251 Acta Cardiol Sin 2013;29:251-260 Basic Science
Fenofibrate Modulates HO-1 and Ameliorates
Endothelial Expression of Cell Adhesion
Molecules in Systolic Heart Failure
Wei-Hsian Yin,1,2 Jaw-Wen Chen,3,4 Yung-Hsiang Chen6 and Shing-Jong Lin3,5Background: Endothelial activation and dysfunction have been implicated in the pathogenesis and progression of
heart failure (HF). In the present study, we investigated if endothelial expression of cell adhesion molecules (CAMs) is inhibited by fenofibrate, a peroxisome proliferator-activated receptor (PPAR) agonist with anti-inflammatory and vascular protective effects, through the regulation of heme oxygenase-1 (HO-1).
Methods: We recruited a total of 20 patients with advanced systolic HF and 20 healthy volunteers who all provided
blood samples. Cultured human pulmonary artery endothelial cells (HPAECs) were treated with 70% sera obtained from study individuals, with or without pretreatment with fenofibrate. The endothelial expression of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and HO-1 were analyzed by mRNA expression and Western blot.
Results: Stimulation of cultured HPAECs with serum from HF patients significantly activated nuclear factor-B
(NF-B) and increased VCAM-1 and ICAM-1 expression but attenuated HO-1 expression. Immunohistochemistry study also confirmed that CAMs were up-regulated, whereas HO-1 was down-regulated in HF patients. HO-1 small interfering RNA significantly suppressed HO-1 expression and exaggerated the HF serum-induced CAM expression, whereas HO-1 inducer cobalt protoporphyrin IX simultaneously stimulated HO-1 expression and suppressed CAM expression. Pretreatment with fenofibrate prevented the decrease of HO-1 expression and the activation of NF-B as well as the increase of CAM expression that induced by HF patient serum.
Conclusions: Our study demonstrated that fenofibrate may exert beneficial effects in patients with systolic HF
through regulation of HO-1 expression and amelioration of endothelial activation.
Key Words: Cell adhesion molecules ﹒ Endothelial cells ﹒ Heart failure ﹒ Heme oxygenase-1 ﹒ Peroxisome proliferator-activated receptor-
INTRODUCTION
Received: July 18, 2012 Accepted: September 18, 2012 1
Heart Center, Cheng-Hsin General Hospital; 3 2Faculty of Medicine, Endothelial activation and dysfunction have been implicated in the pathogenesis and progression of heart National Yang-Ming University; Department of Medical Research failure (HF).1-5 Markers of endothelial activation (se-and Education, Taipei Veterans General Hospital; 4Institute of
Pharmacology; 5Institute of Clinical Medicine, School of Medicine, National Yang-Ming University, Taipei; 6Graduate Institute of Integrated Medicine, School of Chinese Medicine, China Medical University, Taichung, Taiwan.
Address correspondence and reprint requests to: Dr. Shing-Jong Lin, Department of Medical Research and Education, Taipei Veterans General Hospital, No. 201, Section 2, Shih-Pai Rd., Pei-Tou, Taipei 11212, Taiwan. Tel: 886-2-2875-7511; Fax: 886-2-2826-1242; E-mail: sjlin@vghtpe.gov.tw
lectins and cell adhesion molecules) and endothelial leukocyte adhesiveness were significantly increased in patients with advanced HF and those who develop pul-monary edema.6-11 Significantly higher cell adhesion molecule (CAM) levels and endothelial leukocyte adhe-siveness were associated with adverse clinical outcomes in HF patients.6-8,10,11
Expression of heme oxygenase-1 (HO-1) in the en- dothelium plays a cardioprotective role in HF.12-16 Heart failure may cause increased protein expression and en-zymatic activity of HO-1
in the lung.17
Furthermore, HO-1 may inhibit the expression of CAMs associated with endothelial activation and suppress endothelial cell apoptosis.18,19
Peroxisome proliferator-activated receptor (PPAR) deficiency may lead to impaired functional capacity of the heart.20-22
Recent studies
demonstrated numerous pleiotropic effects of
fenofibrate, a
PPAR activator, on the heart that afford direct myocardial protection besides its lipid lowering effects.23-30 Our previous study also demonstrated that fenofibrate can inhibit endothelial monocyte adhesion in HF through inhibition of cytokine- induced CAM expression,31 suggesting that fenofibrate may ameliorate vascular
inflammation and
endothelial dysfunction and exert beneficial effects in HF patients.
It is well-known that many CAMs possess the regu- latory sequences for the binding of nuclear factor-kappa B (NF-B), activator protein-1
(AP-1), and/or AP-2 in their promoter regions, just as HO-132 and HO-1 can in-hibit the expression of pro-inflammatory genes associ- ated with endothelial activation.18 Furthermore, HO-1
expression is
transcriptionally up-regulated by PPARs.33 Therefore, in the present study, we sought to determine the effect of fenofibrate on the possible regulatory role of endothelial HO-1 in
mediating the
expression of CAMs and the functional importance of this process in patients, which have not previously been well studied.
METHODS
Study population
A total of 20 consecutive outpatients with stable advanced chronic HF were recruited from Cheng-Hsin General Hospital. Patients were included if they had New York Heart Association functional class II or III symptoms of HF, and a lef ventricular ejection fraction (LVEF) of < 35% by echocardiography. The etiology of HF was determined as ischemic when coronary angio-graphy revealed > 70%
luminal diameter
narrowing in at least 2
arteries. In those pa-tients with HF without coronary artery disease for whom the endomyocardial biopsy revealed findings compatible
with dilated
cardiomyopathy, the cause of HF was deter- mined to be dilated cardiomyopathy.
Patients were
excluded if they had hemodynamically
significant obstructive valvular heart disease, cor pul- monale,
restrictive or
hypertrophic cardiomyopathy,
myocarditis, constrictive pericarditis or congenital heart disease. Patients were excluded if there was severe comorbidity, or if there was evidence of systemic in- fection or an inflammatory illness. Those patients taking non-steroid anti-inflammatory drugs, antioxidants, L- arginine, or lipid-lowering agents such as statins and fibrates were also excluded.
Twenty
age-matched healthy
subjects provided blood samples for use as normal controls. No subject in the control group had any clinical signs or symptoms of HF and their LVEFs were all > 50% by echocardiography.
Written informed consent was obtained from all partici- pants, and the study protocol was approved by the local ethics committee.
Blood sampling and isolation of human
Blood samples were obtained from the forearm using standard vein puncture. All samples were placed immediately on ice, centrifuged at 4 C within 2 hours, and then frozen to -20 C and stored at that tempera- ture until use. The time intervals between blood sam-pling and LVEF studies were all within one week.
Measurement of circulating levels of CAMs
Assays for
circulating vascular cell adhesion mole- cule-1
(VCAM-1) and
intercellular adhesion molecule-1 (ICAM-1)
were done
concurrently to
minimize any effects of repeated freeze-thaw cycles. The levels of VCAM-1 and ICAM-1 were measured by means of en- zyme-linked immunosorbent assay by commercial kits (R&D Systems, Inc., Minneapolis, MN, USA
for ICAM-1 and
Biosource International,
Camarillo, CA, USA for VCAM-1). The intra-assay
and inter-assay
coefficients for VCAM-1 and ICAM-1 in our
laboratory were
approxi- mately 5% and
Culture of HPAECs
Human pulmonary artery endothelial cells (HPAECs, Cascade Biologics, Portland, OR, USA) were grown in en-dothelial cell growth medium (medium 200, Cascade Biologics) in a poly-L-lysine-coated culture dish in a
hu-al. midified incubator with
5% CO2 at 37 C. The culture me- dium was renewed every 3 to 4 days. In all experiments, the cell passage number was between 3 and 6.
Isolation of total RNA and real-time PCR
Total RNA was isolated using a RNeasy Mini Kit and a RNase-free DNase set (Qiagen, Valencia, CA, USA). RNA (2 g) was reverse-transcribed using the SuperScriptTM First-Strand Synthesis System for reverse transcription polymerase chain reaction kit (Invitrogen, Carlsbad, CA,
USA). The primers for real-time PCR were designed us- ing Primer Express sofware (Real Quant, Roche) based on published sequences. The following primers were used in the present study: human glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) 5’-AGC CAC ATC GCT CAG ACA-3’ (sense) and 5’-GCC CAA TAC GAC CAA ATC C-3’ (antisense); human VCAM-1 5’-AGG GGA CCA CAT CTA CGC T-3’ (sense) and 5’-ACA GAG CTC CCA TTC
ACG A-3’ (antisense); and human ICAM-1 5’-GGC AAG AAC CTT ACC CTA CG-3’ and 5’-GAG ACC TCT GGC TTC
GTC AG-3’ (antisense). Procedures included an
initial de- naturation at 94 C for 180 s, followed by 40 cycles at 95
C for 30 s, 60 C for 25 s, 72 C for 30 s, and 1 cycle at 72 C for 7 min. Fluorescence data were acquired at the end of amplification. A melt analysis was run for all products to determine the specificity of the amplifica- tion using the Real Quant sofware (Roche). All values were normalized to the constitutive expression of the housekeeping gene GAPDH.
Western blot analysis
The cell lysate was prepared using a cell lysis buffer (Cell Signaling Technology, Beverly, MA, USA) and West- ern blot analyses were performed. Briefly, the cell lysate (25 to 40 g) was subjected to 12% sodium dodecyl sul-fate polyacrylamide gel electrophoresis gel electropho- resis and
transferred onto
polyvinylidene difluoride (PVDF) membranes, which was then blotted. Afer being blocked with 5% skim milk in Tween-20/phosphate- buffered saline (PBS), blots were incubated with various primary antibodies, including anti-human VCAM-1, ICAM-1, HO-1, and -tubulin (Chemicon, Temecula, CA, USA). Blots were then incubated with
the horseradish
peroxidase-conjugated secondary antibodies. The signal was detected using Chemiluminescence Reagent Plus (NEN, Boston, MA, USA). The intensity of each band was
scanned and quantified using a densitometer linked to computer sofware (ImageQuant; Amersham, Amer- sham, UK).
Nuclear extracts preparation and electrophoretic
mobility shif assay (EMSA)
The following are protocols for nuclear protein ex- tracts preparation. Briefly, afer washing with PBS, the cells were scraped off the plates in 0.6 ml of ice-cold buffer A 10 mM N-(2-hydroxyethyl) piperazine-N’-(2-ethenesulfonic acid) (HEPES), pH 7.9, 10 mM KCl, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluo- ride (PMSF), 1.5 mM MgCl2, and 2 g/ml each of ap- rotinin, pepstatin, and leupeptin. Afer centrifugation at 300 g for 10 min at 4 C,
the cells were
resuspended in buffer B (80 l of 0.1% Triton X-100 in buffer A), lef on ice for 10 min, then centrifuged at 12,000 g for 10 min at 4 C. The nuclear pellets were resuspended in 70 L of ice-cold buffer C (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 1 mM DTT, 0.2 mM EDTA, 1 mM PMSF, 25% glycerol, and 2 g/ml each of
aprotinin,
pepstatin, and
leupeptin), then incubated for 30 min at 4 C, followed by centrifugation at 15,000 g for 30 min at 4 C.
The resulting
supernatant was stored at -70 C as the nuclear extract. Protein concentrations were determined by the Bio-Rad method.
EMSA was
performed according to DIG gel shif kit instruction manual (Roche, USA). Gel shif oligonucle- otides were labeled with terminal transferase and DIG- 11-dUTP (Roche, USA).
Immunohistochemical staining
Human coronary arteries of 3 recipient hearts ob- tained from
3 dilated
cardiomyopathy
patients under- went heart transplantation (2 males and one female, aged 25, 48 and 52 years, respectively) and one carotid artery from one healthy male donor (aged 18 years)
were used in
immunohistochemical study.
The vessels were rinsed with ice-cold PBS, immer- sion-fixed
in 4% buffered
paraformaldehyde, paraffin- embedded, and then
cross-sectioned for further study. The arterial
sections were
deparaffinized,
rehydrated, and washed with PBS, then non-specific binding was blocked by pre-incubation for 1 h at room temperature with PBS containing 5 mg/ml of bovine serum albumin. Sequential serial sections were incubated with goat
anti-human von Willebrand factor (vWF, a marker of en- dothelial integrity), HO-1, VCAM-1, and ICAM-1 primary antibody (R&D systems, U.S.A.). The sections were then incubated with biotinylated conjugated horse anti-goat IgG for 1 h at room temperature and antigen-antibody complexes detected by incubation with avidin-biotin- horseradish peroxidase complex for 1.5 h at room tem-perature, followed by 0.5 mg/ml of 3,3- diaminoben-zidine/0.01% hydrogen peroxide in 0.1 M Tris-HCl buffer, pH 7.2, as chromogen (Vector Lab, USA). Negative controls were performed by non-specific primary antibody IgG.
The effects of small interfering RNA (siRNA) for HO-1 and HO-1 inducer cobalt protoporphyrin-IX
(COPPIX) on endothelial activation
The siRNA
nucleotide sequences for human HO-1 and control GAPDH were designed and synthesized us- ing the computer sofware and SilencerTM siRNA con- struction kit from Ambion (Austin, TX, USA) according to the manufacturer’s
instructions. HPAECs grown in
100-mm dishes were
transfected with HO-1 or GAPDH siRNA at selected concentrations with the use of Oligofec- tamine reagent (Invitrogen) in a total transfection vo-lume of 6 ml medium. Afer incubation at CO2 at 37 C, 5% CO2 for 5 hours, 3 ml of normal growth medium was added and incubated with HPAECs for 48 hours.
Exposure of HPAECs to COPPIX, a synthetic pro- toporphyrin that induces the expression of HO-1 (10
M; Sigma, Saint Louis, MO, USA) for 12 h was used to increase the expression of HO-1 to measure the inhibi- tory effect of HO-1 on endothelial activation. Pharmacological treatments with fenofibrate Fenofibrate was obtained from Laboratories Fournier S.A. (Fontaine Les Dijon,
France) and was
dissolved in dimethyl sulfoxide (DMSO) as a stock solution. The con-fluent HPAECs were pretreated with the
growth medium
supplemented with 50 M fenofibrate for 18 hours, fol- lowed by 70% sera obtained from patients with HF or normal individual for 12 hours at 37 C. The endothelial expression of ICAM-1,
VCAM-1, HO-1, and transcriptional activation of NF-B was then confirmed by mRNA expres- sion and Western
blot, and EMSA,
respectively.
Statistics
All values were expressed as mean standard error
of mean. Comparisons of clinical and biochemical char- acteristics between two groups were made with the Wilcoxon rank-sum test for quantitative data and with Fisher’s exact test for qualitative data. Comparisons be- tween multiple groups were determined by means of a one-way analysis of
variance (ANOVA)
followed by Dunnett’s test. A p value of less than 0.05 was con-sidered statistically significant.
RESULTS
Baseline clinical characteristics and the circulating levels of CAMs of the study patients
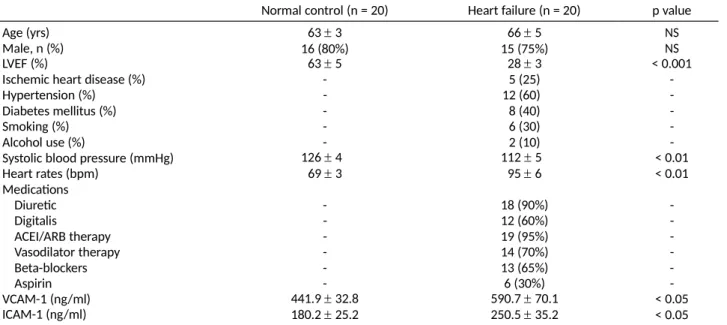
The baseline
characteristics of the 20 patients with advanced HF and the 20 healthy controls are shown in Table 1.
Significant
differences in LVEF, systolic blood pres- sure, and heart rates were detected between the pa- tients with HF and normal controls. There were more men than women in this sample. Patients were included with both ischemic as well as non-ischemic etiology in the HF group.
al. Induction of CAMs on HPAECs by serum obtained from HF patients Incubation for 12 h of HPAECs with serum from HF patients (HFS) markedly increased the protein and mRNA expression of VCAM-1
and ICAM-1 as
compared with those incubated with serum from normal subjects (NRS) and control medium (Figure 1A and B). Although the protein and mRNA expression of VCAM-1 was also elevated to some extent by serum from normal sub- jects, the ICAM-1 was not significantly elevated. More- over, the induction of both VCAM-1 and ICAM-1 by se- rum from HF patients was evident as early as 3 h, and the augmentation lasted for at least 24 h, whereas se- rum from normal subjects only time-dependently in- duced VCAM-1 expression to a lesser extent (Figure 1C).
Down-regulation of HO-1 expression by serum obtained from HF patients
Incubation for 12 h of HPAECs with 70% serum from HF patients significantly decreased the protein and mRNA expression of HO-1 as
compared with those
incu-Table 1. Baseline clinical characteristics of the study population
Normal control (n = 20) Heart failure (n = 20) p value Age (yrs) Male, n (%) 63 3 16 (80%) 66 5 15 (75%) NS NS LVEF (%)
Ischemic heart disease (%)
63 5 -28 3 5 (25) < 0.001 -Hypertension (%) - 12 (60) -Diabetes mellitus (%) - 8 (40) -Smoking (%) - 6 (30) -Alcohol use (%) - 2 (10)
-Systolic blood pressure (mmHg) Heart rates (bpm) Medications 126 4 69 3 112 5 95 6 < 0.01 < 0.01 Diuretic - 18 (90%) -Digitalis - 12 (60%) -ACEI/ARB therapy - 19 (95%) -Vasodilator therapy - 14 (70%) -Beta-blockers - 13 (65%) -Aspirin - 6 (30%) -VCAM-1 (ng/ml) ICAM-1 (ng/ml) 441.9 32.8 180.2 25.2 590.7 70.1 250.5 35.2 < 0.05 < 0.05 ACEI/ARB, angiotensin converting enzyme inhibitors or angiotensin II receptor blockers; ICAM-1, intercellular adhesion molecule-1; LVEF, lef ventricular ejection fraction; NS, non-significant; VCAM-1, vascular cellular adhesion molecule-1.
al. bated with serum from
normal subjects and control me- dium (Figure 2A). The down-regulation of HO-1 expres- sion by serum from HF patients was evident at 12 h, and the HO-1 expression decreased further at 24 h, whereas serum from normal subjects did not significantly affect HO-1 expression (Figure 2B).
Immunohistochemical staining for HO-1 and CAMs (Figure 2C)
In vessels of normal subjects, HO-1 protein
was de- tected
predominantly in the endothelial vWF-positive cells, relative to the thin layer of endothelial cells. In vessels of patients with HF, HO-1 protein expression was much attenuated in the vWF-positive cells in the endo- thelium. By contrast, increased expression of VCAM-1
and ICAM-1 was
presented mainly in both endothelium and tunica media of vessels from HF patients.
Modulation of HF serum-induced CAM expression via HO-1 regulation
As shown in Figure 3A, HO-1 siRNA dose-depend- ently attenuated
HO-1 expression.
Attenuation of HO-1 by HO-1 siRNA potentiated
the induction of VCAM-1 and ICAM-1 expression by serum from HF patients and normal subjects (Figure 3B).
HPAECs were exposed to CoPPIX dose (2.5-20
M)-and time (3-24 h)-dependently induced high levels of HO-1 expression (Figure 3C).
The increased
endothelial expression of VCAM-1 by serum from HF patients and normal subjects and the expression of ICAM-1 by serum from HF patients were significantly inhibited by CoPPIX (Figure 3D). Fenofibrate inhibits HF serum-induced CAM expression and HO-1 down-regulation and attenuates serum-induced activation of NF-B HPAECs were
pretreated for 18 h with fenofibrate before the addition of serum from HF patients and nor- mal subjects. Fenofibrate
caused a
dose-dependent de- crease in VCAM-1 expression induced by serum from HF patients and normal subjects, and dose-dependently decreased HF serum-induced ICAM-1 expression (Figure 4A). By contrast, fenofibrate treatment at 25 and 50 M significantly inhibited HF serum-induced HO-1 down-regulation (Figure 4B).
Gel shif assays showed that HF serum
treatment resulted in strong activation of NF-B, whereas serum from normal subjects only slightly increased the shifed band, compared to control medium. Pretreatment with fenofibrate reduced the density of the NF-B shifed bands induced by serum both from HF patients and nor- mal subjects (Figure 4C).
al. A
B
C
Figure 1. Induction of adhesion
molecules and down-regulation of heme-oxygenase l (HO-1) expression on human pulmonary artery endo- thelial cells (HPAECs) by serum obtained from HF patients. (A and B) In-cubation for 12 h of HPAECs with serum from HF patients (HFS) mark- edly increased the protein and mRNA expression of VCAM-1 and ICAM-1 as compared with those incubated with serum from normal subjects (NRS) and control medium (Control). (C) The induction of both VCAM-1 and ICAM-1 by serum from HF patients was evident as early as 3 h, and the augmentation lasted for at least 24 h, whereas serum from normal subjects only time-dependently induced VCAM-1 expression to a lesser extent. Densitometric analysis was conducted with software to semiquantify Western blot data. Three independent experiments gave
similar results. *p < 0.05, compared to control group; #p < 0.05,
com-pared to NRS group.
DISCUSSION
In the present study,
we showed that
stimulation of cultured HPAECs with HF patient serum significantly down-regulated HO-1 expression, activated
redox-sensi- tive
transcription factor NF-B, and increased VCAM-1
A
B
C
Figure 2. (A) Incubation for 12
h of HPAECs with 70% HFS significantly decreased the protein and mRNA expression of HO-1 as compared with those incubated with serum from NRS and control medium. (B) The down-regulation of HO-1 expression by serum from HF patients was evi- dent at 12 h, and the HO-1 expression decreased further at 24 h, whereas serum from normal subjects did not significantly affect HO-1
expression. Three independent experiments gave similar results. The summarized data (mean 主 standard error of mean) from 3 separate ex-periments is shown in the bar graph. *p < 0.05, compared to control group. (C) Immunohistochemical analysis revealed the distribution of vWF, HO-1, VCAM-1, and ICAM-1 in the arteries from the normal subject and HF patients. In vessel of the normal subject (NR), HO-1 protein was detected predominantly in the endothelial vWF-positive cells, relative to the thin layer of endothelial cells. In vessels of patients with heart fail- ure (HF), HO-1 protein expression was much attenuated in the vWF-pos- itive cells in the endothelium. By contrast, increased expression of VCAM-1 and ICAM-1 was
presented mainly in both endothelium and tunica media of vessels from HF patients.
and ICAM-1 expression. Immunohistochemistry study of coronary and
carotid arteries
confirmed that VCAM-1 and ICAM-1 were up-regulated, whereas HO-1 was down- regulated in HF patients. HO-1 inducer CoPPIX simulta-neously stimulated HO-1
expression and
suppressed VCAM-1 and ICAM-1 expression, whereas HO-1 siRNA significantly exaggerated HF patient serum-induced
A
B
C
D
Figure 3. Modulation of HF serum-induced adhesion molecules expression via HO-1 regulation. (A) HO-1 small interfering RNA (siRNA) dose-de-
pendently attenuated HO-1 expression. (B) Attenuation of HO-1 by HO-1 siRNA potentiated the induction of VCAM-1 and ICAM-1 expression by HFS and NRS. (C) Exposure of HPAECs to CoPPIX dose (2.5-20 µM)- and time (3-24 h)-dependently induced high levels of HO-1 expression. (D) The in-creased endothelial expression of VCAM-1 by serum from HF patients and normal subjects and the expression of ICAM-1 by serum from HF patients were significantly inhibited by CoPPIX. Three independent experiments gave similar results. The summarized data (mean 主 SEM) from 3 separate ex-periments is shown in the bar graph. *p < 0.05, compared to control group; #p < 0.05, compared to NRS/HFS group.
A
B C
Figure 4. Fenofibrate prevents
HF serum-induced HO-1 down-regula- tion and inhibits adhesion molecule expression. HPAECs were pretreated for 18 h with PPAR-α agonist fenofibrate (Feno) before addition of
se-rum from HF patients (HFS) and normal subjects (NRS). (A) Fenofibrate (12.5, 25, and 50 µM) caused a dose-dependent decrease in VCAM-1 ex-pression induced by serum from HF patients and normal subjects, and dose-dependently decreased HF serum-induced ICAM-1 expression. (B)
Fenofibrate treatment at 25 and 50 µM significantly inhibited HF
se-rum-induced HO-1 down-regulation. Three independent experiments gave similar results. (C) Gel shift assays showed that HFS treatment re-sulted in the strongly activation of NF-KB, whereas NRS only slightly
in-crease the shifted band, compared to control medium. Pretreatment with fenofibrate reduced the density of the NF-KB shifted bands induced
by serum both from HF patients and normal subjects. The summarized data (mean 主 SEM) from 3 separate experiments is shown in the bar
graph. *p < 0.05, compared to control group; #p < 0.05, compared to NRS/HFS group.
VCAM-1 and ICAM-1 expression on HPAECs.
Further- more,
pretreatment with fenofibrate may prevent the decrease of HO-1, the activation of NF-B, as well as the increase of VCAM-1 and ICAM-1 induced by HF serum.
Endothelial
activation and
dysfunction may con-tribute to exercise intolerance, impaired myocardial per- fusion,
lef ventricular
remodeling, cardiogenic shock, and pulmonary edema.1-5,34,35 CAMs induced by inflam-matory cytokines may play a direct and potentially critical role in the pathophysiology of HF.6-11,34,35 We and others have reported that patients with advanced HF
have elevated circulating levels of CAMs, irrespective of the cause of HF.6-8 Significantly higher circulating CAM levels and endothelial leukocyte adhesiveness were pre- dictors of adverse clinical outcomes in HF patients.6-8,10,11 HO-1 exhibits anti-inflammatory property to pro-vide endothelial protection during atherogenesis,
restenosis, and other inflammatory
cardiovascular disor-ders.12-16,36,37 Moreover, HO-1 may inhibit the expression of CAMs
associated with
endothelial activation via a mechanism that is associated with the inhibition of NF-B activation18 and suppress endothelial cell apop-tosis via the activation of p38 MAPK.19 Recently,
it has been
demonstrated that HO-1 expression is trans-criptionally regulated in human endothelial cells and vascular smooth muscle cells by PPAR, indicating a mechanism of anti-inflammatory action of PPAR ligands via up-regulation of HO-1.33 In this study, our results fur- ther confirmed that the endothelial activation
and vas- cular
inflammatory processes are systemic and can be modified through
regulation of HO-1 in HF. Beyond its regulatory effects on cardiac energy and control of myocardial lipid metabolism,20,24
PPARalso exert
numerous effects by
interaction with
different transcription factors to repress pro-inflammatory
genes.24,25,29 The activators of PPARhave been demon- strated to exert cardiovascular antioxidant and anti-inflammatory effects by interfering negatively with transcription factor pathways such as NF-B, signal transducers and activators of transcription (STAT), and AP-1.24,25,29 Among these, the transcription factor NF-B is critical for the induction of the VCAM-1 and ICAM-1 examined in this study. Furthermore, fenofibrate has an additional potential to prevent the induction and pro- gression of hypertensive heart damage, cardiac hyper-trophy, heart failure, myocarditis, lipotoxic cardiomyo- pathy and vascular endothelial dysfunction-associated cardiovascular
abnormalities.26-30 Our results support this hypothesis because fenofibrate counteracted HO-1 down-regulation
and suppressed
endothelial CAM ex-pression that were
al. induced by
pro-inflammatory stimuli of HF serum via inhibition of NF-B signaling pathway. Since a daily dosage of fenofibrate 200 mg produced plasma concentrations within the range of 5-35 mg/l (14-100 uM) in 12 dyslipidaemic patients receiving the drug over a 3-month period,38 the doses we studied in
this experiment are clinically relevant.
al.
CONCLUSION
In conclusion, although our study does not exclude a role for the
effects of
PPARactivators on the expres- sion of other factors involved in endothelial activation
and
leukocyte-endothelial interaction, nor does it rule out the possibility that the molecules tested may act through unrelated mechanisms in addition to PPARac- tivation, this work does identify HO-1 as a target gene for PPARand provides a basis for further investigation of HO-1
modulation by
fenofibrate as a therapeutic strategy for endothelial activation and dysfunction in HF as well.
ACKNOWLEDGMENT
This work is support by grants from the Cheng-Hsin General Hospital (Grant 95-10 and Grant 96-03 to Dr. Yin) and the National Science Council (NSC 95-2314- B-350-001 to Dr. Yin). DISCLOSURES N o n e . REFERENCES
1. Wrigley BJ, Lip GY, Shantsila E. The role of monocytes and inflam- mation in the pathophysiology of heart failure. Eur J Heart Fail 2011;13:1161-71.
2. Chong AY, Blann AD, Patel J, et al. Endothelial dysfunction and damage in congestive heart failure: relation of flow-mediated dilation to circulating endothelial cells, plasma indexes of endo- thelial damage, and brain natriuretic peptide. Circulation 2004; 110:1794-8.
3. Chong AY, Freestone B, Patel J, et al. Endothelial activation, dys- function, and damage in congestive heart failure and the relation to brain natriuretic peptide and outcomes. Am J Cardiol 2006; 97:671-5. 4. Verma A, Bulwer B,
Dhawan I, et al. Aldosterone receptor an-tagonist and heart failure following acute myocardial infarction. Acta Cardiol Sin 2010;26:203-15.
5. Chen CN, Chen HR, Chang HI, et al. Relationship between the antiplatelet effect of aspirin and serum VCAM-1 concentration in patients at high risk for cardiovascular events. Acta Cardiol Sin 2010;26:28-36.
6. Tsutamoto T, Hisanaga T, Fukai D, et al. Prognostic value of plasma soluble intercellular adhesion molecule-1 and endo-thelin-1 concentration in patients with chronic congestive heart failure. Am J Cardiol 1995;76:803-8.
7. Tousoulis D, Homaei H, Ahmed N, et al. Increased plasma ad-hesion molecule levels in patients with heart failure who have ischemic heart disease and dilated cardiomyopathy. Am Heart J 2001;141:277-80. 8. Yin WH, Chen JW, Jen HL,
et al. The prognostic value of circulat- ing soluble cell adhesion molecules in patients with chronic con-gestive heart failure. Eur J Heart Fail 2003;5:507-16. 9. Geppert A, Zorn G, Heinz
G, et al. Soluble selectins in the pulmo- nary and systemic circulation in acute cardiogenic and non- cardiogenic pulmonary failure. Intensive Care Med 2001;27: 521-7.
10. Yin WH, Jen HL, Chiang MC, et al. Circulating soluble tumor ne- crosis factor- and cell adhesion molecules in patients with acute cardiogenic pulmonary edema. J Emerg Crit Care Med 2004;15:9-19. 11. Yin WH, Chen JW, Young
MS, Lin SJ. Increased endothelial monocyte adhesiveness is related to clinical outcomes in chronic heart failure. Int J Cardiol 2007;121:276-83. 12. Lakkisto P, Kytö V, Forsten
H, et al. Heme oxygenase-1 and carbon monoxide promote
neovascularization afer myocardial infarc- tion by modulating the expression of HIF-1alpha, SDF-1alpha and VEGF-B. Eur J Pharmacol 2010;635:156-64.
13. Wang G, Hamid T, Keith RJ, et al. Cardioprotective and anti- apoptotic
effects of heme
oxygenase-1 in the failing heart. Circulation 2010;121:1912-25. 14. Tang JM, Wang JN, Zhang L,
et al. VEGF/SDF-1 promotes cardiac stem cell
mobilization and
myocardial repair in the
infarcted heart.
Cardiovasc Res
2011;91:402-11.
15. Lakkisto P, Siren JM, Kytö V, et al. Heme oxygenase-1 induction protects the heart and modulates cellular and extracellular
remodeling afer
myocardial infarction in rats. Exp Biol Med (Maywood)
2011;236:1437-48. 16. Wu ML, Ho YC, Yet SF. A
central role of heme oxygenase-1 in car-diovascular protection. Antioxid Redox Signal 2011;15:1835-46.
17. Lam CF, Croatt AJ, Richardson DM, et al. Heart failure increases protein expression and enzyme activity of heme oxygenase-1 in the lung.
Cardiovasc Res
2005;65:203-10.
18. Soares MP, Seldon MP, Gregoire IP, et al. Heme oxygenase-1 modulates the expression of adhesion molecules
associated with
endothelial cell activation. J Immunol 2004;172:3553-63.
19. Brouard S, Otterbein LE, Anrather J, et al. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med 2000;192:1015-26. 20. Luptak I, Balschi JA, Xing
Y, et al. Decreased contractile and meta-bolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation 2005;112:2339-46. 21. Labinskyy V, Bellomo M, Chandler MP, et al. Chronic activation of peroxisome proliferator-activated receptor-alpha with
fenofib-Acta Cardiol Sin 2013;29:251-260 260 al.
Acta Cardiol Sin 2013;29:251-260 261 rate prevents alterations in
cardiac metabolic phenotype with- out changing the onset of decompensation in pacing-induced heart failure. J Pharmacol Exp Ther 2007;321:165-71. 22. Sweets PJ, Teunissen BE,
Willemsen PH, et al. Cardiac hyper- trophy is enhanced in PPAR alpha -/-mice in response to chronic pressure overload. Cardiovasc Res 2008;78:79-89. 23. Balakumar P, Rohilla A, Mahadevan N. Pleiotropic actions of fenofibrate on the heart. Pharmacol Res 2011;63:8-12.
24. Schiffrin EL. Peroxisome proliferator-activated
receptors and
cardiovascular
remodeling. Am J Physiol Heart Circ Physiol 2005;288:H1037-43. 25. Sweets PJ, de Vogel-van
den Bosch HM, Willemsen PH, et al. Transcriptomic analysis of PPARalpha-dependent alterations during cardiac hypertrophy. Physiol Genomics 2008;36:15-23.
26. Ogata T, Miyauchi T, Sakai S, et al. Stimulation of peroxisome- proliferator-activated receptor alpha (PPAR alpha) attenuates cardiac fibrosis and endothelin-1 production in pressure-over- loaded rat hearts. Clin Sci (Lond) 2002;103:284S-8S. 27. Ogata T, Miyauchi T, Sakai
S, et al. Myocardial fibrosis and dia- stolic dysfunction in deoxycorticosterone acetate-salt hyperten- sive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate,
partly by suppressing inflammatory responses associated with the nuclear factor- kappa-B pathway. J Am Coll Cardiol 2004;43:1481-8.
28. Diep QN, Benkirane K, Amiri F, et al. PPAR alpha activator fenofibrate inhibits myocardial inflammation
and fibrosis in
angiotensin II-infused rats. J Mol Cell Cardiol 2004;36:295-304.
29. Ichihara S, Obata K, Yamada Y, et al. Attenuation of cardiac dys- function by a PPAR-alpha agonist is associated with down-regula- tion of redox-regulated transcription factors. J Mol Cell Cardiol 2006;41:318-29.
30. Wayman NS, Hattori Y, McDonald MC, et al. Ligands of the
peroxisome proliferator-activated receptors (PPAR-gamma and PPAR-alpha) reduce myocardial infarct size. FASEB J 2002;16: 1027-40.
31. Huang WP, Yin WH, Chen JW, et al. Fenofibrate attenuates endo- thelial monocyte adhesion in chronic heart failure: an in vitro study. Eur J Clin Invest 2009;39:775-83. 32. Lavrovsky Y, Schwartzman
ML, Levere RD, et al. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci U S A 1994;91:5987-91. 33. Krönke G, Kadl A,
Ikonomu E, et al. Expression of heme oxygenase-1 in human vascular cells is regulated
by peroxisome
proliferator-activated receptors. Arterioscler Thromb Vasc Biol 2007;27:1276-82. 34. Oikonomou E, Tousoulis
D, Siasos G, et al. The role of inflam- mation in heart failure: new therapeutic approaches. Hellenic J Cardiol 2011;52:30-40.
35. Picano E, Morales MA, del Ry S, Sicari R. Innate
inflammation in
myocardial perfusion and its implication for heart failure. Ann N Y Acad Sci 2010;1207:107-15. 36. Chen YH, Lin SJ, Lin MW,
et al. Microsatellite
polymorphism in
promoter of heme oxygenase-1 gene is associated with suscep-tibility to coronary artery disease in type 2 diabetic
patients. Hum Genet 2002;111:1-8.
37. Chen YH, Chau LY, Lin MW, et al. Heme oxygenase-1
gene pro- motor
microsatellite
polymorphism is
associated with angio-graphic restenosis afer coronary stenting. Eur Heart J 2004; 25:39-47. 38. Balfour JA, McTavish D,
Heel RC. Fenofibrate: a
review of its
pharmacodynamic and pharmacokinetic
properties and
thera-peutic use in
dyslipidaemia. Drugs 1990;40:260-90.