell fusing agent virus 6 Coronavirus Porcine epidemic diarrhea virus 20 Flavivirus Deer tick virus 7 Coronavirus SARS coronavirus 20 Flavivirus Deer tick virus 7 Coronavirus SARS coronavirus 21 Flavivirus Apoi virus
8 Flavivirus Japanese encephalitis virus 21 Flavivirus Apoi virus 8 Flavivirus Japanese encephalitis virus 22 Flavivirus Rio Bravo virus 9 Flavivirus Murray Valley encephalitis virus 22 Flavivirus Rio Bravo virus 9 Flavivirus Murray Valley encephalitis virus 23 Flavivirus Modoc virus 10 Flavivirus West Nile virus 23 Flavivirus Modoc virus
11 Flavivirus Louping ill viru 24 Flavivirus Montana myotis leukoencephalitis virus 12 Flavivirus Yellow fever virus 24 Flavivirus Montana myotis leukoencephalitis virus 12 Flavivirus Yellow fever virus 25 Flavivirus Tamana bat viru
13 Flavivirus Degus virus type 1 25 Flavivirus Tamana bat viru 14 Flavivirus Degus virus type 2 26 Flavivirus Alkhurma virus