Deprotonation Pathway in the Reaction of Me
6
Si
2
with
MeLi
Ling-Kang Liu*
,†,‡and Lung-Shiang Luh
†Institute of Chemistry, Academia Sinica, Nankang, Taipei, Taiwan 11529, ROC,
and Department of Chemistry, National Taiwan University, Taipei, Taiwan 10767, ROC
Received October 13, 1999
Summary: A mixture of 3 mol of Me6Si2
and 1 mol of
MeLi in the presence of P(O)(NMe2)3
produced Me3SiLi
as the initial product. This then deprotonated excess Me
6-Si2
and started an unprecedented transformation
lead-ing to (Me3Si)2(SiMe2H)CLi (3), whose quench by
[(η
5-C5H5)Fe(CO)2PPh3
+] produced
{
η
4-exo-[(Me3Si)2C-(SiMe
2H)]C
5H
5}
Fe(CO)
2(PPh
3) (2e). In the literature the
reaction of Me6Si2
and MeLi gives Me3SiLi and/or
Me3-SiSiMe2Li (4). The lithium compound 3 is the major
product when a previously unnoticed deprotonation
pathway is enhanced by use of an excess of Me6Si2
and
a longer reaction time.
Organosilanes have been used to enhance reactivity
and selectivity in chemical transformations.
1In one
synthetic approach, the addition of silyl anions to a
variety of organic electrophiles results in formation of
the needed Si-C bond.
2Among such silyl anions, R
3
-SiLi can be generated in situ by reaction of R
3SiCl with
Li, of (R
3Si)
2Hg with Li, or of R
3SiSiR
3with R
′
Li.
3We
have used the electrophile [(η
5-C
5
H
5)Fe(CO)
2PPh
3+] (1)
to quench the generated silyl anions. The results
revealed an unprecedented conversion of Me
3SiLi to
(Me
3Si)
2(SiMe
2H)CLi and gave retro-chemical evidence
for a deprotonation pathway in the Me
6Si
2reaction with
MeLi.
The R
3SiLi anions were generated by reaction of the
respective disilane with MeLi. The resulting solutions
were cooled to -78 °C and transferred dropwise by
cannula to a solution of 1:1 (η
5-C
5
H
5)Fe(CO)
2I/PPh
3in
THF at -78 °C, the practical equivalent to [1][I], after
chemical initiation with a trace of lithiated reagent,
4which in the present cases is the first few drops of R
3-SiLi. The color of the solution changed gradually from
black to orange-red during the addition of R
3SiLi,
sometimes with formation of a yellow precipitate that
redissolved as the reaction proceeded. For the silyl
anions with at least one aryl group, the Cp ring
silylation products (η
4-exo-R
3
SiC
5H
5)Fe(CO)
2(PPh
3) (R
3) Ph
3(2a, 51%), MePh
2(2b, 36%), Me
2Ph (2c, 45%))
were isolated as major products after column
chroma-tography.
5Thus, the Si-based nucleophiles add at the
Cp ring of 1, similar to what occurred with C-based
nucleophiles
6and different from O-based nucleophiles
* To whom correspondence should be addressed. E-mail: liuu@ chem.sinica.edu.tw.
†Academia Sinica.
‡National Taiwan University.
(1) (a) Fleming, I. In Comprehensive Organic Chemistry; Barton, E., Ollis, W. E., Eds.; Pergamon Press: Oxford, U.K., 1979. (b) Colvin, E. Silicon in Organic Synthesis; Butterworth: Boston, 1981. (c) Weber, W. P. Silicon Reagents in Organic Synthesis; Springer-Verlag: New York, 1983. (d) Patai, S., Rappoport, Z., Eds. The Chemistry of Organic Silicon Compounds; Wiley: New York, 1989. (e) Hwu, J. R.; Wang, N. Chem. Rev. 1989, 89, 1599. (f) Rappoport, Z.; Apeloig, Y., Eds. The Chemistry of Organic Silicon Compounds; Wiley: New York, 1998; Vol. 2.
(2) Oshima, K. In Advances in Metal-Organic Chemistry; Liebeskind, L. S., Ed.; JAI Press: London, 1991; Vol. 2, pp 101-141.
(3) (a) Tamao, K.; Kawachi, A. Adv. Organomet. Chem. 1998, 38, 1 and references cited herein. (b) Wiberg, E.; Stecher, O.; Andrascheck, H. J.; Kreubichler, L.; Staude, E. Angew. Chem., Int. Ed. Engl. 1963, 2, 507. (c) Fujita, M.; Hiyama, T. J. Synth. Org. Chem. Jpn. 1984, 42, 293. (d) Vyazankin, N. S.; Razuvaev, G. A.; Gladyshev, E. A.; Korneva, S. P. J. Organomet. Chem. 1967, 7, 353. (e) Gladyshev, E. N.; Fedorova, E. A.; Yuntila, L. A.; Razuvaev, G. A.; Vyazankin, N. S. J. Organomet. Chem. 1975, 96, 169.
(4) Gipson, S. L.; Liu, L.-K.; Soliz, R. U. J. Organomet. Chem. 1996, 526, 393.
(5) Manipulations were carried out under N2with dry degassed reagents. Preparation of 2a (typical): a 100 mL two-necked flask was charged with Ph3SiCl (5 mmol), and fine-cut Li wire (20 mmol) and then THF (30 mL) was added. The solution became turbid after stirring for several minutes and the color changed gradually from yellow to brown to black. After it was stirred for 6 h, the resulting solution was cooled to -78 °C and filtered through a pad of Celite. The filtrate was added dropwise to a mixture of (η5-C
5H5)Fe(CO)2I (3 mmol) and PPh3 (3 mmol) in THF (100 mL), also at -78 °C. The color of solution changed from black to orange-red during the addition, accompanied by the formation of a yellow precipitate, which redissolved when the addition was completed. The reaction mixture was quenched with H2O (200 mL) and extracted with Et2O (100 mL× 2) after it was stirred overnight. The organic layers were combined, dried over MgSO4, and then evaporated to dryness under vacuum. The oily residue was purified by SiO2column chromatography with 1/15-20 EtOAc/hexane as eluent to give yellow-orange 2a (51%). IR (CH2Cl2): νCO1962 (s), 1903 (s) cm-1.1H NMR (C
6D6): δ 2.71 (b, 2H), 3.90 (b, 1H), 5.10 (b, 2H), 6.96-7.53 (m, 30H). 31P NMR (C
6D6): δ 72.1 (s). 29Si NMR (C6D6): δ -22.6 (s). FAB MS: m/z 698 (M+). Anal. Calcd for C43H35 -FeO2PSi: C, 73.92; H, 5.05. Found: C, 73.60; H, 4.99. 2b: yield 36%. IR (CH2Cl2): νCO1963 (s), 1902 (s) cm-1.1H NMR (C6D6): δ 0.32 (s, 3H), 2.57 (b, 2H), 3.51 (b, 1H), 5.18 (b, 2H), 6.97-7.52 (m, 25H).31P NMR (C6D6): δ 71.6 (s).29Si NMR (C6D6): δ -17.3 (d, JPSi) 10.0 Hz). FAB MS: m/z 636 (M+). Anal. Calcd for C
38H33FeO2PSi: C, 71.70; H, 5.23. Found: C, 71.58; H, 5.22. Orange side product (η5-C
5H5 )Fe(CO)C-(O)SiMePh2(PPh3): yield 12%. IR (CH2Cl2): νCO1906 (s), 1574 (m) cm-1.1H NMR (C 6D6): δ 1.22 (s, 3H), 4.07 (s, 5H), 6.96-7.77 (m, 25H). 31P NMR (C 6D6): δ 75.7 (s).29Si NMR (C6D6): δ -36.5(s). Anal. Calcd for C38H33FeO2PSi: C, 71.70; H, 5.23. Found: C, 71.87; H, 5.15. 2c: yield 45%. IR (CH2Cl2): νCO1960 (s), 1901 (s) cm-1.1H NMR (C6D6): δ -0.04 (s, 6H), 2.46 (b, 2H), 3.02 (b, 1H), 5.18 (b, 2H), 6.95-7.48 (m, 20H).31P NMR (C
6D6): δ 71.8 (s).29Si NMR (C6D6): δ -11.5 (d, JPSi) 8.0 Hz). FAB MS: m/z 574 (M+). Anal. Calcd for C
33H31FeO2PSi: C, 69.00; H, 5.44. Found: C, 69.16; H, 5.33.
(6) (a) Liu, L.-K.; Luh, L.-S. Organometallics 1994, 13, 2816. (b) Luh, L.-S.; Liu, L.-K. Bull. Inst. Chem., Acad. Sin. 1994, 41, 39. (c) Liu, L.-K.; Luh, L.-S.; Chao, P.-C.; Fu, Y.-T. Bull. Inst. Chem., Acad. Sin.
1995, 42, 1. (d) Luh, L.-S.; Eke, U. B.; Liu, L.-K. Organometallics 1995,
14, 440.
374
Organometallics 2000, 19, 374-376
10.1021/om9908175 CCC: $19.00 © 2000 American Chemical Society Publication on Web 01/21/2000
Downloaded by NATIONAL TAIWAN UNIV on September 10, 2009 | http://pubs.acs.org
which react at the CO ligand.
7The single-crystal X-ray
structure of 2b confirmed that the MePh
2Si group is exo
to Fe (Figure 1),
8indicative of a direct silyl attack.
Compounds 2a-c were desilylated upon acidification
with HBF
4(aq) or HCl(aq) to re-form the cationic
com-pound [1][X] (X ) BF
4, Cl), as shown by IR and
1H and
31P NMR spectroscopic studies.
Me
3SiLi was prepared by treating Me
6Si
2with MeLi
in the presence of P(O)(NMe
2)
3for 15 min at 0 °C.
9The
reagent solution was then quenched with 1:1 (η
5-C
5H
5)-Fe(CO)
2I/PPh
3at -78 °C in THF, with the expectation
that
{
η
4-exo-(Me
3
Si)C
5H
5}
Fe(CO)
2(PPh
3) (2d) would be
formed. IR monitoring of the reaction solution indicated
a quantitative formation of an η
4product. However,
after column chromatography, only a low-yield product
(<10%) was obtained with spectroscopic data as follows
(cf. 2a-c): IR ν
COstretching bands at 1967 (s) and 1908
(s) cm
-1, a
31P NMR resonance (C
6
D
6) at δ 76.1 (s), and
1H NMR resonances (C
6
D
6) at δ 2.58 (b, 2H), 3.65 (b,
1H), and 5.03 (b, 2H). Nevertheless, the proton
integra-tion for silyl-Me was not correct, nor was the elemental
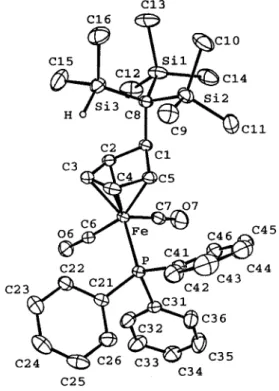
analysis. A single-crystal X-ray analysis confirmed the
structure as being
{
η
4-exo-[(Me
3
Si)
2C(SiMe
2H)]C
5H
5}
-Fe(CO)
2(PPh
3) (2e), which is actually a Cp ring
alkyla-tion product (Figure 2).
10It seemed possible that 2d might have been produced
as the initial product but was desilylated or decomposed,
since 2a-c are acid-sensitive. However, the generation
of Me
3SiLi by the literature procedure
9must have also
produced, at the same time, a small amount of (Me
3-Si)
2(SiMe
2H)CLi (3) before the quench (vide infra). We
suggest that the in situ generation of deep red Me
3SiLi,
from MeLi and Me
6Si
2, is facile because a strong
nucleophilic base easily breaks the Si-Si bond.
11When
the reaction is carried out on a larger scale, the C-Si
bond in Me
6Si
2is often observed to be cleaved in a side
reaction, which leads to the formation of Me
3SiSiMe
2Li
(4)
12(Scheme 1).
13Obviously, the negative charge on
the silicon atom is stabilized by the second silicon atom
in the R-position.
14Still another pathway that has not
(7) Liu, L.-K.; Eke, U. B.; Mesubi, M. A. Organometallics 1995, 14, 3958.
(8) Crystal data for 2b: C38H33FeO2PSi, triclinic, P1h, a ) 9.619(1) Å, b ) 10.424(1) Å, c ) 16.566(3) Å, R ) 87.22(1)°, β ) 77.06(1)°, γ ) 82.19(1)°, V ) 1603.5(3) Å3, Z ) 2, D
calcd) 1.318 g/cm3, 4739 reflections (I > 2.0 σ(I)), 389 parameters, R ) 0.031, Rw) 0.039, GOF ) 1.92.
(9) Still, W. C. J. Org. Chem. 1976, 41, 3063.
(10) Preparation of 2e. A solution of Me3SiSiMe3 (5 mmol) in anhydrous P(O)(NMe2)3(4 mL) was cooled to 0 °C. MeLi (4 mmol) was added via syringe, and the resulting deep-red solution was stirred for 15 min. THF (30 mL) was added, and the solution was cooled to -78 °C. The solution was transferred dropwise via cannula into the mixture of (η5-C
5H5)Fe(CO)2I (3 mmol) and PPh3(3 mmol) in THF (100 mL) at -78 °C. The color of solution gradually changed from black to orange, accompanied by the formation of a yellow precipitate that redissolved when the addition was completed. The solution was warmed to room temperature and stirred overnight before the mixture was quenched with H2O (200 mL) and extracted with Et2O (100 mL × 2). The combined organic layers were dried over MgSO4and then evaporated to dryness under vacuum. The oil-like residue was purified by SiO2 column chromatography with 1:12 (v/v) EtOAc/hexane as eluent to give yellow-orange 2e (6%). Improved procedure: The treatment of Me3SiSiMe3(15 mmol) in anhydrous P(O)(NMe2)3(12 mL) with MeLi (5 mmol) and stirring for 2 h, (otherwise the same procedure as above) resulted in 2e (60%). IR (CH2Cl2): νCO1967(s), 1908(s) cm-1.1H NMR (C6D6): δ 0.14 (s, 18H), 0.21 (d,3JHH= 4 Hz, 6H), 2.58 (b, 2H), 3.65 (b, 1H), 4.30 (hept,3J HH= 4 Hz, 1H), 5.03 (b, 2H), 6.98-7.53 (m, 15H). 31P NMR (C 6D6): δ 76.1(s).29SiNMR (C6D6): δ -15.9 (s), -16.5 (s). FAB MS m/z: 656 (M+). Anal. Calcd for C34H45FeO2PSi3: C, 62.18; H, 6.90. Found: C, 61.99; H, 6.81. Crystal data of 2e: C34H45FeO2PSi3, triclinic P1h, a ) 11.283(2) Å, b ) 12.591(2) Å, c ) 14.276(3) Å, a ) 66.63(1)°, β ) 72.17(2)°, γ ) 76.70(1)°, V ) 1758.5(5) Å3, Z ) 2, D
calcd ) 1.240 g/cm3, 5238 reflections (I > 2.5σ(I)), 374 parameters, R ) 0.033, Rw) 0.045, GOF ) 2.31.
(11) Marschner, C. Eur. J. Inorg. Chem. 1998, 221.
(12) (a) Hudrlik, P. F.; Waugh, M. A.; Hudrlik, A. M. J. Organomet. Chem. 1984, 271, 69. (b) Nadler, E. B.; Rappoport, Z. Tetrahedron Lett.
1990, 31, 555. (c) Allred, A. L.; Smart, R. T.; Van Beek, D. A., Jr.
Organometallics 1992, 11, 4225. (d) Hwu, J. R.; Wetzel, J. M.; Lee, J. S.; Butcher, R. J. J. Organomet. Chem. 1993, 453, 21. (e) Krohn, K.; Khanbabaee, K. Angew. Chem., Int. Ed. Engl. 1994, 33, 99.
(13) It is believed that the C-based nucleophile MeLi is transformed to the Si-based nucleophile Me3SiLi before other second-stage reactions. As an indirect clue, the formation of 4 was only observed in large-quantity preparations.
(14) Armitage, D. A. In Comprehensive Organometallic Chemistry: The Synthesis, Reactions and Structures of Organometallic Compounds; Wilkinson, G., Stone, F. G. A., Abel, E. W., Eds.; Pergamon Press: Oxford, U.K., 1982; Vol. 2, pp 1-203.
Figure 1. Molecular plot of 2b.
Figure 2. Molecular plot of 2e.
Communications
Organometallics, Vol. 19, No. 4, 2000
375
Downloaded by NATIONAL TAIWAN UNIV on September 10, 2009 | http://pubs.acs.org
yet been reported for Me
6Si
2is deprotonation of a
methyl substituent, which results in a lithiated
carban-ion Me
3SiSiMe
2CH
2Li (5) (Scheme 2). The R-SiMe
2group stabilizes the polar C-Li bond. The β-SiMe
3group, however, destabilizes the C-Li bond (a β-Si atom
normally stabilizes a carbonium ion).
14Thus,
intramo-lecular Me
3Si group migration
15from the silicon atom
to the carbon atom results, followed by a 1,2-proton
shift. This gives the lithiated carbanion Me
3Si(SiMe
2H)-CHLi (6), which is isomeric with 5 and is stabilized by
two R-silyl groups. An extra 1 equiv of Me
6Si
2is
attacked by 6 to cleave the Si-Si bond and regenerate
Me
3SiLi, which deprotonates the (Me
3Si)
2(SiMe
2H)CH
thus formed to give the final lithiated species, 3, which
is stabilized by three R-silicon atoms. The nucleophilic
alkylation of the Cp ring of 1 by 3 affords 2e.
16As the characteristic deep red color of Me
3SiLi in
solution is clearly observed, the base effecting
deproto-nation in Scheme 2 must be Me
3SiLi. Thus, it takes
overall 3 mol of Me
6Si
2in order for 1 mol of MeLi to
produce 1 mol of 3; therefore, an increase in
stoichio-metric ratio between Me
6Si
2and MeLi should favor the
deprotonation pathway. The isolated yield of 2e was
improved to 60% when a 3:1 mixture of Me
6Si
2/MeLi
was allowed to react for a longer time (2 h), resulting
in a color change from deep red to orange before the
quench. To our knowledge, this is the first example of
the transformation of a Me anion to a silyl anion and
then back to a carbanion, starting with a simple
disilane. The present deprotonation pathway in the
reaction of Me
6Si
2with MeLi is intermolecular. The
known intramolecular transfer of the organolithium
function in 1-Me
3Si-8-Li-C
10H
6to form 1-Me
2SiCH
2Li-C
10H
717is a similar process (C
10H
6) 1,8-disubstituted
naphthalene skeleton; C
10H
7) 1-substituted
naphtha-lene skeleton).
The speculative mechanism shown in Scheme 2 was
tested with different organic electrophiles in order to
provide evidence that 3 actually is formed under the
reaction conditions. When a 3:1 mixture of Me
6Si
2/MeLi
was quenched with Me
3SiCl, for instance, the expected
(Me
3Si)
3CSiMe
2H
18could be isolated (ca. 30%, not
optimized) (
29Si NMR (C
6
D
6) δ -16.4 (SiMe
3) and -16.1
(SiMe
2H);
1H NMR (C
6D
6) δ 0.24 (s, SiMe
3, 27H), 0.29
(d,
2J
HH
) 4.0 Hz, SiMe
2H, 6H), 4.31 (hept,
2J
HH) 4.0
Hz, SiMe
2H, 1H)). Spectroscopic evidence for the
forma-tion of Me
3SiH also was obtained. In a sealed NMR tube
experiment, a 3:1 mixture of Me
6Si
2/MeLi with
P(O)-(NMe
2)
3in d
8-THF gave
1H NMR peaks at δ 4.61 (hept,
2J
HH
) 4.0 Hz), assigned to the unique SiH of 3, and at
δ 4.00 (decatet,
2J
HH
) 4.0 Hz), assigned to the unique
SiH of HSiMe
3, in the correct molar ratios. The
corre-sponding
29Si NMR data were δ -28.5 (SiMe
3) and
-27.9 (SiMe
2H) for 3 and δ -16.1 for Me
3SiH.
In conclusion, the reaction of Me
6Si
2and MeLi results
in Me
3SiLi (and 4), plus the previously unnoticed 3. The
latter is the more important product when excess Me
6-Si
2is used.
Acknowledgment. Thanks are due to the National
Science Council of the ROC for financial support, to Mr.
Yuh-Sheng Wen for X-ray data collection, and to Prof.
Jih-Ru R. Hwu and Prof. Hiromi Tobita for fruitful
discussions.
Supporting Information Available: Details of the single-crystal structure analyses for 2b,e including tables of posi-tional parameters and bond lengths and angles. This material is available free of charge via the Internet at http://pubs.acs.org. OM9908175
(15) (a) Brook, A. G.; Bassindale, A. R. In Rearrangements in Ground and Excited States; De Mayo, P., Ed.; Academic Press: New York, 1980; Vol. 2, pp 149-227. (b) Eisch, J. J.; Tsai, M.-R. J. Organomet. Chem.
1982, 225, 5.
(16) Occasionally{η4-exo-[(Me
3Si)CH(SiMe2H)]C5H5}Fe(CO)2(PPh3), the speculative Cp-ring alkylation product of 6 and 1, could be detected in trace amount in the1H NMR spectrum of 2e. The pure complex has not been isolated for complete characterization.
(17) Wroczynski, R. J.; Baum, M. W.; Kost, D.; Mislow, K.; Vick, S. C.; Seyferth, D. J. Organomet. Chem. 1979, 170, C29.
(18) Dua, S. S.; Eaborn, C.; Happer, D. A. R.; Hopper, S. P.; Safa, K. D.; Walton, D. R. M. J. Organomet. Chem. 1979, 178, 75.
Scheme 1
Scheme 2
376
Organometallics, Vol. 19, No. 4, 2000
Communications
Downloaded by NATIONAL TAIWAN UNIV on September 10, 2009 | http://pubs.acs.org