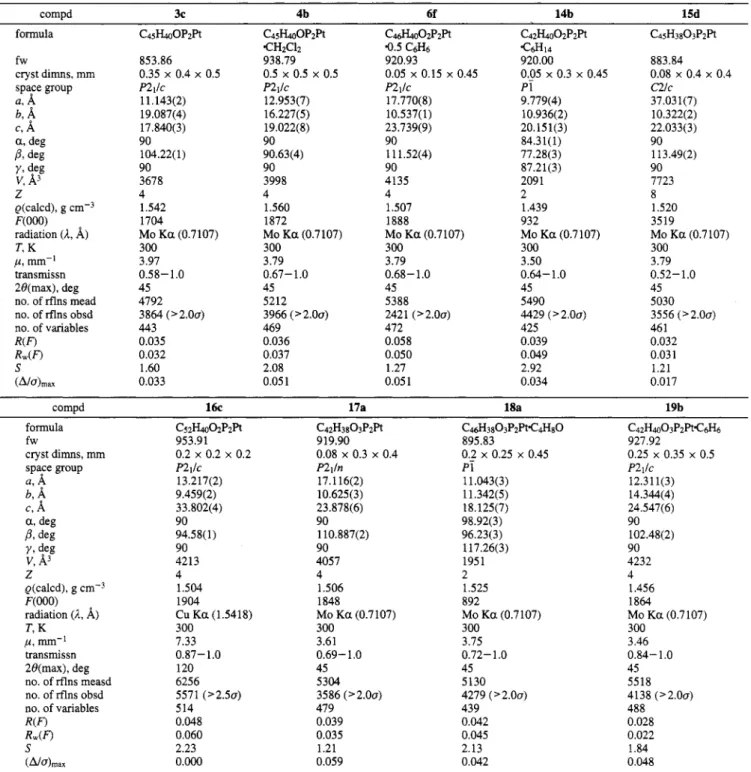
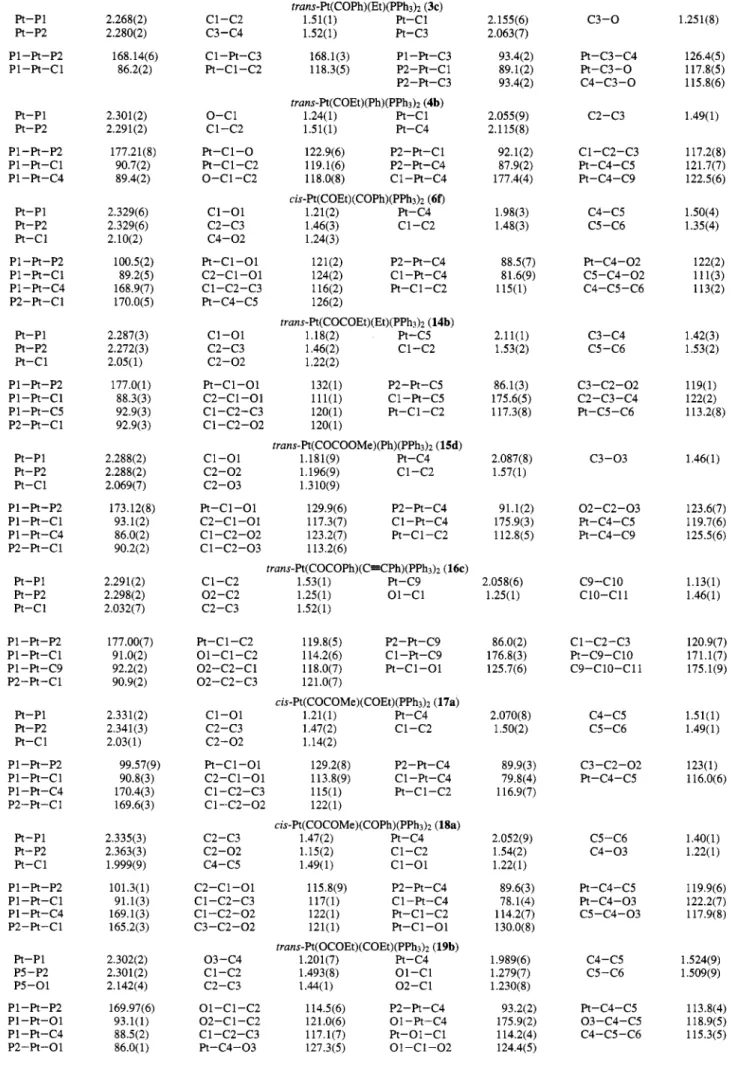
4804 Organometallics 1994, 13, 4804-4824
Insertion Reactions
of
Square-Planar Diorganoplatinum.
Carbonylation, Decarbonylation, and Relevant Reactions
of Alkyl Acyl, Aryl Acyl, Alkyl a-Ketoacyl, Aryl
a-Ketoacyl, Diacyl, and Acyl a-Ketoacyl Complexes?
1.
Scope and Mechanisms of Stereoselective
Jwu-Ting Chen,* Yu-Sung Yeh, Ching-Shuenn Yang, Fu-Yu Tsai,
Gwo-Liang Huang, Bor-Chyr Shu, Tsang-Mia0 Huang, Yan-Su Chen,
Gene-Hsiang Lee, Ming-Chu Cheng, Chih-Chieh Wang, and Yu Wang
Department of chemistry, National Taiwan University, Taipei, Taiwan 106, Republic of China Received August 16, 1994@
Transmetalation between truns-Pt(COR)(X)(PPh3)2
(R
= Me, Et, Ph; X = halide) and R2-Zn ( R = Me, E t , Ph) in benzene or toluene at 25 "C readily results in the formation of trans-
Pt(COR)(R)(PPh3)2 in high yields. Alkylation of truns-Pt(COCOR)(OTf(PPh3)2 or trans- [Pt(COCOR)(L)(PPh3)21+ (L = H20, THF; R = Me, Et, Ph, OMe) with use of R2Zn (R = Me, Et,
Ph)
provides trans-Pt(COCOR)(R)(PPh3)2. Carbonylation of trun~-Pt(cOR)(R')(PPh3)~exclusively gives cis-Pt(COR)(COR)(PPh3)2 in quantitative yields with its reactivity following the order
R
= E t >> Ph > Me. An alternative synthetic route to the cis diacyl complexes, albeit in low yields, is by means of t h e nucleophilic addition of L i R or RMgX to the cis-[Pt(COR)(CO)(PPh3)21+ cation. With the assistance of CO,
c~s-P~(COE~)(COM~)(PP~~)~
undergoes reversible intermolecular acyl scrambling to form cis-Pt(COMe)2(PPh3)2 and cis-Pt(COEt)2(PPh3)2, reaching the equilibrium ratio 2 : l : l . CO also promotes isomerization of
trun~-Pt(Me)(CO2Me)(PPh3)2
to its cis derivative, which suffers substitution of CO for PPh3to produce (SP-4-2)- and (SP-4-4)-Pt(PPh3)(COzMe)(CO)(Me). The stereoselective carbon- ylation of trans-Pt(COCOR)(R')(PPh3)2 exclusively affords novel cis-Pt(COCOR)(COR)(PPh3)2
in quantitative yields. The reactivity also follows the order
R
= E t >> P h > Me. The mechanism of carbonylation of the trans diorganoplatinum complexes is likely led by a reversible substitution of CO for a PPh3 ligand, followed by alkyl (or aryl) migration from metal t o CO and subsequent recoordination of PPh3. At 25 "C, truns-Pt(COCOR)(R)(PPh3)2can spontaneously transform to trans-Pt(COR)(COR)(PPh3)2. In a comparable sense, trans- Pt(COR)(R)(PPh3)2 species (R = Me,
R
= Et; R = Ph,R
= Me, Et) convert to trans-Pt- (COR)(R)(PPh& irreversibly at 50 "C. Such reactions likely proceed via a five-coordinate intermediate by intramolecular CO transfer. The methoxyoxalyl derivatives trans-Pt(COCO2- Me)(R)(PPh& are also subject to decarbonylation, resulting in ci~-Pt(COzMe)(R)(PPh3)2 instead. Thermolysis of t h e cis diacyl complexes leads to the formation of C(0)-C(0) bonds, yielding vicinal diketones. The cis acyl a-ketoacyl complexes first suffer decarbonylation to form the trans diacyl complexes, which succeedingly convert to trans acyl carboxylato complexes, truns-Pt(COR)(OCOR)(PPh3)2 and truns-Pt(OCOR)(COR)(PPh3)2, in solutions with moisture, as well as organic ketones or diketones. The X-ray crystallographic analyses for all title species and a carboxylato complex are provided.Introduction
The reactions of carbonylation and decarbonylation of the ds square-planar complexes of group 10 metals are of considerable importance to organic functional- ization and cata1ysis.l Relevant mechanistic studies of Partially based on the Ph.D. thesis of Y.-S.Y., National Taiwan University, 1992.
@ Abstract published in Advance ACS Abstracts, October 15, 1994. (1) (a) Colquhoun, H. M.; Thompson, D. J.; Twigg, M. V. Carbon- ylation: Direct Synthesis of Carbonyl Compounds; Plenum Press: New York, 1991. (b) Comprehensive OrganometalEic Chemistry; Wilkinson, G., Ed.; Pergamon Press: Oxford, England, 1982; Vol. 6 and 8. (c) Heck, R. F. Organotransition Metal Chemistry; Academic Press: New York, 1974. (d) Tsuji, T. Organic Synthesis by Means of Transition Metal Complexes; Springer-Verlag: Berlin, Heiderberg, New York,
1975. (0 Collman, J. P.; Hegedus, L. S.; Norton, J. R.; Finke, R. G. Principles & Applications of Organotransition Metal Chemistry; Uni- versity Science Books: Mill Valley, CA, 1987.
0276-7333194123 13-48Q4$Q4.5Q/Q
such reactions have been largely focused on monoorgano complexes. For instance, it has been shown that the typical species M(R)(X)L2 (M = Ni, Pd, Pt; R = alkyl, aryl; X = halide; L = monodentate phosphine, arsine, etc.) react with CO to yield acyl species M(COR)(X)L2 with only the thermodynamically stable trans configu- ration.2 There has been no example of cis acyl deriva- tive that results from carbonylation, unless bidentate ligand is i n ~ o l v e d . ~
(2) (a) Booth, G.; Chatt, J. J . Chem. SOC. A 1966,634. (b) Clark, H. C.; Puddephatt, R. J. Inorg. Chem. 1970, 9, 2670. (c) Glyde, R. W.; Mawby, R. J. Inorg. Chem. 1971, 10, 854. (d) Wilson, C. J.; Green, M.; Mawby, R. J. J . Chem. SOC. A 1974,421, 1293. (e) Garrou, P. E.; Heck, R. F. J . Am. Chem. SOC. 1976, 98, 4115. (0 Sugita, N.; Minkiewicz, J. V.; Heck, R. F. Inorg. Chem. 1978, 17, 2809. (g)
Anderson, G. K.; Cross, R. J. J . Chem. SOC. A 1980, 712, 1434. (h) Anderson, G. K.; Cross, R. J. Acc. Chem. Res. 1984, 17, 67 and
references therein.
0 1994 American Chemical Society
Downloaded by NATIONAL TAIWAN UNIV on August 5, 2009
Reactions Square-Planar Diorganoplatinum Scheme 1
0
r R' IIV
Lt
\
/
ICarbonylation and decarbonylation in square-planar diorgano systems, particularly those containing acyl ligands, has been nearly ~ n e x p l o r e d . ~ Well-character- ized acyl alkyl and diacyl complexes of such systems were actually unprecedented prior to our communica- t i ~ n , ~ although involvement of these species in the group 10 metal-mediated reactions of carbonylation and double carbonylation has been considered. The Pd-catalyzed coupling reactions of acid chloride with organotin re- agents have been proved to be very useful for the synthesis of ketones.6 The mechanistic studies sug- gested that acyl(alky1)palladium complexes (I in Scheme 11, presumably resulting from transmetalation between the acylhalopalladium and organotin compounds, might be a crucial intermediate in these reactions. However, no detection of I or its platinum analog has been made. Tanaka also reported the reactions of carbonylative coupling between organozinc and acid halide, yielding diketones and related products, and proposed a mecha- nism involving an intermediate diacyl species (I1 in Scheme 11, still without direct e v i d e n ~ e . ~
The paucity of trans diorgano complexes, particularly those containing acyl ligands, has been partially as- cribed to the fact that the complexes having ligands with
(3)(a) Bryndza, H. E.; Tam, W. Chem. Reu. 1988, 88, 1163. (b) Bryndza, H. E.; Kretchmar, S. A.; Tulip, T. H. J. Chem. Soc., Chem.
Commun. 1986,977. (c) Bryndza, H. E. Organometallics 1985,4,406.
(d) Bennett, M. A,; Rokicki, A. Organometallics 1986, 4, 180. (e) Bennett, M. A.; Robertson, G. B.; Whimp, P. 0.; Yoshida, T. J. Am.
Chem. SOC. 1973,95,3028. (0 van Leeuwen, P. W. N. M.; Roobeek, C.
F.; Wife, R. L.; Frijns, J. H. G. J . Chem. Soc., Chem. Commun. 1986, 31. (g) van Leeuwen, P. W. N. M.; Roobeek, C. F.; Frijns, J. H. G.; Orpen, A. G. Organometallics 1990, 9, 1211. (h) Dekker, G. P. C.; Buijs, A.; Elsevier, C. J.; Vrieze, K.; van Leeuwen, P. W. N. M.; Smeets, W. J. J.; Spek, A. L.; Wang, Y. F.; Stam, C. H. Organometallics 1992, 11, 1937. (i) Davcid, Y. B.; Portnoy, M.; Milstein, D. J . Am. Chem.
Soc. 1989,111, 8742. (j) Dekker, D. P. C. M.; Buijs, A.; Elsevier, C. J.; Vrieze, K.; van Leeuwen, P. W. N. M.; Smeets, W. J. J.; Spek, A. L.; Wang, Y. F.; Stam, G. H. Organometallics 1992, 11, 1937. (k) Portnoy, M.; Milstein, D. Organometallics 1993, 12, 1655.
(4) (a) Ozawa, F.; Yamamoto, A. Chem. Lett. 1981,289. (b) Ozawa, F.; Ito, T.; Yamamoto, A. J. Am. Chem. SOC. 1980,102, 6457.
( 5 ) A preliminary communication has been published Chen, J.-T.;
Huang, T.-M.; Cheng, M.-C.; Wang, Y. Organometallics 1990, 9, 539. (6) (a) Tanaka, M. Tetrahedron Lett. 1979, 2601. (b) Labadie, J. W.; Stille, J. K. J. Am. Chem. Soc. 1983, 105, 6129.
(7)Yamashita, H.; Kobayashi, T.; Sakakura, T.; Tanaka, M. J . Organomet. Chem. 1988,356, 125.
high trans influence at the trans coordination sites are congenitally u n f a ~ o r e d . ~ ~ ~ Besides, the synthetic strat- egy for such species is still confined to conventional transmetalation with main-group organometallic among species, which the acyl reagents are not readily avail- able.1° Our previous results on the formation of trans- Pt(COPh)(COEt)(PPh& from trans-Pt(COCOPh)(Et)-
(PPh3)2 have demonstrated that trans diorgano complexes
can be reasonably stable under suitable conditionsll and has prompted us t o extend this synthetic work t o other trans alkyl a-ketoacyl and alkyl acyl derivatives. It has been found that these four-coordinate trans diorgano complexes can undergo efficient stereoselective carbon- ylation under rather mild conditions, leading to novel cis diacyl and cis acyl a-ketoacyl complexes, respectively, and eventually to diketones and ketones.12 Such results not only reveal the intriguing reactivity of new diorgano complexes of platinum(I1) but also provide a good model for the Pd-catalyzed coupling and carbonylative coupling reactions of acid halides with organometals. From a mechanistic viewpoint, the stereoselective carbonylation and decarbonylation in such diorgano complexes are also worthy of a closer look. In this article, we report the synthesis, structures, and reactivity of the new acyl alkyl, alkyl a-ketoacyl, diacyl, acyl a-ketoacyl, and related complexes of platinum(I1). The reaction scope and mechanisms of carbonylation, decarbonylation, and the relevant reactions are discussed.
Results
Synthesis and Characterization of trans-Pt- (COR)(R')(PPh&. Synthesis of the trans acyl alkyl and acyl aryl complexes has been conducted by conve- nient transmetalation between trans-Pt(COR)(X)(PPh3)z and diorganozinc reagents. The starting acyl halo complexes were prepared according to documented methods.13 In typical experiments, treatment of trans- Pt(COR)(Cl)(PPh3)2 (R = Me (la), Et (lb), Ph (IC)) with R2Zn in benzene or toluene under a nitrogen atmo- sphere at 25 "C readily resulted in the formation of yellow tran~-Pt(COR)(R)(PPh3)2 ( R = Me, R = Me (2a), E t (2b), Ph (2c); R = Et, R = Me (3a), E t (3b), Ph (3c);
R = Ph, R = Me (4a), E t (4b), Ph (4c)), as illustrated in Scheme 2. Most of the acyl alkyl complexes were precipitated by n-hexane from the reaction solutions to give excellent yields. The methyl derivatives were generally formed along with substantial amounts of cis- Pt(Me)z(PPh&. The desired products 2a-c were puri- fied satisfactorily by silica gel column chromatography with EtzOhenzene solutions as the eluent. In chloro- form, trans-Pt(COR)(R)(PPh3)2 slowly transforms to
trans-Pt(COR)(Cl)(PPh3)2, presumably due to the reac- tion with HC1 in the solvent.
( 8 ) Appleton, T. G.; Clark, H. C.; Manzer, L. E. Coord. Chem. Rev.
1973, 10, 335.
(9) (a) Yamamoto, A. Organotrunsition Metal Chemistry: Funda- mental Concepts and Applications; Wiley: New York, 1986 Chapter
6. (b) Anderson, G. K.; Clark, H. C.; Davies, J. A. Inorg. Chem. 1981, 20, 3607.
(10) (a) Wakefield, B. J. Organolithium Methods; Academic Press: London, San Diego, CA, 1988. (b) Seyferth, D.; Weinstein, R. M.; Wang, W.-L.; Hui, R. C.; Archer, C. M. Isr. J . Chem. 1984,24, 167.
(11) Chen, J.-T.; Huang, T.-M.; Cheng, M.-C.; Wang, Y. Organome-
tallics 1991, 10, 2838.
(12) Huang, G.-L.; Huang, T.-M.; Chen, J.-T.; Lee, G.-H.; Wang, Y.
Inorg. Chem. 1992, 30, 4034.
(13) Kubota, M.; Rothrock, R. IC; Geibel, J. J . Chem. SOC. A 1973, 1267.
Downloaded by NATIONAL TAIWAN UNIV on August 5, 2009
4806 Organometallics, Vol. 13, No. 12, 1994 Scheme 2 R'2Zn/benzene 7Ph3 RC-Pt-CI
-
I
25 "C PPh3 Chen et al. 0 PPh3 III
R'= M e , R = Me, Et, Ph 2 , ~ RC-Pt-R' R ' = E t , R = Me,Et,Ph&c R ' = Ph, R = Me, Et, Ph 4 6 cI
PPh3 Scheme 3 PPh3I
AgOTVCO Me-R-I-
I
25 'C PPh3 2d P P h 3 1+
0 PPh3I
NaOMe/MeOH III
25 "CI
I
Me-Pt-CO OTT MeOC-Pt-Me PPh3 PPh3 Scheme 4 0 PPh3 0 PPh3 II
I
PhCIGH/EtaN III
I
25 "CI
PhC-Pt-Cl P- PhC-Pt-CICPh PPh3 PPh, 5cThe acyl carbonyls in trans-Pt(COR)(R)(PPh3)~ often exhibit stretching absorptions lower than 1600 cm-l in their infrared spectra. The relatively low stretching frequency of the C=O bond in comparison with that of the acyl C=O in truns-Pt(COR)(X)(PPh3)~ is ascribed t o
the fact that the R groups of a-donor, unlike halides, lack of competition with the R-acyl bond for x-interac- tion. The 13C NMR spectra show that the a-carbon of the acyl ligand of
trun~-Pt(COPh)(Me)(PPh3)~
is further downfield than that of tran~-Pt(COPh)(Cl)(PPh3)~ (6 262.2 versus 212.5). Such data indicate that the back- x-donation from metal to the acyl group in trans-Pt- (COR)(R)(PPh& may be significant. TheJH-R
values corresponding to the a-hydrogen of R are in the region of 40-50Hz.
The methoxycarbonyl methyl derivative trans-Pt(CO2- Me)(Me)(PPh& (2f) was acquired via nucleophilic ad- dition of methoxide ion to
trans-[Pt(Me)(CO)(PPh3)~1-
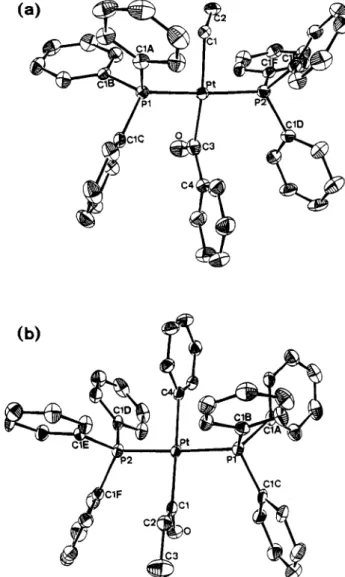
(OTf) (2e), as shown in Scheme 3. Complex 2e was prepared by abstraction of iodide ion from trans-Pt(Me)- (IKPPh3)z (2d) with use of silver trifluoromethane- sulfonate (AgOTf) in the presence of CO. The acetylide derivative truns-Pt(COPh)(C~CPh)(PPh3)~ (5c) was pre- pared by the reaction of IC and phenylacetylene with the assistance of Et3N (Scheme 4).The single-crystal structures of complexes 3c, 4b,c, and 5c were determined by X-ray diffraction. The ORTEP drawings of the two "isomers)' 3c and 4b (vide supra) are shown in Figure 1. Their crystallographic data and important bond parameters are collected in the Experimental Section. The rest of the data are supplied in the supplementary material. The distances of M-C(alky1) and M-C(ary1) bonds are slightly longer than the M-C(acy1) bonds. The acyl plane (Pt-C-0) is roughly perpendicular to the molecular plane, with
0
Figure 1. ORTEP drawings of transpositional isomers (a) trans-Pt(COPh)(Et)(PPh& (3c) and (b) trans-Pt(COEt)(Ph)- (PPh3)2 (4b). (All H atoms are omitted for clarity.) the dihedral angle being 88.3(6)' for 3c, 76.1(7)' for 4b, 99.3(7)' for 4c, and 94.5(5)' for 5c. The acetylide ligand of 5c is linear with LPt-C8-C9 = 174.9(6)' and LC8- C9-C10 = 176.8(7)'. The length of the Pt-C(acety1ide) bond (2.077(5)
A)
falls in the long range for platinum acetylides (1.9-2.1A),
suggesting that the trans influ- ence of the benzoyl ligand is rather strong.* The distance of the CEC bond (1.098(8)A)
thus is relatively short among platinum acetylides (1.07-1.22A),14
Transpositional Isomerization of trans-Pt(COR) (R)(PPh&. At 50 "C in benzene, trans-Pt(Et)(COMe)- (PPh3)2 (3a) was found to transform to trans-Pt(C0Et)- (Me)(PPhs)z (2b). The conversion was .over 80% within 2 h, and the rest of 3a decomposed to MeC(0)Et (Scheme 5). Complex 2b did not revert to 3a under similar conditions. Complexes 2b and 3a are the isomers with the methyl and ethyl groups transposed between the adjacent carbonyl carbon and Pt atom. An analogous transformation from 2c to 4a along with formation of acetophenone was also observed at 54 "C. However, the inorganic and organic species only reached the ratio 5:l:l after 5 h. After a 9-h heating, over 90% of 2c decomposed to PhC(O)Me, with only a trace of 4a
(14) Furlani, A,; Licoccia, S.; Russo, M. V. J. Chem. Soc., Dalton Trans. 1982, 2449.
Downloaded by NATIONAL TAIWAN UNIV on August 5, 2009
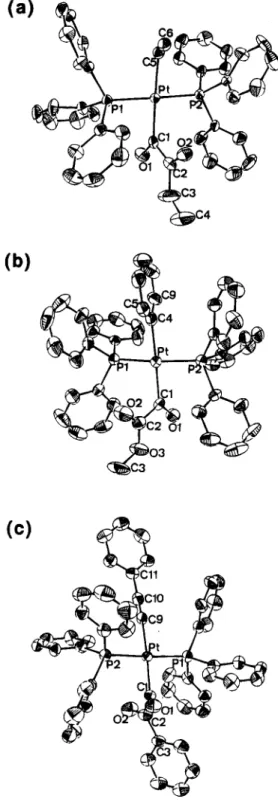
Figure 2. ORTEP drawings of trans-Pt(COCOR)(R)- (PPh&: (a) 14b; (b) 15d (c) 16c. (All H atoms are omitted for clarity.) Scheme 5 0 PPh3 0 PPh,
e
l
A (50 "C) III
MeC---Et-
EtC---Me+
benzene I PPh, I PPh, 80% 20%being detected. The reverse reaction of 4a to 2c never had a chance to build up to a detectable extent before 4a decomposed t o PhC(0)Me. When complex 3c was heated to 50 "C, 4b and PhC(0)Et appeared at a rate similar to that for the analogous reaction of 2c. The amount of 4 b was never over 30% of the starting
Organometallics, Vol. No. 12, 1994 4807
Scheme 6 0 PPh3 A II
I
CD3C-Pt-MMe MeC-Pt-CD, I I PPh3 PPh,amounts of 3c through the course of the reaction. PhC- (0)Et was the only end product. While the reaction was carried out in the presence of acrylonitrile, the rate of ketone formation from 4b markedly increased.15 How- ever, the transformation of 3c to 4b was not influenced by the addition of CH2CHCN. The isomerization of
tran~-Pt(COCD3)(Me)(PPha)z
(d3-acetyl-2a) to trans-Pt-(COMe)(CD3)(PPh& (ds-Me-2a) at 44 "C reached a 1:l ratio within 1 h. The relative amounts of these two complexes remained unchanged after 4 h. However, trans- and
cis-Pt(Me)(ds-Me)(PPh3)2
continued to in- crease at the expense of total d3-2a. These results indicate the reversibility of transposition between d3- acetyl-2a and ds-Me-2a (Scheme 6). Similar ethyl (and d5-ethyl) scrambling in tran~-Pt(COC2Ds)(Et)(PPh3)2 was never observed.Formation of cis-Pt(COR)(COR)(PPh& via Ste-
reoselective Carbonylation of trans-Pt(R)(COR)-
(PPh&. When a &-benzene solution containing trans-
Pt(COPh)(Et)(PPh& (3c) was exposed to CO, the immediate measurement of the 31P NMR exhibited complete conversion from a singlet to a pair of doublets at 6 14.9 and 15.3 with Jpt-p = 1695 and 1561
Hz,
respectively. Longer exposure t o CO (for instance, bubbling CO through the solution for 15 min or even longer) did not cause any further change. The reaction of trans-Pt(COEt)(Ph)(PPh& (4b) with CO gave the same results but took nearly 1 h to complete. Both reactions were exclusive. The doublets of doublets of31P resonances indicate that the product (designated as
60 contains two phosphines occupying the cis sites in a
square-planar geometry. The values of Jpt-p suggest that both phosphines are trans to the acyl ligand^.^ The IR spectrum of 6f, which comprises two carbonyl absorption lines a t 1604 and 1622 cm-l, and its lH NMR spectrum also support the existence of two acyl ligands. The reaction of 3c and 13C0 showed that external 13C0 was incorporated only in the resulting propionyl ligand. Such carbonylation reactions could be substantially retarded when PPh3 was present; increasing CO en- hances the reaction rate.
The reaction of
trans-Pt(COEt)(Et)(PPh3)2
(3b) with CO led to a single product, for which the 31P resonance was a singlet at 6 14.9 with Jpt-p = 1568 Hz, indicating that the two PPh3 ligands are magnetically equivalent. The 31P NMR data of the products obtained from the reactions of trans-Pt(COMe)(Me)(PPh& (4a) or trans- Pt(COPh)(Ph)(PPh& with CO were observed at 6 14.0(Jpt-P = 1558
Hz)
and 6 14.1 (Jpt-P = 1600 Hz). They are thus identified as cis-Pt(COR)2(PPh3)2 (R = Me (6a),Et (6b), Ph (612)) (Scheme 7). The carbonylation rate for the phenyl derivative is slightly faster than that for the methyl complex but markedly slower than for all ethyl derivatives under the same conditions. No car- bonylation of the acetylide complex 5c was observed.
(15) (a) Tatsumi, K.; Hoffman, R.; Yamamoto, A.; Stille, J. K. Bull. Chem. SOC. Jpn. 1981, 54, 1857. (b) Yamamoto, A. Organotransition Metal Chemistry: Fundamental Concepts and Applications; Wiley: New York, 1986; Chapter 6.
Downloaded by NATIONAL TAIWAN UNIV on August 5, 2009
4808 Organometallics, Vol. 13, No. 12, 1994 Scheme 7 0 7Ph3 II benzene RC-Pt-R'
+
CO-
I 25 "C PPh, Chen et al. 0 R' II I c : o R = R ' = M e , Et, Ph6a-c R - C \ / R = M e , W = P h 6 e R = Et, R' = Ph 6f Pt / \ Ph,P PPh3 Scheme 8 EtC- Pt-
Me I 11
2 5 0 c 0 PPh3 I1I
MeC-
Pt-
EtMeC, ,C:O
-
MeC\ ,C:O El-C, ,C:O Pt+
Pt Pt / \ / . -\ / \ Ph,P PPh, Ph3P PPh3 Ph3P PPh3 6d 6a 6b 2 1 1When a solution of truns-Pt(COMe)(Et)(PPhs)z (3a) was briefly exposed to trace CO, transformation of 3a
to
cis-Pt(COMe)(COEt)(PPh3)2
(6d) was accomplished. Bubbling CO through a solution of 3a for 10 min afforded not only 6d, as expected, but surprisingly also 6a,b in the ratio 2:l:l. Similar treatment of trans-Pt- (COEt)(Me)(PPh& (2b) in the presence of sufficient CO led to the same results of acyl scrambling. Direct reaction of 6d with CO or mixing of equal amounts of 6a and 6b with CO for 10 min also resulted in the three diacyl complexes with the same relative abundance. These reactions are summarized in Scheme 8. Noreaction ever occurs, if CO is not provided. The carbon- ylation of
truns-Pt(COPh)(Me)(PPhs)z
(2c) similarly yielded 6e, 6a, and 6c, however, in the ratio 5.5:1.5:1 in over 90% total conversion. The addition of PPh3 t othe reaction solutions severely retarded such carbon- ylative acyl scrambling. A detailed study of such reactions will be reported in a separate paper.
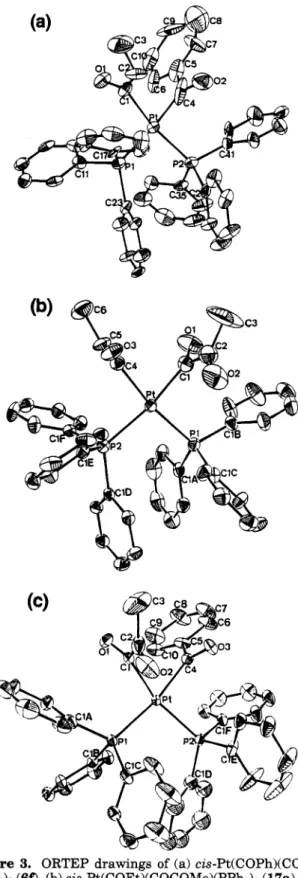
X-ray diffraction confirmed the structure of 6f, and its ORTEP diagram is shown in Figure 3a. The four ligands and the metal are in a distorted-square-planar configuration, and the two acyl ligands are in a cis disposition, with LCl-Pt-C4 being only 81.6(9)". Such a small angle causes the distance between the C1 and C4 atoms to be only 2.67(3)
A.
Both acyl planes (Pt- C1-01 and Pt-C4-02) are nearly perpendicular to the molecular plane (Pl-P2-C4-C1), with the dihedral angles being 97(2) and 93(2)", respectively. The two acyl carbonyls are s-trans to each other with a torsional angle (Ol-Cl-C4-02) of 138(3)". Other bonding parameters are similar to those of the dibenzoyl ana10g.~Instead of undergoing carbonylation, trans-Pt(CO2- Me)(Me)(PPh& (20 isomerizes t o its cis derivative 2f in over 80% yield within a few hours in the presence of
Figure 3. ORTEP drawings of (a) cis-Pt(COPh)(COEt)- (PPh& (60, (b) cis-Pt(COEt)(COCOMe)(PPh& (17a), and (c) cis-Pt(COPh)(COCOMe)(PPh& (18a). (All H atoms are omitted for clarity.)
CO. No reaction was observed after 1 day, when external CO was not provided. When 13C0 was used, the complexes 2f and the labeled cis (13C-2f) and trans (13C-2f) derivatives in the ratio 23:60:17 were acquired (based on the data of NMR integrations) (Scheme 9). Further bubbling of CO into the solution causes the
occurrence of (SP-4-2)- and ( S P - ~ - ~ ) - P ~ ( P P ~ ~ ) ( C O Z M ~ ) - (CO)(Me) (8b and 8b) (vide supra, Scheme 15).
Formation of
cis-Pt(COR)(COR')(PPh3)2
byNu-
Downloaded by NATIONAL TAIWAN UNIV on August 5, 2009
Scheme 9 0 PPh3 i l l 13co MeOC-Pt-Me ___) I 25 "C PPh3 2f / ' .\ I 2T 23% "C-2f 60% 'aC.2fl?% Ph3P PPh3 Ph3P' -'PPh3 PPh3 Scheme 10
!
TPh3
AgBF4,CH2C12 Ph$C-Pt-CI-
I
-AgCI, -30 "C PPh3 9c 7 R = Ph 8~ 34% R = "Bu 6h 60% R = allyl 6g 38% Scheme 11 0 -70 "C ri / \ Ph3P PPh3 7 a M e 0 0 i i l II I1 PhC\ /C=O PhC\ PhC, /Me Pt+
Pt+
Pt / '.\
/ ' .\ Ph3P PPh3 Ph3P PPh3 OC' 'PPh3 W 16% 212 46% 8a 38%cleophilic Addition of Carbanion to cis-[Pt(COR)- (CO)(PPhs)al+. Abstraction of chloride from trans- Pt(COCOPh)(Cl)(PPh3)2 with AgBF4 in dichloromethane
at -29 "C led to formation of
cis-[Pt(COPh)(CO)(PPh3)21-
(BF4) (7).16 The reactions of complex 7 with PhLi orRMgCl (R = allyl, "Bu) provide a complementary route to
cis-Pt(COPh)(COR)(PPh3)2
(R = Ph (6c), allyl (6g), "Bu (6h), respectively), albeit in low yields (generally<40%) (Scheme 10). The single-crystal X-ray structure of 6c was determined and reported in a prior com- m ~ n i c a t i o n . ~ In the reaction of 7 and MeLi, cis-Pt- (COPh)(Me)(PPh& (2c') and (sP-4-3)-Pt(PPh3)(CO)- (COPhI(CH3) (8a) were formed along with 6e in a ratio 2.5:3:1 in 55% conversion (Scheme 11). When this reaction was carried out in the presence of a n extra 2 equiv of PPh3, only 2c' and 8a were obtained in 1:3.5 relative yields, and the total conversion was raised to 90%. Both 8a and 2c' have been isolated and charac- terized spectroscopically and cry~tallographically.~ Ad- dition of PPh3 to a solution of 8a readily results in complete conversion to 2c'. Bubbling CO into a solution
(16) (a) Huang, T.-M.; Chen, J.-T.; Lee, G.-H.; Wang, Y. Organome-
tallics 1991, IO, 175. (b) Sen, A.; Chen, J.-T.; Vetter, W. M.; Whittle,
R. R. J. Am. Chem. SOC. 1987, 109, 148.
Organometallics, Vol. No. 4809 Scheme 12 0 PPh3 0 PPh3 AgOTf/CH2C12/-10 "C II
I
-AgCII
R$C-Pt- CI
I1I
R$C-Pt-OTf 6Ph3 / o PPh3 sad/
l l a dof 2c' slowly caused the appearance of 8a and its isomer
(SP-4-4)-Pt(PPh3)(CO)(COPh)(CH3)
(8a'). The intercon- version between 6e and 2c' has never been detected. Instead, both complexes thermally decomposed to ac- etophenone.Synthesis and Characterization of trans-Pt(C0- COR)(R')(PPhs)z. I t has been previously found that ethylation of
[truns-Pt(COCOPhXTHF)(PPh3)2l(BF4)
(104 with EtzZn results in trans-Pt(Et)(COCOPh)(PPh& (14c).11 Due to the unfortunate solubility of the THF derivatives in common organic solvents, a modified procedure in which trans-Pt(COCOR)(OTf)(PPh3)2 (OTf = OS02CF3; R = Me ( l l a ) , E t ( l l b ) , Ph ( l l c ) , OMe ( l l d ) ) or[trans-Pt(COCOR)(H20)(PPh3)230
(12a- d)17 were used instead has been established for the synthesis of other derivatives of 14c. Ethylation of complexes lla-d or 12a-d with use of EtaZn led totruns-Pt(COCOR)(Et)(PPh3)2 (14a-d) in high yield
( > 70%). Transmetalation between trans-Pt(COC0R)- (Cl)(PPh3)2 (9a-c) and EtsZn also feasibly affords the a-ketoacyl ethyl complexes 14a-c in good yields (-70%). However, the reaction of
tran~-Pt(COCO2Me)(Cl)(PPh3)2
(9d) with EtzZn was complicated. trans-Pt(COC0R)- (Me)(PPh& (13a-d) and trans-Pt(COCOR)(Ph)(PPh& (15a-d) were prepared from the reactions of lla-d or 12a-d with PhzZn or MenZn, respectively. Relatively low yields were obtained in such cases (Scheme 12). In the reactions of 9a-c with MezZn or PhsZn, the desired products 13a-c and 15a-c resulted along with the dimethyl or diphenyl products ci~-PtR2(PPh3)2, which severely interfered with the purification and lowered the yields of the target compounds. The acetylide complex
trans-Pt(COCOPh)(C=CPh)(PPh3)2
( 1 6 ~ ) was prepared according to a procedure similar to that used for preparing the benzoyl analog 5c.A feature of trans-Pt(COCOR)(R)(PPh& (except R = OMe) that distinguishes these species from most of the known organoplatinum complexes is their striking purple appearance. Complexes 13- 15(a-c) generally exhibit a long-wavelength MLCT band in the region over 530 nm, which is about 200 nm longer than the corresponding absorption of the acyl analogs 2-4(a- c). The infrared spectra of such complexes, as for other a-ketoacyl complexes, comprise two well-resolved car- (17) (a) Chen, J.-T.; Yeh, Y.-s.; Tzeng, W.-H.; Huang, T.-M.; Cheng, M.-C.; Lee, G.-H.; Wang, Y. J. Chin. Chem. Soc. 1991, 38, 573. (b) Chen, J.-T.;Yeh,Y.-S.;Lee, G.-H.; Wang,Y. J. Organomet. Chem. 1991, 414, C64.
Downloaded by NATIONAL TAIWAN UNIV on August 5, 2009
4810 Organometallics, Vol. 13, No. 12, 1994 Scheme 13 Chen et al. 1 0 PPh, R' = Me 138-c R ' = E t 148-C R = M , R = E t 6 d ' R ' = P h l S b c R = M , R = P h W (R = Me 8, Et b, Ph C) R' = Ph 81 R = R' = Me, Et, Ph 8d-d R = Et,
bony1 stretching bands. The frequency with respect to a-CO is lower than 1600 cm-l, which is similar to those of tran~-Pt(COR)(R)(PPh3)2. The 13C NMR spectrum of 13d consists of the resonances of a-CO a t 6 237.1
(Jc-R = 844 Hz), of p-CO at 6 168.7 (Jc-R = 129 Hz), and of the methyl ligand a t 6 -8.6 (Jc-p = 9.1 Hz, Jc-pt = 398 Hz). The methoxyoxalyl derivatives are distin- guishable from other a-ketoacyl complexes in several respects. Complexes 13d-15d are yellow. Indeed, their MLCT bands show a blue shift of about 100 nm relative to those of other a-ketoacyl analogs. The IR absorptions of the p-CO of the methoxyoxalyl ligands are in the region of higher wavenumbers (> 1700 cm-l), and the a-CO absorptions are similar to those of other a-ketoacyl ligands.
The molecular structures of 13a, 14b,d, 15d, and 16c have been determined by X-ray crystallographic analy- sis. The ORTEP drawings of the three representatives 14b, 15d, and 16c with ellipsoids at the 20% probability level are shown in Figure 2. They have a trans square- planar geometry, and the bond parameters of trans-Pt- (COCOR)(R)(PPh& are similar to those corresponding ones of trans-Pt(COR)(R)(PPh3)2. The oxalyl moiety in 14b or 15d is in a planar s-trans configuration with the Ol-Cl-C2-02 torsion angle being 178(2) or 178(2)", respectively, and is substantially distorted from the planar mold in 16c (141( 1)'). The oxalyl C-C bonds of the a-ketoacyl ligands are generally ca. 0.1
A
longer than the standard C(sp2)-C(sp2) bond (ca. 1.45A).17,18
These data indicate that electronic delocalization in the a-ketoacyl ligands is of limited importance. The wide span of the a-ketoacyl conformation may be readily influenced by either steric or environmental conditions, such as the ligand size, crystal packing, etc.Transpositional Isomerization of truns-Pt(C0- COR) (R') (PPh3)2 Leading to trans-Pt(COR) (COR')- (PPhs)z. A typical reaction of trans-Pt( COCOR)(R)- (PPh3)2, which reflects the weak oxalyl C-C bond of the a-ketoacyl ligands, is spontaneous decarbonylation of such complexes in solutions at room temperature, leading to trans diacyl complexes. As shown in Scheme 13, trans-Pt(COCOR)(R)(PPh3)2 species (13c, 14a-c, 15a,b) exclusively transform to trans-Pt(COR)(COR)- (PPh3)2 (R = R' = Et (6b'); R = Me, R = Et or R = Et,
R = Me (6d); R = Me, R = Ph or R = Ph, R = Me (6e'); R = Ph, R = Et or R = Et, R = Ph (6f)). (Our previous attempts to prepare the trans diacyl complexes by the reactions of trans-Pt(COR)(CO)(PPh3)2+ and nucleophilic reagents such as RMgX and R L i only resulted in unidentified decomposed residue.) The reactions of Scheme 13 are facile in noncoordinating solvents. Coordinating solvents such as diethyl ether, THF, acetone, acetonitrile, and water or a ligating compound such as PPh3 severely hindered these reac-
(18) Chen, J.-T. J. Chin. C h e m . SOC. 1992, 39, 603.
tions to varied extents. The reaction of 15c resulted in trans-Pt(COPh)2(PPh3)2 (6c') as the major product along with unidentified side products. The products from the reactions of 13a and 13b were complicated, although the expected tran~-Pt(COMe)z(PPh3)2 (6a') and 6 d in <20% yields were indeed detected by NMR techniques. The reactivity shown in Scheme 13 apparently depends on the variation of R , following the order Et >> Ph > Me. Besides, when complexes have a common R ligand, the reactivity of benzoylformyl derivatives (Pt-CO- COPh) appears to be more facile than that of the derivatives with COCOMe and COCOEt.
At 25 "C in the presence of trace I3CO in CDC13, exclusive transformation from 14b to 6b' without in- corporation of 13CO was still found within 1 h. However, when the reaction was carried out at -10 "C, a new product (designated as lsC-17b, vide supra) instead of 6b' was acquired after 12 h. The 31P NMR spectrum of 13C-17b consists of a pair of doublets at 6 14.0 (Jc-P = 112 Hz, Jp-pt = 1577 Hz) and 6 13.7 (Jc-p FZ 16 Hz,
J p - p t = 1861 Hz), and its lH NMR spectrum has two
sets of ethyl resonances a t 6 2.00,0.22 and 6 1.74,0.68, respectively. The NMR data for 13C-17b resemble those for the cis diacyl complex, and the incorporation of 13C0 completely takes place in the resulting propionyl ligand. The complex lSC-17b is thus assigned as the novel acyl a-ketoacyl complex
cis-Pt(l3COEt)(C0COEt)(PPh3)2
with the labeled CO completely incorporated in the propionyl ligand. The coexistence of 14b and sufficient I3CO in solution at 25 "C also afforded 13C-17b exclusively within a few minutes. The results of the labeling experiments as shown in Scheme 14 strongly support that the reactions of Scheme 13 are intramolecular and that the carbonylation of 14b is intermolecular.The alkyl (or aryl) methoxyoxalyl derivatives trans- Pt(COC02Me)(R)(PPh3)2 follow a reaction pattern dif- ferent from that of other a-ketoacyl analogs. Heating
tran~-Pt(COCOzMe)(Me)(PPh3)2
(13d) at 65 "C for 5 minfirst resulted in cis-Pt(CO2Me)(Me)PPh3)2 (2f) in >90% yield. Complex 2f' then suffered displacement of PPh3 by CO, yielding (SP-4-2)- and (SP-4-4)-Pt(PPh3)(CO2- Me)(CO)(Me) (8b and 8b').l9 At 45 "C in &-acetone, complex 14d decomposed to ci~-Pt(COzMe)(Et)(PPh3)2
(30 in 30% conversion within 10 min. After 30 min, the starting 14d completely disappeared, and complex 3f and the new compound cis-Pt(COCO2Me)(COEt)- (PPh3)z (17d) in the ratio 1.2:l were found. The latter product was apparently formed via facile carbonylation of 14d. The incorporated CO has to be released from -COC02Me of 14d during the formation of 3f. Complex 3f was isolated by recrystallization from acetonitrile. Complex 3f undergoes substitution of CO for a PPh3 ligand to form (SP-4-2)- and (SP-4-4)-Pt(PPh3)(CO2Me)- (CO)(Et) (8c and 8c') in the ratio 2.6:l with 60% conversion (Scheme 15). The rest of 3f decomposed, and identification of the residue was not attempted.
Formation of
cis-Pt(COCOR)(COR')(PPhs)2
via Carbonylation of trans-Pt(R')(COCOR)(PPh3)2. Re- actions of trans-Pt(COCOR)(R)(PPh3)2 ( R = Et, R = Me, Et, Ph, OMe (14a-d); R = Ph, R = Me, Et, Ph, OMe (15a-d) with CO readily provided the bright yellow acyl a-ketoacyl complexes cis-Pt(COCOR)(COR)(PPh& ( R= Et, R = Me, Et, Ph, OMe (17a-d); R = Ph, R = Me, (19)Hsu, B.-C.; Huang, T.-M.; Chen, J.-T.; Cheng, M.-C.; Lee, G.- H.; Wang, Y. J. Chin. Chem. SOC., in press.
Downloaded by NATIONAL TAIWAN UNIV on August 5, 2009
Reactions of Square-Planar Diorganoplatinum Organometallics, Vol. No. Scheme 14 0 PPh3 II
I
1 ;
EtC-
Pt-
CEt (6b' >95%) 0 PPh3 ItI
E%C-Pt-Et+
tracel3c0
52
pp;I
Elf;lC, yC.0 PPh3 -10°C 0 p t (17b >95vo) / \ 14b I Ph3P PPh3 sufficient l3CO/25 "Ct
Scheme 15 0 PPh3 III
MeO$C-Pt- Mel
PPh3 13dII
MOC\ /M Pt / \ Ph3P PPhsz?
S O Y O 0 R = Me 8b W o R = Et 8C 24%+
?
MOC\ /R Pt / \ Ph3P CO R = Me 8b' 30% R = Et 8c' 9% 14d 17d 45% 3f 55%I
Scheme 16 n PPh3 O ? i i I benzene R:C, ,C:O R$C-Pt-R+
CO-
25 "CA
Ph,P PPh,I
PPh, R ' = Et 1- R = E t 1 7 4 R'=Ph 15a-d R' = Ph 18ad(R = Me, Et, Ph, OMe ad)
Et, Ph, OMe (Ma-d)) (Scheme 16). Further exposure t o CO at 1 atm did not cause any change to the acyl a-ketoacyl products. The carbonylation of ethyl com- plexes 14a-d (ca. 20 mg and monitored by NMR) at 0 "C in CO-saturated chloroform is quantitative, and the reactions could be done within 1 h. It took over 1 day for the phenyl derivatives to finish the reactant under the same conditions. During the long course of carbo- nylation of complexes 15a-d, the products of cis benzoyl a-ketoacyl complexes have already started to decompose (vide supra). The carbonylation of methyl derivatives could be observed in benzene, but the reactions were far from completion. The acetylide derivatives have never been detected under atmospheric pressure of CO. Adding PPh3 t o the systems and decreasing the con- centration of CO have markedly retarded such carbo- nylation.
The NMR data for the new cis acyl a-ketoacyl complexes are similar to those for the cis diacyl com- plexes. In the infrared spectra of 17 and 18, three prominent carbonyl bands could be clearly identified. The MLCT bands are generally (30-50 nm) lower than those of trans alkyl a-ketoacyl complexes. The X-ray structures of 17a and 18a have been obtained and are shown in parts b and c of Figure 3, respectively. Like the diacyl species, the cis acyl a-ketoacyl complexes have a distorted-square-planar geometry with C-Pt-C = 79.8(4)" in 17a and 78.1(4)" in Ma. The distance between the two a-carbon atoms is 2.63(1)
A
in 17a and2.55(1)
A
in Ma, which fall in the shortest realm for the nonbonded vicinal a-hydrocarbyls.16a.20 Their a-ke- toacyl ligands are in a n s-trans configuration with the torsion angle Ol-Cl-C2-02 being 158(1)" in 17a and 148(1)O in Ma. The Pt-C(a-ketoacyl) bond is slightly shorter than the Pt-C(acy1) bond in each of the com- plexes. The Pt-P bond trans t o the a-ketoacyl ligand is slightly longer than the one trans to the acyl ligand. Thermolysis of cis-Pt(COR)(COR)(PPh& andcis-Pt(COCOR)(COR)(PPhs)2. The aforementioned diorganoplatinum species are generally subjected to thermolysis mainly via reductive coupling of the two organic ligands. Specifically, ketone formation was obtained from trans acyl alkyl or trans acyl aryl complexes; cis diacyl complexes tend to decompose to both ketone and diketone with the former as the major product. However, when sufficient CO was applied, diketones became predominant. The decomposition of 6d resulted in crossover ketones and diketones. The relative yields of the organic products from various diacyl complexes are collected in Table 1. Facile decar- bonylation of the a-ketoacyl group and the strong Pt-C bonds in the cis acyl a-ketoacyl complexes make our attempts at "triply carbonylative" coupling unfeasible. Thermolysis of 17a in the presence of CO at 66 "C gave a result comparable to the reaction of 6d, also listed in Table 1.
Detailed NMR investigation indicates that cis-Pt- (COCOR)(COR)(PPh& species (17a-c, 18a-c) are subjected to exclusive decarbonylation, first yielding the trans diacyl complexes 6 b - f quantitatively (Scheme 17). Such a reaction affords a pragmatic route for the synthesis of 6c', since it could not be satisfactorily obtained from 16c. The presence of excess PPh3 sig- nificantly suppressed such processes. When the reac- (20) Protasiewicz, J. D.; Masschelein, A.; Lippard, S. L. J . Am. Chem. Soc. 1993,115, 809.
Downloaded by NATIONAL TAIWAN UNIV on August 5, 2009
4812 Organometallics, Vol. 13, NO. 12, 1994
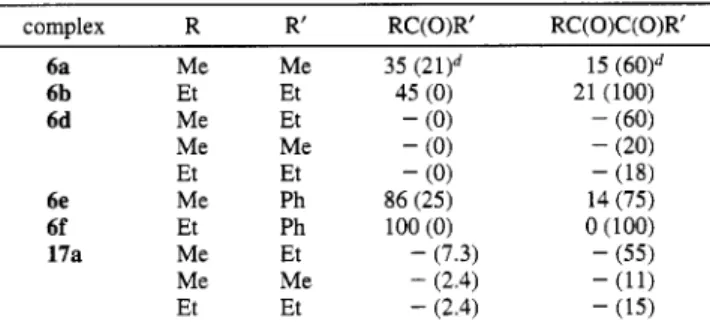
Table 1. Relative Abundance of Organic Product@ from Thermally Decomposed cis-Pt(COR)(COR')(PPh3)26 and
cis-Pt(COEt)(COCOMe)(PPh3)f Chen et al. complex R R' RC(0)R' RC( 0 ) C (0)R' 6a Me Me 6b Et Et 6d Me Et Me Me Et Et 6e Me Ph 6f Et Ph 17a Me Et Me Me Et Et 35 (2l)d 45 ( 0 ) - (0) - (0) - (0) 86 (25) 100 (0) - (7.3) - (2.4) - (2.4) 15 (60)d 21 (100) - (60) - (20) - (18) 14 (75) 0 (100) - (55) - (11) - (15)
a All data are based on the integration data of 'H NMR spectra.
Reactions were carried out in &-benzene at 50 "C. Reaction was carried out in &-benzene at 66 "C. Data in parentheses were obtained from the reactions in the presence of saturated CO.
Scheme 17 0 ?I I 1 0 PPh3 R$C, ,C:o
-
A II1
R C - P t - C R + CO O IPt\I ; ;
Ph3P PPh3 PPh, R ' = Et 17ac R ' = P h 18a-C 6b- 6P >95%tion solution was exposed to air, the trans diacyl complexes would further decompose to the acyl carbox- ylato complexes. In a typical case, when 17a was dissolved in undried &-benzene, exclusive transforma- tion from 17a to 6d' was completed after 1 day according
to NMR spectroscopy. A few days later, two singlets at
6 19.48 (Jp-pt = 3655 Hz) and 19.55 (Jp-pt = 3655 Hz) in the 31P NMR spectrum increased slowly a t the expense of 6 d . In the lH NMR spectrum, the cor- responding growing resonances were observed at d 0.24
(t, 3H,
JH-H
= 7.2 Hz), 1.25 (s, 3H), and 1.80 (9, 2H,JH-H
= 7.2 Hz) and a t 6 0.66 (t, 3H,JH-H
= 7.4 Hz),1.40 (s, 3H), 1.51 (9, 2H,
JH-H
= 7.4 Hz), which have been identified astrans-Pt(COMe)(OCOEt)(PPh3)2
(19d) and trans-Pt(COEt)(OCOMe)(PPh& (19d), respec- tively. The relative abundances of 19d and 19d were roughly 1:1, and total conversion from 17a was 50%. In a labeling experiment, the reaction of 14c with 13C0 forms cis-Pt( 13COEt)(COCOPh)(PPh3)2 (13C-17c), which spontaneously transforms to trans-Pt(13COEt)(COPh)- (PPh& (13C-6f') via decarbonylation of the benzoyl- formyl group. Then,tran~-Pt(~~C0Et)(OCOPh)(PPh3)2
(13C-19f) and
tran~-Pt(0~~C0Et)(COPh)(PPh&
(13C- 19f') were detected (Scheme 18).The reaction starting directly from 6 d (in other words, without the presence of CO) gave the same results. Under ambient conditions, trans diacyl com- plexes slowly transform to mixed trans acyl carboxylato complexes: trans-Pt(COR)(OCOR)(PPh3)2 or trans-Pt- (OCOR)(COR)(PPh& (R = R = Me (19a), E t (19b), Ph ( 1 9 ~ ) ; R = Me, R = Et (19d); R = Et, R = Me (19d); R = Me, R' = Ph (19e); R = Ph, R = Me (19e'); R = Et, R
= Ph (19f); R = Ph, R' = E t (198)) (Scheme 19). When halogenated solvent (CHCl3 or CH2C12) was used, trans- Pt(COR)(Cl)(PPh& and tran~-Pt(COR)(Cl)(PPh3)2 were the major products. The carboxylato products became the minor ones. Organic ketone was detected as well, however, only in a small quantity. When the trans diacyl complexes were kept under a dry nitrogen atmosphere, ketone was the major product along with
many unidentified inorganic species. In any event, no carboxylato complex was detected in such cases.
Platinum(I1) complexes with hard oxygen-donor ligands are relatively rare.17,21 The single-crystal X-ray struc- ture of complex 19b has been determined and consti- tutes the first example of a structurally characterized platinum acyl carboxylato species, although a perfluoro analog is known.22 As is shown in the ORTEP picture in Figure 4, complex 19b has a trans square-planar configuration. The carboxylato ligand is in an oxygen- bound monodentate mode. The metal-bound 01-C1 bond is slightly longer than the metal-detached 02- C 1 bond (1.279(7) and 1.230(8)
A,
respectively). Oxygen- donor ligands are usually weakly bonded to the PMII)center. Indeed, the Pt-01 bond is 2.142(4)
A.
The Pt- C4 bond (1.989A)
trans to the carboxylato ligand is thus shorter than the Pt-C(acy1) bond in trans-Pt(COR)(R)- (PPh& or other acylplatinum complexes. The carbox- ylato plane Pt-01-C1 forms a dihedral angle of 68.2- (4)" with the molecular plane Pl-Ol-P2-C4. The angle between the acyl plane Pt-C4-03 and the molecular plane is 98.0(6)", again being close to a vertical feature. The torsion angle 03-C4-01-C1 is thus 29.8(7)".Discussion
Transmetalation of Organoplatinum with Dior- ganozincs. Alkylation of platinum complexes via transmetalation has been one of the most extensively used strategic methods to prepare organoplatinum species.23 The nucleophilic alkylating reagents em- ployed for such a purpose comprise a wide domain of organometallics, including the Grignard reagents, or- ganolithium, and reagents containing other main-group metals such as Hg, Cu, Cd, T1, Sn, etc.lb Curiously, the use of diorganozinc reagents for such a purpose has been uncommon. The convenient and efficient synthesis of the new trans alkyl (aryl) acyl and trans alkyl (aryl) a-ketoacyl complexes of platinum(I1) with use of dior- ganozinc compounds provides a pragmatic extension for the usage of the transmetalation reactions of organo- platinum.
When the Grignard reagents and organolithium were used, the desired diorganoplatinum products were recovered along with significant amounts of dihaloplati- num(I1) and Pt(0) residue, which caused severe prob- lems in separation. It seemed that mild reagents for alkylation and reduction could be more appropriate. On the other hand, neither organomercury nor organotin species show any reactivity with complexes la-c at ambient temperature. Increased temperature had en- hanced the decarbonylation rate of the starting acyl- platinum, and failed alkylation. Our survey ended with the satisfactory emoployment of diorganozincs. The
(21) (a) Hartley, F. R. Chem. Rev. 1973, 73, 163. (b) Hartley, F. R.; Davies, J. A. Rev. Inorg. Chem. 1982,4,27. (c) Davies, J. A.; Hartley,
F. R. Chem. Rev. 1981,81,79. (d) Davis, J. A,; Hartley, F. R.; Murray, S . G. Inorg. Chem. 1980,19,2299. (e) Bryndza, H. E.; Tam, W. Chem. Rev. 1988, 88, 1163. (0 Siedle, A. R.; Gleason, W. B.; Newmark, R.
A.; Pignolet, L. H. Organometallics 1986, 5, 1969. (g) Siedle, A. R.; Newmark, R. A.; Gleason, W. B. J . A m . Chem. SOC. 1986,108,767. (h) Demma, A,; Lukehart, C. M.; McPhail, A. T.; McPhail, D. R. J . A m . Chem. SOC. 1989,111, 7615. (i) Kim, Y. J.; Osakada, K.; Takenaka,
A,; Yamamoto, A. J . A m . Chem. SOC. 1990,112, 1096.
(22) Blake, D. M.; Shields, S.; Wyma, L. Inorg. C h m . 1974,13,1595.
(23) Hartley, F. R. The Chemistry of Platinum and Palladium;
Applied Science: London, 1973.
Downloaded by NATIONAL TAIWAN UNIV on August 5, 2009
Organometallics, Vol. Scheme 18 6 b - 6t' R = R'I Me 19a R = R' = Ph 1 9 ~ R = M e , R ' = E t l O d R=Et, R ' = P h l W R = Me, R' = Et 19d' R = Et, R' = Ph 1 W R =R' =Et 19b R = Me, R'= Ph 1%' R =Me, R'= Ph 19e ""L 0 Et 25 "C I'
I
PPh, 14C '3C-6t' dry benzene/N2 6Figure 4. ORTEP drawing of trans-Pt(OCOEt)(COEt)- (PPh3)z (19b). (All H atoms are omitted for clarity.) reaction conditions used for transmetalation of organo- haloplatinum with RzZn were mild, and the yields for the desired diorganoplatinum products had been excel- lent. In the reactions of truns-Pt(COR)(X)(PPh3)2 or trans-Pt(COCOR)(L)(PPh&+ with R'zZn, trans diorgano derivatives result nearly quantitatively. However, in the reaction of trans-Pt(R)(X)(PPh3)2 with R'zZn, cis-Pt- (R)(R')(PPh& becomes the predominant product.lg The stereochemical discrepancy is presumably not simply due to thermodynamic factors, since both trans and cis diorganoplatinum isomers exist. Further research is still needed to acquire kinetic rationales.
Trans Influence of a-Ketoacyl Ligands. 31P NMR spectroscopy affords a powerful tool for structural characterization of the diorganoplatinum( 11) species. The phosphorus-platinum coupling is assumed t o be dominated by the Fermi contact term and is suitable
13,
No.
12,1994 4813for probing the strength of the Pt-P bond.8 In a pragmatic sense, the value of lJp-pt with respect to a
phosphino ligand is sensitive to its trans ligand. In comparable chemical environments, the greater value of
V-pt
reflects a stronger P-Pt bond, thus indicating a smaller trans influence applied to this phosphine by its trans ligand. Bennett and his associates have measured the value of lJp-pt for Pt(Me)(L)(dppe) (dppe = bis(diphenylphosphino)ethane), in which L represents a ligand with a a-carbon donor, to correlate lJp-pt withthe trans influence of L. The results assert that the alkyl, aryl, and acyl groups are all in the category of
strong trans influence and follow the order C(O)CsHg > CsH9 FZ Et > Ph > CHzPh
The synthesis of cis acyl a-ketoacyl complexes pro- vides new examples of organic ligands with strong trans influence. In
cis-Pt(COCOR)(COR')(PPh3)2,
the PPh3 trans to the acyl ligand always has a smaller lJp-ptvalue than that of PPh3 trans t o an a-ketoacyl ligand (Table 2). Such data suggest that the a-ketoacyls generally have a smaller trans inflence than the acyls but a larger influence than the aryl and the alkyl groups. Meanwhile, the length of the P-Pt bond trans
to the a-ketoacyl group is indeed slightly shorter than that trans t o the acyl ligand in the structures of 17a,d and 18a. An extended list for some a-hydrocarbyls with decreasing trans influence is thus arranged as C(0)Et > C(0)Me > C(0)Ph > E t FZ COCOEt, COCOMe, COCOPh > Ph > COCOzMe
=-
COzMe > Me.Mechanism and Reactivity of Transpositional Isomerism of truns-Pt(COR)(R)(PPhs)z and truns-
Pt(COCOR)(R)(PPh& One may conceive that the reactions in both Schemes 5 and 13 result in two groups transposing between the adjacent Pt and C atoms:
Me.24
(24) (a) Appleton, T. G.; Bennett, M. A. Znorg. Chem. 1978,17, 738. (b) Bennett, M. A.; Rokicki, A. J. Organomet. Chem. 1983,244, C31.
(c) Bennett, M. A.; Rokicki, A. Organometallics 1986, 4 , 180.
Downloaded by NATIONAL TAIWAN UNIV on August 5, 2009
4814 Organometallics, Vol. 13, No. 12, 1994 Table 2. 31P NMR Data for cis-Pt(RMR'MPPh&
Chen et al. Scheme 20 111 ~~ ~ complex R R' ~(PR)(JP-R, H Z ) ~ ~ ( P R ~ ) ( ~ H z ) 2c' Me COPh 24.4 (2196) 20.0 (1423) 2 f Me C02Me 22.5 (1987) 21.4 (1879) 3 f Et CO2Me 22.6 (1987) 21.4 (1770) 6a C(0)Me C(0)Me 14.0 (1558y
6b C(0)Et C(0)Et 14.9 (1568) 6c C(0)Ph C(0)Ph 14.1 (1600y 6d C(0)Me C(0)Et 13.2 (1517) 14.8 (1582) 6e C(0)Me C(0)Ph 13.9 (1676) 15.5 (1568) 6f C(0)Et C(0)Ph 14.9 (1695) 15.3 (1516) 6g C(O)CsH5 C(0)Ph 14.7 (1598) 14.3 (1652) 6h C(0)"Bu C(0)Ph 15.4 (1520) 14.2 (1690) 17a C(O)C(O)Me C(0)Et 14.1 (1877) 14.3 (1571) 17b C(O)C(O)Et C(0)Et 13.7 (1861) 14.0 (1577) 17c C(O)C(O)Ph C(0)Et 14.8 (1860) 14.1 (1571) 17d C(O)C02Me C(0)Et 14.0 (1949) 14.3 (1533) 18a C(O)C(O)Me C(0)Ph 14.0 (1804) 14.7 (1702) 18b C(O)C(O)Et C(0)Ph 13.6 (1786) 14.9 (1706) 18c C(O)C(O)Ph C(0)Ph 14.3 (1786) 15.8 (1691) 18d C(O)C02Me C(0)Ph 13.7 (1882) 14.2 (1646)
Data were measured in &-benzene.
X Y Y X
1
1
I
I
L,Pt-C(O)
-
kPt-C(O)case 1: X, Y = alkyl or aryl case Ii: X = alkyl or aryl, Y = acyl
PPh3"
In the case of trans acyl alkyl (or aryl) complexes, two alkyl (or aryl) groups exchange to give another acyl alkyl isomer. In Scheme 13, an alkyl (or aryl) ligand switches with an acyl fragment of the a-ketoacyl ligand in truns- Pt(COCOR)(R)(PPh3)2 to give truns-Pt(COR)(COR)- (PPh3)2. These two types of isomerization are unprec- edented for square-planar platinum(I1) systems. We name it transpositional isomerism.
From a mechanistic point of view, since there is no incorporation of external CO in both reactions, they may comprise an intramolecular CO transfer between two trans organic ligands. Using the reaction of the alkyl a-ketoacyl derivative as an illustrative example, a consecutive mechanism is depicted in Scheme 20. The reaction may be preceded by decarbonylation of the a-ketoacyl ligand via an acyl migration from ligand to metal, first resulting in the five-coordinate intermediate 111, which contains an acyl, an alkyl, and a carbonyl ligand on the same metal. The subsequent processes will comprise ligand rearrangement. The alkyl and the carbonyl ligands require cis positions, so that they will be available for migratory CO insertion. For example, the intermediate
IV
may be attained simply by moving the carbonyl in I11 by 90" to the vacant site. It then will constitute a good candidate for the ensuing CO insertion t o finish the formation of the trans diacyl product. Pathways involving other unraveled five- coordinate isomerization are possible, although they may be more complicated.Recalling that such isomerization reactions could be hindered by coordinating species, we must consider a dissociative mechanism involving four-coordinate isomer- ization (Scheme 21). In such a pathway, the dissociation of PPh3 from the starting reactant first opens a coordi- nation site (as in
VI,
and facilitates the migration of the acyl group from the a-ketoacyl moiety to the metal. The four-coordinate intermediate VI, in which the alkyl and the carbonyl ligands are in a trans disposition, results. Successive ligand substitutions lead to intermediate VI1Vlll VI1
and then
VIII,
which contains the alkyl group and the carbonyl in cis positions. The alkyl migration in VI11 and entering of the PPh3 group will result in the trans diacyl complex. There are at least three reasons against this mechanism. First of all, the formation of VI11 from VI1 by ligand substitution would allow the incorporation of external CO into the products, which is not the case. Second, intermediates VI-VI11 are known to be stable species. However, none of them have been detected throughout the course of the reactions. The most critical disproof of Scheme 21 is that carbonylation does not occur for cis-Pt(COPh)(Me)(PPh3)2 (2c'), (SP-4-3)- and(SP-4-4)-Pt(PPh3)(CO)(COPh)(Me)
(8a and Sa'), and the related species (Scheme 15).19Mechanism and Reactivity of Stereoselective Carbonylation. The reactions of trans square-planar diorganoplatinum(I1) complexes with carbon monoxide, leading to the cis carbonylated derivatives, are facile under mild conditions (Scheme 7 or 16). The stereose- lectivity and reactivity of such an unique transformation are peculiarly intriguing. Our preliminary mechanistic studies on the carbonylation of both truns-Pt(COR)(R)- (PPh312 and truns-Pt(COCOR)(R)(PPh3)2 showed that the externally provided CO had been specifically incor- porated into the alkyl (or aryl) ligand. Besides, decreas- ing the concentration of CO and adding PPh3 into the system severely hindered the carbonylation. Such observations may be explained by the mechanism shown in Scheme 22, in which the case of a n a-ketoacyl derivative is again used for representation. Reversible substitution of CO for a PPh3 ligand first results in the intermediate
E,
in which the alkyl ligand is trans to the a-ketoacyl of strong trans influence and possesses the insertion-preferred cis-to-CO arrangement. The ensuing alkyl migration from metal to the carbon atom of the terminal carbonyl establishes the cis acyl a-ke- toacyl structure (as in X). The succeeding occupancyDownloaded by NATIONAL TAIWAN UNIV on August 5, 2009
Scheme 22
IX
0 R'
X
of the vacant coordination site by PPh3 thus accom- plishes the reaction. Since the intermediate
M
has never been detected in any case, such a mechanism presumably follows a steady-state condition. Scheme22 is consistent with the mechanism proposed for the diorganopalladium-mediated carbonylation reactions by Y a m a m ~ t o . ~ ~ An alternative associative mechanism involving a five-coordinate intermediate cannot be arbitrarily excluded, although it would be difficult to elucidate the exclusive trans to cis transformation.
The reactivity of carbonylation in the trans diorga- noplatinum(I1) complexes follows the unusual order E t >> Ph > Me. In fact, the carbonylation of trans-Pt- (COCOR)(Me)(PPh3)2 by no means competes with their decarbonylation reactions, which lead to the trans diacyl derivatives. The peculiarly slow carbonylation of the methyl derivatives is ascribed to the rapid reverse migration of the methyl group from acetyl to metal in the intermediate
X.
Such an assertion is supported by the reaction mechanism of transpositional isomerization of trans-Pt(COR)(Et)(PPh&, in which the decarbon- ylation of the acyl ligand follows the order MeC(0) > PhC(0) >> EtC(0). Consequently, the acetyl derivative 3a readily transforms to 2b. The analogous reactivity of the benzoyl derivative 3c (converting to 4b) is much lower, and none of the propionyl complexes (2b, 3b, or 4b) exhibit any activity t o this kind of isomerization at all. A similar stability of the propionyl group to decar- bonylation has been also observed in other systems and was attributed to the electron-releasing character of the ethyl moiety.25In contrast to the trans diorganoplatinum(II), treat- ment of cis acyl alkyl complexes with CO only leads to ligand substitution instead of carbonylation. For in- stance, the reaction of CO with cis-Pt(COPh)(Me)(PPh3)2 (2c') gives (SP-4-3)- and
(SP-4-4)-Pt(PPh3)(CO)(COPh)-
(Me) (8a and 8a'). It is worth noting that the methyl group in 8a' is cis to the carbonyl and trans to a PPh3 that has high trans influence. Carbonylation of 8a' toform
tran~-Pt(COPh)(COMe)(PPh3)2
(6e') is thought to be a legitimate reaction. In addition, 6e' has been proved to be a thermodynamically stable species. To our surprise, CO insertion has never occurred for 8a'. To explain the distinct reactivities of the acyl analog ofM,
(SP-4-2)-Pt(PPh3)(CO)(COPh)(Me),
and 8a' toward carbonylation, further investigation is needed.Formation of a Cis Diacyl Complex via Addition of a Carbonucleophile to cis-[Pt(COPh)(CO)(P-
(25) (a) Stille, J . IC; Regan, M. T. J. Am. Chem. SOC. 1974,96,1508.
(b) Shie, J.-Y.; Lin, Y.-C.; Wang, Y. J. Organomet. Chem. 1989, 371, 383.
Ph&]+. There are three known electrophilic sites in
cis-[Pt(COPh)(CO)(PPh3)21+
(7): the two metal-bound carbon atoms and the platinum(I1) center. It has been found that complex 7 undergoes addition of hard nu- cleophiles such as alkoxides or amides to result in cis- Pt(COPh)(CONu)(PPh3)2 as the only product, presum- ably via nucleophilic attack at the carbon of the terminal C0.16*26 The reaction of 7 with LiMe without external addition of CO distinguishably yields 6e, 2c', and 8a. Although the transformation between 2c' and 8a may take place via ligand substitution, either 2c' or 8a hasto be first formed from the reaction of Scheme 11.
Seeing that the formation of 8a' has never been estab- lished in this reaction and that the conversion between 6e and 2c' has been disproved by an independent direct approach, we propose a concerted mechanism for the reaction of 7 with MeLi. The crucial addition of the carbonucleophile presumably occurs at the Pt(I1) center, forming the five-coordinate intermediate XI (Scheme
23). Different inorganic products then may be obtained from XI through the independent pathways. The meth- yl migration from metal to CO would lead to 6e. The loss of CO in XI would afford 2c'. Complexes 8a can result from the dissociation of the PPh3 that is trans to
CO. Departure of another PPh3 in XI would produce a benzoyl methyl derivative of
IX
that could transform to 6e according to Scheme 23. However, the facile reverse methyl migration from the resulting acetyl to metal will probably diminish the opportunity of forma- tion of 6e via this route.It may be interesting to take a further look into the mechanisms of certain aforementioned carbonylation or decarbonylation reactions of diorganoplatinum(I1) and their intermediates. As summarized in Scheme 24, a five-coordinate intermediate in the form of
XI1
was detected in the carbonylation reaction of trans-Pt(R)- (XIL2. The stereochemistry has been retained from the alkylhalo complex to the acylhalo product as in case A.2e The other two five-coordinate intermediates XI andIV
in cases B and C are isomers. They undergo migratory CO insertion, leading to the diacyl products of distinct geometry; however, both retain their stereochemistry. Case D comprises the four-coordinate intermediateE.
It leads to the carbonylated product with exclusive cis stereoselectivity, different from the trans reactant.Five-coordinate species have been widely invoked in the carbonylation reactions of group 10 metal com- plexes,2 and intramolecular isomerization of such spe- cies is generally thought to be feasible.27 Those reac- tions containing five-coordinate intermediates in Scheme
24 seem likely to hold their geometrical character. It is possible that the energetic requirement for the migratory CO insertion (or deinsertion) in these five- coordinate species is less critical than that for isomer- ization, particularly with the bulky tertiary phosphines. The intermolecular processes of ligand substitution through four-coordinate species are possibly kinetically favored for the geometrical change of phosphines, compared to the case for the five-coordinate systems. One should not overinterpret the significance of Scheme
24, though, since most of these intermediate species have not been found experimentally.
(26) (a) Huang, L.; Ozawa, F.; Yamamoto, A. Organometallics 1990, 9,2603. (b) Shi, H.-Y. M.S. Thesis, National Taiwan University, 1993.
(27) (a) Anderson, G. K.; Cross, R. J. Chem. SOC. Rev. 1980, 9 , 185.
(b) Cross, R. J. Chem. SOC. Reu. 1986, 197.
Downloaded by NATIONAL TAIWAN UNIV on August 5, 2009