Pergamon Tetrahedron 54 (1998) 9067-9078
TETRAHEDRON
S,S-Dimethyl
dithiocarbonate:
A Novel
Carbonyl Dication Synthon in the Synthesis of Ketones
Chiar-Dy Chert, Jni-Wen Huang, Man-kit Leung*, Huei-hsu Li
Department of Chemistry, National Talwan University, Talpei, Taiwan, R. O. C.
Received 11 February 1998; accepted 10 April 1998
Abstract: We report herein the use o f DMDTC as an effective carbonyl dication equivalent in ketone synthesis. According to our strategy, we also successfully devised a synthetic pathway for S-methyl (trimethylsilyl)thioacetate which may be a potentially useful synthetic reagent in organic synthesis.
© 1998 Elsevier Science Ltd. All rights reserved.
Ketones are an important class of organic compounds whose synthesis and reactions have been actively studied during the last few decades.l In a retrosynthetic analysis for the formation of ketones, one can reasonably disconnect them into two carbanionic fragments as well as a carbonyl dication equivalenL Although this disconnection approach is simple and straightforward, known examples of this approach are rare. 2 In addition, almost all the known carbonyl dication synthons in ketone synthesis are indeed prepared from phosgene. As our continuing efforts in developing mild reagents that can be used to substitute phosgene or its derivatives in organic synthesis, we recently discovered that S,S-dimethyl dithiocarbonate (DMDTC) is a very useful reagent in urea synthesis) With regard to the facts that DMDTC is structually similar to phosgene and could be prepared from methanol, carbon disulfide, and dimethyl sulfate through a two-step sequence, DMDTC is a potential candidate to substitute phosgene and its derivatives as an effective carbonyl dication synthon in
0 II RsC'~R ketone synthesis. 0 II
Q ©c©
O
~, R--M 0 II + MeS s C ~ SMe DMDTCOur primary efforts are focused on developing an appropriate transition-metal catalyst that can effectively mediate the coupling reaction of DMDTC with Grignard or organolithium reagents. Since Fe(acac)3 and CuI are excellent promoters in the conversion of thioesters to ketones, 2a, 4 they are potential candidates to investigate.
In an exploratory investigation carried out in the early stage of our study, we discovered that DMDTC does not react with hexylmagnesium bromide at -78 °C. Gradually warming up the reaction mixture only lead to a complicated product mixture. In addition, our preliminary studies revealed that Fe(acac)3 is not an ideal catalyst. Reaction of hexylmagnesium bromide with DMDTC in the presence of catalytic amounts of Fe(acac)3 gives a product mixture of the corresponding thioester, ketone, and tertiary alcohol. Variation of the reaction 0040-4020198/$19.00 © 1998 Elsevier Science Ltd. All rights reserved.
9068 C.-D. Chen et al. / Tetrahedron 54 (1998) 9067-9078
temperatures or the amounts of Fe(acac)3 do not significantly improve the product ratios. However, dibexylmagnesiocuprate reacts with DMDTC to afford ketone 2 exclusively in high yield (Table 1). The potential efficiency of this reagent attracted our attention and prompted further investigation.
In a typical run for the formation of 2, CH3(CH2)5MgBr was treated with CuI at -50 "C in THF under argon for 2 hours, followed by addition of a solution of DMDTC. The reaction mixture was kept at -50 "C for 0.5 hour and then warmed up gradually to room-temperature for 4 hours. After typical workup and purification procedures, ketone 2 was obtained in pure form. The success of this process is highly dependent upon the extent of complete conversion of hexylbromide to the corresponding Grignard reagent in the previous step. The presence of any residual bexylbromide would destroy the cuprate reagent during the course of its formation and therefore retard the desired reaction. This procedure works well for most of the unhindered primary and secondary alkylmagnesium halides in our trials (Table 1) except in some cases, modifications of the reaction conditions are needed.
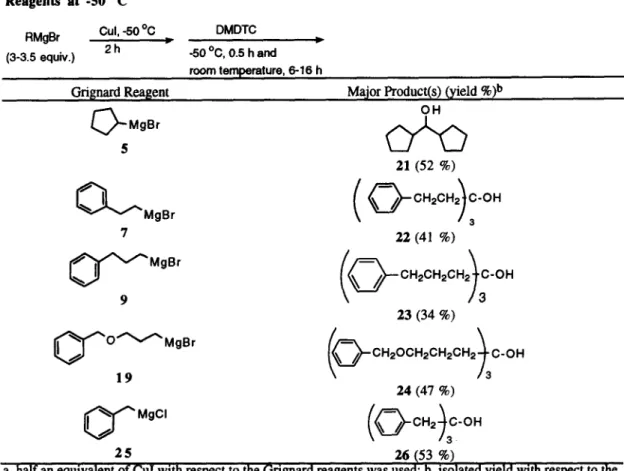
Perhaps due to intramolecular complexation, 5 aryl and alkoxy substituted Grignard reagents are less reactive than bexylmagnesium bromide towards cuprate formation. As a consequence, formation of these kinds of cuprates at -50 oC is sluggish and incomplete. The presence of the unreacted Grignard reagents would consume the newly formed ketones, resulting tertiary alcohols as the final products (Table 2). For reasons of steric hindrance, unreacted cyclopentylmagnesium bromide (5) acts as a reducing agent rather than a nucleophile, transferring a hydride to 6 to provide 21 in moderate yield. In order to improve the yields of the desired ketones, higher reaction temperatures are usually required (entries 3-10 in Table 1) to ensure the cuprate formations
Unlike other aryl substituted cuprates, reaction of dibenzylmagnesio cuprate with DMDTC only afforded alcohol 26 as the product. Increasing the cuprate-formation temperature would not improve the results, only giving significant amounts of dibenzyl as the side product. Nevertheless, we discovered that thioester formation could be mediated by Ni(acac)2 at -78 °C to give 27 in moderate yield.
MgBr Ni(acac)2
,.
DMDTC,-78 °C
25 27
(40 %)
Although the reasons are not immediately obvious, ketone formation from 11 under typical reaction conditions is unsuccessful, giving a product mixture of 12a, 12b, and the corresponding tertiary alcohol. However, the reaction could be stopped at the stage of thioester formation by controlling the reaction temperature at -50"C to give 12a as the major product. In addition, the complication in the formation of 12b could be subdued by employing one more equivalent of CuI in the reaction, affording 12b in acceptable yield.
HO~,,.I ~
1) Cul (2 equiv), -50 °C, 2 h
MgBr 2) DMDTC, -50 °C (0.5 h)
11 (3.9 equiv)
to d (14 h)
H ~
1) CuI (2.2 equiv), -25 °C, 2 h
MgSr 2) DMDTC, -25 °C (0.5 h)
to rt (4 h)11 (2.2 equiv)
O O H ~ , ~ , , SMe0
12a(55 %)
0 12b(51%)
C.-D. Chert et al. / Tetrahedron 54 (1998) 9067-9078 9069
Table 1: F o r m a t i o n o f S y m m e t r i c a l Ketones by Reaction o f D M D T C with various Cuprate R e a g e n t s
Entry Grignard Reagent Cuprate Ketone Major Product(s) (yield %)e formation formation O 1 ~ M g B r -50°C(2h) c - 5 0 ° C ( 0 . 5 h ) ~ A A . J L . ~ ~-~ .- 1 (3.2 eq) (96) to rt (4 h) a
v v v 2 v v v
0 -
/"'X J{. /--'X
O
2 MgBr -50°C(2h) c -50°C ( 0.5 h) to rt (4 h) b ~ " 4 " ~ _ . . . ~ (92) 3 (2.7 eq) 3 MgBr -25°C(2h) c -25°C ( 0.5 h) 5 (3.1 equiv.) to rt (4 h) b (73) 4 - 1 5 ~ ( 6 h ) c -15°(? ( 0.5 h) MgBr to rt (16 h) b (60) 7 (3.0 equiv.) 5 [" I~ ~ Mg Br -25oc(6h) -25oc ( 0.5 h) to rt (16 h) b 9 (4.0 equiv.) 6 ( I " ' 1 -25°C(2h) d -25°C ( 0.5 h) o O to rt (3 h) b H X ~ " " MgB r 11 (2.2 equiv) I I 70 0
-25°C(4h) c -25°C ( 0.5 h) ~ M g B r to rt (5.5 h) a 13 (2.5 equiv.) I I 80.._I-0
-25°(2(4h) c -25oc ( 0.5 h) I ~ MgBr to rt (3 h) a 15 (2.5 equiv) A 9 [ ] -250(2 (6 h) c -25°C (0.5 h),.o
O ~ M g B r to rt (16 h) a 17 (3.9 equiv) 10 A ~ ] ] ~_T O ~ M g B r -15°C(6h) c - 1 5 ° C ( 0 . 5 h) 19 (3.0 equiv.) to rt (16 h) a•
(56) 10ox, ,.hrSMe o. o
H
O 0
0 0 12a [101 f 12b (51)[731 f I I r~ I I~
(53) 14 I I I I0
~
0
(57) O 16 O 18 (56) 20 (46)a, in THF; b, in ether; c, half an equivalent of CuI with respect to the Grignard reagent was used; d, one equivalent of CuI with respect to the Grignard reagent was used; e. isolated yield; f, GC yield.
9070 C.-D. Chen et al. /Tetrahedron 54 (1998) 9067-9078
Table 2: Cul Mediated Reaction of DMDTC with
Reagents at -50 "C
RMgBr
Cul, -50 °C
DMDTC
(3-3.5 equiv.)
2 h
~
-50 °C, 0.5 h and
D,
room temperature, 6-16 h
Gri[nard ReagentAryi or Alkoxy Substituted Grignard
Major ProducKs)
{yield%)b
OH
5 21 (52 %) 7 22 (41%) 9 23 (34 %) 19 24 (47 %)U " 0o,
2s
~ (53 ~)
a. half an equivalent of CuI with respect to the Grignard reagents was used; b, isolated yield with respect to the
amount of DMDTC.
It is worth mentioning that formation of ketone 30 from sterically hindered Grignard reagent 28 is ineffective, giving thioester 29 as the major product. By taking the advantages of this selectivity, we could efficiently synthesize S-methyl (trimethylsilyl)thioacetate 32 in one step. Thioester 32 is a potentially useful synthetic reagent whose higher S-alkyl or aryl homologs and oxo-analogue 33 proved to be a useful reagent in c¢,~-unsaturated thioester synthesis, 6 silylation, 7 nucleophilic addition, 8 as well as 1,1-disubstituted ethylene formation. 9
. ~
1)
Cul,-50°C(2h)
/
'
-
O
~
DMDTC.-50 °C ( 0 . 5 h ~ ' ~ S M e
+
MgBr 2)
to rt (3h)
28 2g (42 %) 30 (<1%)I
1)Cu1,-25°C ~
..--- [ &
~ S i v M g C I
2)
0
/ S i
SMe
I !31 (2.6 equiv.)
Mes'C"sMe
32(680/.)
(DMDTC)
.I
o~ Si~JJ',,,OE t
33An oxo-analogue of 32
C.-D. Chen et al. / Tetrahedron 54 (1998) 9067-9078 9071
In an effort to develop a general synthetic route for unsymmetrical ketones, we examined the reaction of DMDTC with less reactive organocopper reagents. After exploring a number of reaction conditions, we eventually found that reaction of RCu-PPh3 with DMDTC would result R(C=O)SMe selectively in moderate yields. Due to the low reactivity of the organocopper reagents, significant amounts of the starting DMDTC were recovered after reaction.
RMgBr 1) Cul, PP(Ph)3, 0 °C, 1-2 h 2) DMDTC, 0 °C-25 °C, 2-6 h 34 R = octyl 35 R = Ph(CH2)3 36 R = cyclohexyl 0 • R . ~ , , , S M e isolated yield 39 % 3 5 % 37 %
Our experiments first demonstrate that DMDTC can be an effective carbonyl dication equivalent in ketone synthesis. Oxygen-containing functional groups such as acetals and ethers can be tolerated. On the basis of our strategy, we could prepare 32 efficiently in good yield. The applications of 32 are under investigation.
A C K N O W L E D G M E N T
We thank the National Science Council of the Republic of China for the financial support (NSC 86- 2113-M-002-027 and NSC 86-2113-M-002-033)
E X P E R I M E N T A L
General. THF and diethyl ether were distilled from sodium metal under N2 with benzophenone as indicator. All the reactions were carried out under a N2 or Ar atmosphere. DMDTC was prepared according to our recently published method. 3 Reactions were typically monitored using either TLC on commercial silica gel plates or GC. IR spectra were recorded on a Bio-rad FTS 40 FT-IR spectrophotometer as neat film or KBr plate. IH NMR spectra were recorded on a Bruker AC300 (300 MHz) spectrometer. Chemical shifts (5) are reported in ppm downfield from internal standard tetramethylsilane and coupling constants (J) are reported in Hertz (Hz). 13C spectra were either obtained on a Bruker AC300 (75 MHz) spectrometer. Mass spectra were determined using a HP-5989A mass spectrometer. Mps. were measured on a Melt-temp II melting-point apparatus, Laboratory Devices, USA.
A Representative Procedure f o r the Formation o f Symmetrical Ketones f r o m DMDTC: Dihexyl Ketone (2). To a vigorously stirred suspension of Mg (0.58 g, 24 mmol) in THF (1 mL) was added
catalytic amounts of I2. A small portion of l-bromohexane was added to initiate the reaction, followed by dropwise addition of a solution of l-bromohexane (totally 3.3 g, 20 retool) in 20 mL THF. After addition, the reaction mixture was further stirred for 30 rain. The concentration of the hexylmagnesium bromide (1.07 M) was determined by titration, using HC1 (IN) as standard and phenolphthalein as the indicator. The extent of conversion (92 %) was determined by GC analysis.
9072 C.-D. Chen et al. / Tetrahedron 54 (1998) 9067-9078
To prepare dihexyl ketone (2), the hexylmagnesium bromide (1.1 M, 3.6 mL, 3.9 mmol) was slowly added to a suspension of CuI (0.38 g, 2.0 mmol) in THF (2mL) at -50 °C. The mixture was kept at -50 °C for 2 h, followed by addition of a solution of DMDTC (0.15 g, 1.2 mmol) in THF (1 mL). The reaction mixture was further reacted at -50 °C for 0.5 h and gradually warmed to room temperature (rt) for 4 h. The reaction was quenched by using saturated NH4C1 solution. The crude product was extracted with CH2C12, washed with saturated NaCI solution, concentrated and purified through liquid chromatography on silica gel, using a solvent gradient from hexanes to CH2Cl2/hexanes (2:1) as eluent, to afford essentially pure 2 as colorless crystals (0.23 g, 1.16 mmol, 96 % ): mp 30-32 *C [lit. lOa 30 "C]; IH NMR (200 MHz, CDCI3) 8 2.32 (t, J = 7.2 Hz, 4H), 1.53-1.42 (m, 4H), 1.21 (m, 12H), 0.84-0.78 (t, J = 6.7 Hz, 6H) [lit. lOb IH NMR 8 2.28 (t, 4H), 1.00-2.10 (m, 16H), 0.70-1.00 (m, 6H)]; 13C NMR (75 MHz, CDCI3) 8 211.5, 42.7, 31.5, 28.9, 23.8, 22.4, 13.9
[lit. xX I3C NMR 210.8, 42.3, 31.2. 28.5, 23.4, 22.1, 13.5]; IR ~ 1713 cm -I (C=O); MS m/z (rel. intensity, El, 20eV) 199 (M++I, 35), 198 (M +, 100), 169 (5), 155 (5), 141 (10), 128 (15), 113 (30), 85 (8); HRMS (70 eV, M ÷) calcd, for C13H260: 198.1985, found 198.1982.
D i c y c l o h e x y l K e t o n e (4). The cyclohexylmagnesium bromide solution (0.87 M) was prepared from bromocyclohexane (4.0 g, 25 mmol) and Mg (0.72 g, 30 mmol) in Et20 (25mL) according to the representative procedure. The dicyclohexyl ketone (4) was obtained by reaction of DMDTC (0.31 g, 2.5 mmol) in Et20 (2.5 mL) with a mixture of CuI (0.64 g, 3.4 mmol) and cyclohexylmagnesium bromide (0.87 M, 7.7 mL, 6.7 mmol) at -50 °C for 2h and at rt for 4 h. The reaction was worked up as usual and the crude product was purified through liquid chromatography on silica gel, using a solvent gradient from hexanes to CH2C12/hexanes (2:1) as eluent, to afford essentially pure 4 as colorless crystals (0.45 g, 2.3 mmol, 92 %): IH NMR (300 MHz, CDC13) 8 2.46-2.36 (m, 2H), 1.73-1.62 (m, 10H), 1.36-1.14 (m, 10H) [lit. lla IH NMR 8 2.20-2.60 (t, 2H), 1.00-2.00 (m, 20 H)]; 13C NMR (75 MHz, CDC13) 8 217.0, 49.1, 28.5, 25.8, 25.7 [lit. llb 13C NMR 8 216.4, 48.9, 28.4, 25.7, 25.5]; IR ~ 1704 cm -1 (C=O); MS m/z (tel. intensity, EI, 20 eV) 194 (M +, 15), 111 (50), 83 (100); HRMS (70 eV, M++I) calcd, for C13H230: 195.1750, found 195.1750.
Dicyclopentyl Ketone (6). The cyclopentylmagnesium bromide solution (0.82 M) was prepared from 1- bromocyclopentane (7.5 g, 50 mmol) and Mg (1.2 g, 50 mmol) in Et20 (50 mL) according to the representative procedure. The corresponding cuprate reagent was prepared from CuI (1.6 g, 8.2 mmol) and cyclopentylmagnesium bromide (0.82 M, 20 mL, 16 mmol) at -25 °C for 2 h. To the cuprate solution, a solution of DMDTC (0.62 g, 5.1 mmol) in Et20 (5 mL) was added at -25 *C. After reaction at -25 "C for 0.5 h and at rt for 4 h, the mixture was worked up according to the representative procedure to provide a crude oil which was purified through liquid chromatography on silica gel, using a solvent gradient from hexanes to CH2C12/hexane = 112 as eluent to obtain 6 (0.61 g, 3.7 mmol, 72 %) as a colorless solid: IH NMR (300 MHz, CDCI3) 8 2.95-2.79 (m, 2H), 1.71-1.39 (m, 16H) [lit. 12 IH NMR 8 2.98 (2H), 1.30-2.00 (16H)]; 13C NMR (75 MHz, CDCI3) : 5216.0, 50.5, 29.2, 25.9 [lit. l l b 13C NMR 8 215.0, 50.0, 28.6, 25.5]; I R ~ 1706 cm -1 (C=O); MS m/z (rel. intensity, EI, 20 eV) 166 (M +, 10), 97 (50), 69 (100); HRMS (70 eV, M +) calcd, for CllHISO: 166.1359, found 166.1355.
1 , 5 . D i p h e n y l , 3 . p e n t a n o n e (8). The 2-phenylethylmagnesium bromide solution (0.87 M) was prepared from 2-bromoethylbenzene (2.5 g, 14 mmol) and Mg (0.4 g, 17 retool) in Et20 (15 mL) according to the representative procedure. The corresponding cuprate reagent was prepared from CuI (0.83 g, 4.4 mmol) in Et20
C.-D. Chen et al. / Tetrahedron 54 (1998) 9067-9078 9073
(4 mL) and 2-phenylethylmagnesium bromide (0.87 M, 8 mL, 7.2 mmol) at -15 °C for 6 h. To the cuprate solution, a solution of DMDTC (0.27 g, 2.2 mmol) in Et20 (2 mL) was added. After reaction at -15 °C for 0.5 h and at rt for 16 h. the mixture was worked up according to the representative procedure to provide a crude oil which was purified through liquid chromatography on silica gel, using a solvent gradient from hexanes to CH2C12/hexane = 1/2 as eluent to obtain 8 (0.31 g, 1.3 mmol, 60 %) as a colorless solid: 1H NMR (300 MHz, CDC13) 5 7.30-7.25 (m, 4H), 7.21-7.14 (m, 6H), 2.89 (t, J = 7.5 Hz, 4 H ) , 2.70 (t, J = 7.5 Hz, 4H)
lilt. 13 IH NMR ~ 7.20 (2, 10H), 2.70 (m, 8H)]; I3C NMR (75 MHz, CDC13) ~ 209.1, 141.0, 128.5, 128.3,
126.1, 44.5, 29.7; IR ~ 1713 cm -1 (C=O); MS m/z (rel. intensity, El, 20 eV) 238 (M +, 30), 220 (5), 133 (55), 105 (85), 91 (100), 77 (20); HRMS (70 eV, M +) calcd, for C17H180: 238.1359, found 238.1361.
1,7,Diphenyl.4-heptanone (10). The 3-phenylpropylmagnesium bromide solution (1.3 M) was prepared
from l-bromopropylbenzene (6.0 g, 30 mmol) and Mg (0.79 g, 33 retool) in Et20 (30 mL) according to the representative procedure. The corresponding cuprate reagent was prepared from CuI (2.7 g, 14 retool) and 3- phenylpropylmagnesium bromide (1.3 M, 20 mL, 26 mmol) at -25 *C for 6 h. To the cuprate solution, a solution of DMDTC (0.79 g, 6.5 mmol) in Et20 (6 mL) was added at -25 *C. After reaction at -25 °C for 0.5 h and at rt for 16 h, the mixture was worked up according to the representative procedure to provide a crude oil which was purified through liquid chromatography on silica gel, using a solvent gradient from hexanes to CH2C12/hexane = 1/2 as eluent to obtain 10 (0.97 g, 3.7 mmol, 56 %) as a colorless oil: 1H NMR (300 MHz, CDC13) 5 7.35-7.30 (m, 4H), 7.25-7.19 (m, 6H), 2.65 (t, J = 7.5 Hz, 4H), 2.41 (t, J = 7.3 Hz, 4H), 1.99- 1.89 (m, 4H); I3C NMR (75 MHz, CDC13) 5 210.3, 141.5, 128.3, 128.2, 125.8, 41.8, 34.9, 25.0; IR 1711 cm -1 (C=O); MS m/z (tel. intensity, EI, 20 eV) 266 (M +, 8), 175 (20), 162 (8), 147 (6), 104 (100), 91 (45); HRMS (70 eV, M +) calcd, for C19H220: 266.1672, found 266.1669.
3 - ( l , 3 - D i o x a n e . 2 - y l ) t h i o p r o p i o n i c A c i d S - M e t h y l E s t e r ( 1 2 a ) . The 2 - ( l , 3 - D i o x a n e - 2 - yl)ethylmagnesium bromide solution (0.96 M) was prepared from 2-(2-bromoethyl)-l,3-dioxane (2.0 g, 10 retool) and Mg (0.25 g, 10.5 mmol) in THF (10 mL) according to the representative procedure. The corresponding cuprate reagent was prepared from CuI (0.81 g, 4.3 mmol) in THF (4.5 mL) and 2-(1,3-dioxane- 2-yl)ethylmagnesium bromide (0.96 M, 9.0 mL, 8.6 mmol) at -50 °C for 2 h. To the cuprate solution, a solution of DMDTC (0.27 g, 2.2 mmol) in THF (2.2 mL) was added. After reaction at -50 °C for 0.5 h and at rt for 14 h, the mixture was worked up according to the representative procedure to provide a crude oil which was purified through liquid chromatography on silica gel, using a solvent gradient from hexanes/CH2Cl2 = 1/1 to 1/2 as eluent to obtain 12a (0.23 g, 1.2 mmol, 55 %) as a colorless oil: 1H NMR (300 MHz, CDC13) 8 4.52 (t, J = 5.0 Hz, IH), 4.03 (dd, J = 12 Hz, 5 Hz, 2H), 3.70 (dt, J = 12 Hz, 2.2 Hz, 2H), 2.64 (t, J = 8.1 Hz, 2H), 2.24 (s, 3H), 2.04-1.96 (m, 1H) 1.93-1.86 (m, 2H ), 1.30-1.26 (d, J = 12 Hz, 1H ); 13C NMR (75 MHz, CDCI3 ) 5 199.3, 100.5, 66.7, 37.9, 30.4, 25.6, 11.5; IR (neat) ~ 1692 cm -1 (C=O); MS m/z (tel. intensity, EI, 20 eV) 189 (M+-I, 10), 175 (13), 143 (100), 85 (95), 100 (20); HRMS (70 eV, M + ) calcd, for C8H1403S: 190.0664, found 190.0662.
1 , 5 - B i s ( 1 , 3 - d i o x a n e . 2 - y l ) - 3 , p e n t a n o n e (12b). The Grignard solution (0.96 M) was prepared according to the previous procedure. The corresponding cuprate reagent was prepared from CuI (0.98 g, 4.7 mmol) in THF (4 mL) and 2-(l,3-dioxane-2-yl)ethylmagnesium bromide (0.96 M, 5.0 mL, 4.7 mmol) at -25 °C for 4 h. To the cuprate solution, a solution of DMDTC (0.25 g, 2.0 mmol) in THF (2.0 mL) was added. After
9074 C.-D. Chen et al. /Tetrahedron 54 (1998) 9067-9078
reaction at -25 *C for 0.5 h and at rt for 15 h, the mixture was worked up according to the representative procedure to provide a crude oil which was purified through liquid chromatography on silica gel, using hexanes/ethyl acetate -- 1/3 as eluent to obtain 12b (0.26 g, 1.0 mmol, 5 1 % ) as a colorless oil: IH NMR (300 MHz, CDCI3) 5 4.47 (t, J = 5.0 Hz, 2H), 4.01-2.96 (dd, J = 12 Hz, 5 Hz, 4H), 3.65 (dt, J = 12 Hz, 3.3 Hz, 4H), 2.45 (t, J = 7.2 Hz, 4H), 2.04-1.96 (m, 2H), 1.78 (dt, J = 7.2 Hz, 5 Hz, 4H), 1.27-1.22 (d, J = 12.3 Hz, 2H ); 13C NMR (75 MHz, CDCI3 ) 5 209.6, 100.8, 66.7, 36.5, 28.9, 25.6; IR u 1714 cm -I (C=O); MS m/z (rel. intensity, El, 20 eV) 258 (M +, 5), 257 (M+-I, 10), 156 (23), 143 (24), 100 (100), 87 (60); HRMS (70 eV, M+-I ) calcd, for CI2H2104: 258.1468, found 258.1458.
1,7.Bis(2.methyl.l,3.dioxolane.2.yl).4.heptanone (14). To a vigorously stirred mixture of Mg (0.60 g, 25 mmol) and 2-(3-chloropropyl)-2-methyl-l,3-dioxolane (2.0 g, 12 mmol) in THF (15 mL) was added catalytic amounts of I2 and two drops of dibromoethane. The mixture was warmed at 80 *C and two drops of methylmagnesium bromi~de (1.0 M) was added to initiate the reaction. After addition, the reaction mixture was kept at reflux for 2 h to obtain 3-(2-methyl-1,3-dioxolane-2-yl)propylmagnesium chloride (0.90 M). The corresponding cuprate reagent was prepared from CuI (1.0 g, 5.4 mmol) in THF (5 mL) and 3-(2- methyl-l,3-dioxolane-2-yl)propylmagnesium chloride (0.90 M, 12.0 mL, 10.8 mmol) at -25 *C for 4 h. To the cuprate solution, a solution of DMDTC (0.54 g, 4.4 mmol) in THF (4.0 mL) was added. After reaction at -25 °C for 0.5 h and at rt for 5.5 h, the mixture was worked up according to the representative procedure to provide a crude oil which was purified through liquid chromatography on silica gel, using a gradient eluent from hexanes/ethyl acetate = 1/6 to hexanes/ethyl acetate = 1/3 to obtain 14 (0.66 g, 2.31 mmol, 53 %) as a colorless oil: IH NMR (300 MHz, CD3CN) 53.79-3.75 (m, 8H), 2.27 (t, J = 6.8 Hz, 4H), 1.55-1.44 (m, 8H), 1.15 (s, 6H): 13C NMR (75 MHz, CD3CN) 5210.2, 109.5, 64.3, 42.2, 38.0, 23.4, 18.0; IR ~ 1711 cm -I (C=O); MS m/z (tel. intensity, EI, 20 eV) 287 (M++I, 20), 271 (8), 99 (20), 87 (I00); HRMS (70 eV, M +) calcd, for C15H2605: 286.1781, found 286.1788.
1,9.Bis(2.methybl, J.dioxolane.2.yl).5.nonanone (16) To a vigorously stirred mixture of Mg (0.80 g, 33 mmol) and 2-(4-chlorobutyl)-2-methyl-l,3-dioxolane (1.8 g, 9.9 mmol) in THF (13 mL) was added catalytic amounts of I2 and four drops of dibromoethane. After addition, the reaction mixture was kept at reflux for 20 h to obtain 4-(2-methyl-l,3-dioxolane-2-yl)butylmagnesium chloride (0.90 M). The corresponding cuprate reagent was prepared from CuI (0.84 g, 4.4 mmol) in THF (4 mL) and 4-(2-methyl-l,3-dioxolane-2- yl)butylmagnesium chloride (0.90 M, 1.0 mL, 8.8 mmol) at -25 °C for 4 h. To the cuprate solution, a solution of DMDTC (0.43 g, 3.5 mmol) in THF (3.0 mL) was added. After reaction at -25 *C for 0.5 h and at rt for 3 h, the mixture was worked up according to the representative procedure to provide a crude oil which was purified through liquid chromatography on silica gel, using a gradient eluent from hexanes/ethyl acetate = 117 to hexanes/ethyl acetate = 113 to obtain 16 (0.62 g, 2.0 mmol, 57 %) as a colorless oil: IH NMR (300 MHz, CD3CN) 5 3.92-3.82 (m, 8H), 2.41 (t, J = 7.2 Hz, 4H), 1.63-1.46 (m, 8H), 1.39-1.28 (m, 4H), 1.25 (s, 6H); 13C NMR (75 MHz, CD3CN) 8 212.1, 110.8, 65.6, 43.4, 40.0, 25.1, 24.8, 24.4; IR ~ 1711 cm -I (C=O); MS m/z (tel. intensity, EI, 20 eV) 314 (M +, 0.6), 299 (M+-I5, 12), 128 (6), 87 (100); HRMS (70 eV, M +) calcd. for C17H3005: 314.2095, found 314.2101.
1,7-Bis(tetrahydropyran.2.yloxy).4,heptanone (18). The 3 - ( t e t r a h y d r o p y r a n - 2 - y l o x y ) p r o p y l - magnesium bromide solution (0.88 M) was prepared from 2-(3-bromopropoxy)tetrahydrofuran (3.0 g, 14
C.-D. Chen et al. /Tetrahedron 54 (1998) 9067-9078 9075 mmol) and Mg (0.97 g, 40 mmol) in THF (13 mL) according to the representative procedure. The corresponding cuprate reagent was prepared from CuI (1.1 g, 5.9 mmol) in THF (5 mL) and 3- (tetrahydorpyran-2-yloxy)propylmagnesium bromide (0.88 M, 13.0 mL, 11 mmol) at -25 °C for 6 h. To the cuprate solution, a solution of DMDTC (0.36 g, 2.9 mmol) in THF (2.0 mL) was added. After reaction at -25 °C for 0.5 h and at rt for 16 h, the mixture was worked up according to the representative procedure to provide a crude oil which was purified through liquid chromatography on silica gel, using hexanes/ethyl acetate = 1/2 as eluent to obtain 18 (0.51 g, 1.6 mmol, 56 %) as a colorless oil: IH NMR (300 MHz, CDCI3) ~ 4.45 (t, J = 3.3 Hz, 2H), 3.74-3.59 (m, 4H), 3.41-3.29 (m, 4H), 2.45 (t, J = 7.1 Hz, 4H), 1.82-1.42 (m, 16H); 13C NMR (75 MHz, CDC13) 8 210.2, 98.7, 66.4, 62.2, 39.4, 30.5, 25.3, 23.8, 19.5; IR ~ 1713 cm -1 (C=O); MS m/z (rel. intensity, El, 20 eV) 315 (M++I, 0.1), 314 (M +, 0.09-), 213 (M+-101, 7), 129 (50), 111 (10), 85 (100); HRMS (70 eV, M ÷) calcd, for C17H3005: 314.2095, found 314.2087.
1,7-Bis(benzyloxy)heptan-4.one (20). The 3-(benzyloxy)propylmagnesium bromide solution (0.90 M) was prepared from l-benzyloxy-3-bromopropane (2.0 g, 8.7 retool) and Mg (0.42 g, 18 mmol) in THF (8.5 mL) according to the representative procedure. The corresponding cuprate reagent was prepared from CuI (0.70 g, 3.7 mmol) in THF (3 mL) and 3-(benzyloxy)propylmagnesium bromide (0.97 M, 7.5 mL, 7.3 retool). To the cuprate solution, a solution of DMDTC (0.30 g, 2.5 retool) in THF (2.0 mL) was added. After reaction at -15 °C for 6 h and at rt for 16 h, the mixture was worked up according to the representative procedure to provide a crude oil which was purified through liquid chromatography on silica gel, using hexanes/ethyl acetate = 1/2 as eluent to obtain 20 (0.37 g, 1.1 retool, 46 %) as a colorless oil: IH NMR (300 MHz, CDCI3) 8 7.37-7.25 (m, 10H), 4.47 (s, 4H), 3.47 (t, J = 6.2Hz, 4H), 2.52 (t, J = 7.2 Hz, 4H), 1.93-1.84 (m, 4H); 13C NMR (75 MHz, CDCI3) 8 210.1, 138.3, 128.1, 127.4, 127.3, 72.6, 69.1, 39.2, 23.7; IR ~ 1713 cm -1 (C=O); MS m/z (tel. intensity, EI, 20 eV) 327 (M++I, 10), 219 (15), 129 (95), 111 (42), 106 (13), 91 (100); HRMS (70 eV, M +) calcd, for C21H2603: 326.1883, found 326.1880.
Di(cyclopentyl)methanol (21) To a suspension of CuI (1.6 g, 8.2 mmol) in Et20 (8 mL) was added cyclopentylmagnesium bromide (0.82 M, 20 mL, 16 retool) at -50 *C. After addition, the mixture was stirred at -50 °C for 2 h and a solution of DMDTC (0.62 g, 5.1 mmol) in Et20 (5 mL) was added. The mixture was further reacted at -50 °C for 6 h and quenched by addition of saturated NH4CI solution. The crude product was extracted with CH2C12, washed with brine, dried over anhydrous MgSO4, concentrated under reduced pressure, and purified through liquid chromatography on silica gel, using a solvent gradient from hexanes to CH2Cl2/hexanes = 1/1 as eluent to obtain 21 as colorless crystals (0.45 g, 2.7 retool, 52 %): mp 46-48 oC
[lit.N45 *C]; IH NMR (300 MHz, CDC13) 8 3.33-3.27 (t, J = 9 Hz, 1H ), 1.90-1.87 (m, 2H), 1.72-1.28 (m, 16H) [lit. 12 1H NMR ~ 3.35 (1H), 1.10-2.50 (18 H)]; 13C NMR (75 MHz, CDCI3) 8 78.2, 44.7, 29.3, 27.4, 25.6, 25.5; MS m/z (rel. intensity, EI, 20 eV) 168 (M +, 1), 150 (2), 99 (60), 81 (100); HRMS (70 eV, M +) calcd, for CI1H2oO: 168.1515, found: 168.1522. Anal. calcd, for CIlH2oO: C, 78.51%, H, 11.98 %. found C, 78.31%, H, 11.92 %; IR ~ 3396 cm -1 (C-OH).
Tris(2-phenylethyOmethanol (22) Compound 22 was prepared according to the preparative procedure for compound 21 in 41% yield: mp 70-72 *C; IH NMR (300 MHz, CDCI3) ~ 7.33-7.28 (m, 6H), 7.24-7.17 (m, 9H), 2.74-2.68 (m, 6H), 1.93-1.88 (m, 6H) [lit. 15 IH NMR 8 7.20 (s, 15 H), 2.67 (m, 6H), 1.86 (m, 6H), 1.33 (s, 1H); 13C NMR (75 MHz, CDCI3) 8 142.2, 128.5, 128.3, 125.9, 74.2, 41.2, 30.1; MS m/z (tel.
9076 C.-D. Chen et al. / Tetrahedron 54 (1998) 9067-9078
intensity, EI, 20 eV) 326 ( M+-IS, 8), 239 (12), 117 (15), 105 (10), 91 (100); HRMS (70 eV, M+-I8) caicd. for C25H26: 326.2036, found 326.2031. Anal. calcd, for C25H280: C, 87.16%, H, 8.19%, found C, 87.15%, H, 8.19%; IR ~ 3485 cm -1 (C-OH).
Tris(3,phenylpropyl)methanol (23) Compound 23 was prepared according to the preparative procedure for compound 21 in 34 % yield: IH NMR (300 MHz, CDCI3) 8 7.35-7.30 (m, 6H), 7.25-7.19 (m, 9H), 2.61 (t, J = 7.3Hz. 6H), 1.64-1.57 (m, 6H), 1.55-1.44 (m, 6H); 13C NMR (75 MHz, CDCI3) 8 142.3, 128.4, 128.3, 125.7, 74.2, 38.6, 36.2, 25.2; MS m/z (rel. intensity, EI, 20 eV) 368 (M+-IS, 45), 291 (10), 267 (25), 171 (22), 160 (20), 145 (23), 131 (21), 104 (95), 91 (100); HRMS (70 eV, M+-I8) calcd, for C28H32: 368.2496, found 368.2506; IR ~ 3549 cm-l(C-OH).
Tris(3.(benzyioxy)propyOmethanol (24) Compound 24 was prepared according to the preparative procedure for compound 21 in 47 % yield: IH NMR (300 MHz, CDCI3) 8 7.35-7.24 (m, 15H), 4.51 (s, 6H), 3.50-3.47 (t, J = 5.9 Hz, 6H), 1.72-1.64 (m, 6H), 1.58-1.53 (m, 6H); 13C NMR (75 MHz, CDCI3) ~i 138.3, 128.2, 127.5, 127.4, 73.0, 72.8, 70.8, 35.8, 23.9; MS m/z (tel. intensity, EI, 20 eV) 327 (M++I, 10), 219 (15), 129 (95), 111 (42), 106 (13), 91 (100); HRMS (70 eV, M +) calcd, for C31H4004: 476.2928, found 476.2930; IR ~ 3445 cm -1 (C-OH).
Tris(phenylmethyOmethanol (26) Compound 26 was prepared according to the preparative procedure for compound 21 in 53 % yield: mp 112-114 °C [lit. 16 110-114 °C] ; IH NMR (300 MHz, CDCI3) 5 7.40-7.25 (m, 15H), 2.83 (s, 6H): 13C NMR (75 MHz, CDC13) 8 137.3, 130.8, 128.1, 126.4, 73.9, 45.8; MS rrdz (rel. intensity, EI, 20 eV) 284 (M +- 18, 1), 211 (45), 193 (30), 91 (100); HRMS (70 eV, M +- 18 ) calcd, for C22H20: 284.1566, found 284.1565; IR ~ 3569 cm -1 (OH).
(2.Phenyl)thioacetic Acid S.Methyl Ester (27). To a flask with Ni(acac)2 (0.77 g, 3.0 mmol)was added benzylmagnesium chloride (1.16 M, 13 mL, 15 mmol) at -78 "C under argon. The mixture was stirred at -78 °C for lh, and a solution of DMDTC (0.61 g, 5.0 mmol) in THF (5 mL) was added. The reaction was allowed to proceed at -78 °C for 3 h and quenched by addtion of a saturated NH4C1 solution. The crude product was extracted with CH2C12, dried over anhydrous Na2SO4, concentrated under reduced pressure and purified through liquid chromatography on silica gel, using a solvent gradient from hexanes to CH2Cl2/hexanes = 1.5 as eluent to obtain 27 (0.34 g, 2.0 mmol, 40 %) as a slightly yellowish oil: IH NMR (300 MHz, CDCI3) 8 7.36- 7.24 (m, 5H), 3.82 (s, 2H), 2.27 (s, 3H) [lit. 17 1H NMR 7.20 (s, 5H), 3.65 (s, 2H), 2.10 (s, 3H)]; 13C NMR (75 MHz, CDCI3) 8 197.7, 133.6, 129.5, 128.6, 127.3, 50.3, 11.8; IR ~ 1691 cm -1 (C=O for thioesters); MS m/z (rel. intensity, El, 20 eV) 167 (M++I, 60), 166 (M +, 78), 139 (40), 91 (100) ; HRMS (70 eV, M ÷) calcd. t'or CgHIoOS 166.0453, found 166.0425.
(3,3.Dimethyl)thiobutanoic A c i d S . M e t h y l Ester (29) The neopentylmagnesium chloride solution
(0.40 M) was prepared from neopentylchloride (2.5 g, 23 mmol) and Mg (1.5 g, 62 mmol) in Et20 (15.0 mL) according to the representative procedure. The corresponding cuprate reagent was prepared from CuI (1.2 g, 6.0 mmol) in Et20 (6 mL) and neopentylmagnesium chloride (0.4 M, 30 mL, 12 mmol) at -50 "C for 2 h. To the cuprate solution, a solution of DMDTC (0.51 g, 4.2 mmol) in Et20 (4.0 mL) was added. After reaction at -50 "C for 0.5 h and at rt for 2 h, the mixture was worked up according to the representative procedure to
C.-D. Chert et al. /Tetrahedron 54 (1998) 9067-9078 9077 provide a crude oil which was purified through liquid chromatography on silica gel, using a eluent gradient from hexanes to CH2Cl2/hexanes = 1/2 to obtain 29 (0.26 g, 1.8 retool, 42 %) as a colorless oil: IH NMR (300 MHz, CDCI3) 8 2.43 (s, 2H), 2.26 (s, 3H), 1.01 (s, 9H); 13C NMR (75 MHz, CDCI3) 8 198.2, 56.6, 31.4, 29.6, 11.7; IR ~ 1713 cm -1 (C=O); MS m/z (rel. intensity, EI, 20 eV) 146 (M ÷, 5), 131 (3), 99 (75), 89 (10), 57 (100); HRMS (M ÷) calcd, for C7HI4OS: 146.0766, found 146.0770.
(2,Trimethylsilyl)thioacetic Acid S-Methyl Ester (32). The trimethylsilylmethylmagnesium chloride solution (0.97 M) was prepared from chloromethyltrimethylsilane (3.0 g, 24 mmol) and Mg (0.6 g, 25 mmol) in Et20 (24 mL) according to the representative procedure. The corresponding cuprate reagent was prepared from Cul (1.5 g, 7.8 mmol) in Et20 (8 mL) and trimethylsilylmethylmagnesium chloride (16 mL, 16 mmol) at -50 °C for 2 h. To the cuprate solution, a solution of DMDTC (0.73 g, 6.0 mmol) in Et20 (6 mL) was added. After reaction at -50 °C for 0.5 h, 0 °C for 1.5 h and at rt for 2 h, the mixture was worked up according to the representative procedure to provide a crude oil which was purified through liquid chromatography on silica gel, using a solvent gradient from hexanes to CH2Cl2/hexanes = 1/3 as eluent to obtain 32 (0.67 g, 4.1 mmol, 68 %) as a colorless oil: IH NMR (300 MHz, CDCI3) 8 2.30 (s, 2H), 2.24 (s, 3H), 0.10 (s, 9H) ; 13C NMR (75 MHz, CDC13) : 8 196.9, 38.2, 11.9, -1.5; IR a) 1681 cm -1 (C=O); MS m/z (rel. intensity, El, 20 eV) 147 (M ÷- 15, 5), 115 (95), 73 (100); HRMS (70 eV, M÷-15)calcd. for CSHllOSSi: 147.0294, found 147.0298.
A Representative Procedure f o r the Formation o f Thioesters from D M D T C and Alkylcopper Reagents : Thiononanoic Acid S-Methyl Ester (34). To a slurry of CuI (1.8 g, 9.1 mmol) and PPh3 (2.4 g, 9.1 mmol) in Et20 (5 mL) was added an ethereal solution of CH3(CH2)7MgBr (0.91 M, 10 mL) at 0 °C under argon. The mixture was allowed to react at 0 °C for 40 min, followed by addition of an ethereal solution (4 mL) of DMDTC (0.5 g, 4.1 mmol). The reaction mixture was kept at 0 °C for 6 h and quenched by addition of saturated aqueous NH4CI solution. The crude product was extraced with CH2C12 and purified through liquid chromatography on silica gel, using a solvent gradient from hexanes to CH2C12/hexanes (1/4) as eluent, to provide essentially pure 23 (0.3 g, 1.6 mmol, 39 %) as a colorless oil: IH NMR (300 MHz, CDC13) 8 2.53 (t, J = 7.5 Hz, 2H), 2.57 (s, 3H), 1.63 (m, 2H), 1.24 (m, 10 H), 0.85 (t, J = 6.6 Hz, 3H); 13C NMR (75 MHz, CDCI 3) : 5 200.1, 44.0, 31.8, 29.2, 29.1, 29.0, 25.7, 22.6, 14.1, 11.5; IR ~ 1696 cm -I (C=O for thioesters); MS m/z (rel. intensity, El, 70 eV) 189 (M++I, 72), 188 (M +, 20), 141 (M÷-SCH3, 100); HRMS (70 eV, M ÷) calcd, for C10H20OS: 188.1236, found 188.1233.
(4.Phenyl)thiobutanoic Acid S.Methyl Ester (35). Compound 35 was obtained according to the representative procedure as a colorless oil (35 %): IH NMR (300 MHz, CDCI3) 8 7.26-7.31 (m, 2H), 7.16- 7.22 (m, 3H), 2.65 (t, J = 7.5 Hz, 2H), 2.57 (t, J = 7.5 Hz, 2H), 2.29 (s, 3H), 1.95-2.05 (m, 2H) ; 13C NMR (75 MHz, CDCI3) 199.5, 141.1, 128.4, 128.3, 126.0, 43.1, 34.8, 27.1, 11.5 [lit. 18 199.0, 140.9, 128.2, 128.1, 125.8, 42.8, 34.6, 26.9, 11.2]; IR u 1688cm -1 (C=O for thioesters); MS m/z (rel. intensity, El, 70 eV) 195 (M++I, 20), 194 (M +, 5), 147 (M+-SCH3, 100), 91 (25); HRMS (70 eV, M +) calcd, for CI1HI4OS: 194.0766, found 194.0766.
(Cyclohexane)thiocarboxylic Acid S-Methyl Ester (36) Compound 36 was obtained according to the representative procedures as a colorless oil (37 %): 1H NMR (300 MHz, CDCI3) 8 2.42-2.51 (m, IH),
9078 C.-D. Chen et aL / Tetrahedron 54 (1998) 9067-9078
1.35 (m, 3H) [lit. t7 1.1-2.8 (m, IlH), 2.2 (s, 3H)]; 13C NMR (75 MHz, CDCI3) 203.5, 52.5, 29.5, 25.6, 25.5, 11.1; IR ~) 1692 cm -1 (C=O for thioesters); MS m/z (tel. intensity, EI, 70 eV) 158 (M +, 6), 143 (M +- CH3, I0), 111 (M+-SCH3, 58); 83 (C6H11 +, 100), 55 (53); HRMS (70 eV, M +) calcd, for C8H14OS:
158.0767, found 158.0770
REFERENCES
(1) Larock, R. C. Comprehensive Organic Transformations ; VCH, New York, 1989.
(2) For examples, see: (a) Cardellicchio, C.; Fiandanese, V.; Marchese, G.; Ronzini, L. Tetrahedron Lett. 1985, 26, 3595; (b) Aoki, S.; Nakamura, E.; Kuwajima, I. Tetrahedron Lett, 1988, 29, 1541; (c) Caniez, G; Normant J. F. Bull. Soc. Chim. Ft. 1977, 570; (d) Seetz, J. W. F. L.; Tol, R. ; Akkerman, O. S.; Bickelhaupt, F. Synthesis 1983, 721; (e) Hlasta, D. J.; Court, J. J. Tetrahedron Lett 1989, 30,1773; (f) Whipple, W. L.; Reich, H. J. J. Org. Chert 1991, 56, 2911.
(3) Leung, M.-k; Lai, J.-L.; Lau, K.-H.; Yu, H.-h.; Hsiao, H. J. J. Org. Chert 1996, 61, 4175.
(4) For reviews, see: (a) O'Neill, B. T. Comprehensive Organic Synthesis Vol. 1, Trost, B. M.; Fleming, I. Schreiber, S. L. Eds.; Pergamon: Oxford, 1991, 433-435; (b) Bonini, B. F.; Capperuci, Comes- Franchini, M.; Degl'Innocenti, A.; Mazzanti, G.; Ricci, A.; Zani, P. Synlett 1993, 937.
(5) Sworin M. and Neumann, W. L. Tetrahedron Lett. 1987, 28, 3217.
(6) (a) Lucast, D. H.; Wemple, J. Tetrahedron Lett. 1977, 1103; (b) Tajima, Y.; Yoshida, A.; Tekeda, N. Oida, S. Tetrahedron Lett. 1985, 26, 673.
(7) Nakamura, E; Hashimoto, K.; Kuwajima, I Bull. Chert Soc. Jpn.. 1981, 54, 805. (8) Nakamura, E; Shimizu, M; Kuwajima, I. Tetrahedron Lett. 1976, 1699.
(9) Larson, G. L.; Hern~mdez, Tetrahedron Lett. 1982, 23, 1035.
(10) (a) SaviUe, M. B.; Shearer, G. Jr. Chem. Soc. 1925, 592; (b) Tanaka, K.; Matsui, S.; Kaji, A. Bull.
Chert Soc. Jpn. 1980, 53, 3619.
(11) (a) Krapcho, A. P.; Kashdan, D. S.; Jahngen, Jr. E. G. E., J. Org. Chem. 1977,42, 1189; (b) Devasagayari, A.; Lakshmi, M.; Rao, N.; Periasamy, M. J. Organomet. Chert 1991, 421, 147. (12) Funke, C. W,; de Boer, L. M.; Geenevasen, J. A. J.; Ceffontain, H. J. Chem. Soc. Perkin Trans. 2
1976, 1083.
(13) Savoia, D.; Trombini, C.; Umani-Ronchi. A. Z Org. Chert 1978, 43, 2907.
(14) Hughes, R. J.; Ncube, S.; Pelter, A.; Smith, K. J. Chem. Soc. Perkin Trans. 1 1977, 1172. (15) Komatsu, K. ; Shirai, S.; Tomioka, I.; Okamoto, K. Bull. Chert Soc. Jpn. 1984, 57, 1377. (16) Ruggli, P.; Hegudus, B. Heir. Chirt Acta 1942, 25, 1285.
(17) Seebach, D.; Burstinghans, R. Synthesis 1975, 461.