Synthesis of Thermally Stable and Highly Active Bimetallic Au-Ag
Nanoparticles on Inert Supports
Xiaoyan Liu,†,‡Aiqin Wang,† Xiaofeng Yang,†,‡,§Tao Zhang,*,†Chung-Yuan Mou,| Dang-Sheng Su,⊥and Jun Li§
State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Zhongshan Road 457, Dalian 116023, P. R. China, Graduate UniVersity of the Chinese Academy of Sciences, Beijing 100049, P. R. China, Department of Chemistry, Tsinghua UniVersity, Beijing 100084, P.
R. China, Department of Chemistry, National Taiwan UniVersity, Taipei 106, Taiwan, and Fritz-Haber Institute of the Max Planck Society, Berlin D-14195, Germany
ReceiVed October 11, 2008. ReVised Manuscript ReceiVed NoVember 20, 2008
A general two-step approach has been developed for the synthesis of very small and sintering-resistant bimetallic gold-silver nanoparticles on inert supports including commercial silica and alumina. In this approach, gold particles were formed in the first step on amino-functionalized silica or alumina support. Our density functional theory (DFT) calculations on selected model clusters show that the surface atoms of the gold particles formed in the first step carry slightly negative charges, which facilitates the subsequent Ag+adsorption on the gold particle surface. Upon Ag+adsorption and reduction by NaBH4in the second
step, specific nanoparticles with gold-silver alloy core and a silver nanoshell have been formed, as confirmed by our ultraviolet-visible spectroscopy (UV-vis), high-resolution transmission electron microscopy (HRTEM), and X-ray photoelectron spectroscopy (XPS) characterizations. Such particles have been found to be highly thermally stable, and their sizes remain substantially unchanged (∼3 nm) even upon calcination in air at 500°C. After the final reduction treatment in H2, a randomly distributed
alloy composed of gold and silver is formed, and the resultant Au-Ag alloy particles are highly catalytically active for CO oxidation, even superior to Au/TiO2. The role of Ag in stabilizing the particles has been
discussed.
1. Introduction
Supported gold nanoparticles have attracted intensive research interest in heterogeneous catalysis and fuel-cell electrocatalysis in the past two decades because of their exceptionally high activities for low-temperature CO oxidation1-4 and distinguished selectivities in a variety of
redox reactions.5-8These unique features are very different
from the traditional platinum group metals. One of the most important features of gold catalysis is the striking size-dependence. As bulk gold is highly noble, reactivity of gold-based catalysts is critically dependent on the particle size; only those particles with sizes below 5 nm show high
reactivities.9-11Preparation of small-sized gold-based
nano-partciles on various supports is therefore highly desirable for catalysis. Typically, such small particles are obtained on reducible supports (TiO2, Fe2O3, Co3O4, etc.) via a
deposition-precipitation (DP) or a codeposition-precipitation (CP) method. In the case of nonreducible or “inert” supports such as SiO2,
however, both DP and CP method fail to work because of the low isoelectric point of silica. From the viewpoint of surface science, because of a lack of anchoring sites (e.g., oxygen vacancies) on the silica support,12,13silica-supporting
gold particles are susceptible to aggregation upon thermal treatment, which causes a dramatic loss of activity. To prevent or reduce this sintering effect, many attempts have been made, including confining gold particles in the nanop-ores of a mesoporous support,14-17 trapping gold particles
* Corresponding author. Fax: 86-411-84691570. Tel: 86-411-84379015. E-mail: taozhang@dicp.ac.cn.
†Chinese Academy of Sciences.
‡Graduate University of the Chinese Academy of Sciences. §Tsinghua University.
|
National Taiwan University.
⊥Fritz-Haber Institute of the Max Planck Society.
(1) Haruta, M. Catal. Today 1997, 36, 153.
(2) Schubert, M. M.; Hackenberg, S.; van Veen, A. C.; Muhler, M.; Plzak, V.; Behm, R. J. J. Catal. 2001, 197, 113.
(3) Kung, H. H.; Kung, M. C.; Costello, C. K. J. Catal. 2003, 216, 425. (4) Comotti, M.; Li, W. C.; Spliethoff, B.; Schu¨th, F. J. Am. Chem. Soc.
2006, 128, 917.
(5) Grisel, R. J. H.; Nieuwenhuys, B. E. J. Catal. 2001, 199, 48. (6) Hughes, M. D.; Xu, Y. J.; Jenkins, P.; McMorn, P.; Landon, P.; Enache,
D. I.; Carley, A. F.; Attard, G. A.; Hutchings, G. J.; King, F.; Stitt, E. H.; Johnston, P.; Griffin, K.; Kiely, C. J. Nature 2005, 437, 1132. (7) Gonza´lez-Arellano, C.; Corma, A.; Iglesias, M.; Sa´nchez, F. J. Catal.
2006, 238, 497.
(8) Wang, X. K.; Wang, A. Q.; Wang, X. D.; Yang, X. F.; Zhang, T.
Gold Bull. 2007, 40, 52.
(9) Chen, M. S.; Goodman, D. W. Science 2004, 306, 252. (10) Haruta, M. Chem. Rec. 2003, 3, 75.
(11) Falsig, H.; Hvolbæk, B.; Kristensen, I. S.; Jiang, T.; Bligaard, T.; Christensen, C. H.; Nørskov, J. K. Angew. Chem., Int. Ed. 2008, 47, 4835.
(12) Min, B. K.; Wallace, W. T.; Goodman, D. W. Surf. Sci. 2006, 600, L7.
(13) Yan, Z.; Chinta, S.; Mohamed, A. A.; Fackler, J. P.; Goodman, D. W.
J. Am. Chem. Soc. 2005, 127, 1604.
(14) Yang, C. M.; Liu, P. H.; Ho, Y. F.; Chiu, C. Y.; Chao, K. J. Chem.
Mater. 2003, 15, 275.
(15) Bore, M. T.; Pham, H. N.; Switzer, E. E.; Ward, T. L.; Fukuoka, A.; Datye, A. K. J. Phys. Chem. B 2005, 109, 2873.
(16) Zhu, H.; Liang, C.; Yan, W.; Overbury, S. H.; Dai, S. J. Phys. Chem.
B 2006, 110, 10842.
(17) Sun, J.; Ma, D.; Zhang, H.; Liu, X.; Han, X.; Bao, X.; Weinberg, G.; Pfa¨nder, N.; Su, D. J. Am. Chem. Soc. 2006, 128, 15756.
10.1021/cm8027725 CCC: $40.75 2009 American Chemical Society Published on Web 12/23/2008
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
in the matrix of the silica support by coassembly,18,19and
grafting a reducible oxide layer on the silica surface to adhere to the gold particles.20-22
Recently, we developed a facile two-step method for synthesis of Au-Cu alloy particles, where the particle size was well-controlled at 2-3 nm in the nanochannels of SBA-15.23The employment of SBA-15 is presumably to confine
the nanoparticles in the orderly arranged channels to prevent their sintering or agglomeration, as reported earlier by various groups.14-17 However, we found that even with a harsh
treatment, e.g., calcination at 500°C in air for 6 h followed by reduction at 550°C in H2for 1 h, the particle size of the
Au-Cu alloy was only around 3 nm, which is far less than the pore size (∼6 nm) of the SBA-15 support. This result seems to suggest that the nanopores of the SBA-15 support have little effect in stabilizing the gold alloy particles. Potentially, the method can be developed into a general approach to synthesize very small and thermally stable gold alloy particles on a ubiquitous commercial support, such as silica and alumina. In the present work, we will demonstrate this approach by synthesizing highly dispersed Au-Ag nanoparticles on a variety of inert supports. The Au-Ag bimetallic systems are chosen based on the following reasons. First, there is no report so far on synthesis of highly dispersed and thermally stable Au-Ag alloy nanoparticles on com-mercial silica or alumina supports in spite of its widespread applications in optics, electronics, and catalysis. In most cases, synthesis of Au-Ag bimetallic nanoparticles is conducted in a solution based on a wet chemical reduction method and good control in both size and morphology can therefore be accomplished under the presence of protective agents (surfactants or polymers).24-28 However, when the
nanoparticles with well defined structure and size in solution are deposited on a support and are subject to a thermal treatment for removing the protective agents, the original nanostructure, its size, and morphology are hardly retained due to heat- and atmosphere-induced sintering and restructur-ing. For example, in our earlier work,29-32 Au-Ag alloy
particles with sizes smaller than 7 nm were prepared in an aqueous solution under the protection of hexadecyl trimethyl ammonium bromide (CTAB). However, upon deposition on
MCM-41 support and calcination at 500°C for removal of CTAB, the particles were remarkably enlarged to 20-30 nm. Although such large alloy particles are very active for CO oxidation, which is quite different from pure gold, they are completely deactivated by the presence of H2in the reacting
stream. We wonder if this deactivation is related to the large particle size, which stimulates us to search for a new method to synthesize very small and uniform Au-Ag alloy particles on a commercial silica support. Second, although silver belongs to the same group as gold, it has very different properties from gold with regard to oxygen adsorption. As gold binds oxygen very weakly, oxygen activation is generally thought as a rate-limiting step for most oxidation reactions catalyzed by gold nanoparticles.33-35 In contrast,
silver adsorbs oxygen very well and different oxygen species that are activated on silver surface have been identified.36,37
The intrinsically different properties of gold and silver toward oxygen activation have great potential to be fine-tuned for their synergetic effect in catalyzing various oxidation reactions.30,38Third, and most importantly, in our recently
reported Au-Cu case,23the presence of Cu was found to
greatly limit the growth of the prepared particles, which led to a much smaller particle size for Au-Cu than for pure gold. We wonder if the same trend exists in other gold-alloy systems, e.g., Au-Ag.
On the basis of these observations, we are interested in investigating if it is of general significance that the presence of a second metal stabilizes the gold particles to a relatively small size. The understanding of such questions may open up a new route for preparing highly thermally stable gold or gold alloy particles. In the present work, we have synthesized small sized and thermally stable Au-Ag alloy particles on commercial silica and alumina support. We have shown that our approach is in principle applicable to a variety of supports provided that the support surface possesses rich hydroxyl groups and the presence of Ag plays a key role in stabilizing the particles. To uncover the underlying reason for the stabilizing effect, detailed investigations, step by step, into the structural changes occurring during the preparation and thermal treatment are conducted by employing HRTEM, UV-vis, and XPS techniques. In addition, theoretical investigations based on selected cluster models are also used to provide understanding of the stabilities and electronic properties of the nanoparticles formed. Finally, catalytic tests have shown that the Au-Ag alloy nanoparticles on inert silica supports are very active for CO oxidation with or without the presence of H2, even more active than Au/TiO2.
2. Experimental Section
2.1. Synthesis of Oxide-Supported Au-Ag Particles. The synthesis of Au-Ag nanoparticles on silica support started with
(18) Budroni, G.; Corma, A. Angew. Chem., Int. Ed. 2006, 45, 3328. (19) Liu, H.; Ma, D.; Blackley, R. A.; Zhou, W.; Bao, X. Chem. Commun.
2008, 2677.
(20) Yan, W.; Mahurin, S. M.; Chen, B.; Overbury, S. H.; Dai, S. J. Phys.
Chem. B 2005, 109, 15489.
(21) Horva´th, A.; Beck, A.; Sa´rka´ny, A.; Stefler, G.; Varga, Z.; Geszti, O.; To´th, L.; Guczi, L. J. Phys. Chem. B 2006, 110, 15417. (22) Qian, K.; Huang, W.; Jiang, Z.; Sun, H. J. Catal. 2007, 248, 137. (23) Liu, X.; Wang, A.; Wang, X.; Mou, C. Y.; Zhang, T. Chem. Commun.
2008, 3187.
(24) Mallin, M. P.; Murphy, C. J. Nano Lett. 2002, 2, 1235.
(25) Pande, S.; Ghosh, S. K.; Praharaj, S.; Panigrahi, S.; Basu, S.; Jana, S.; Pal, A.; Tsukuda, T.; Pal, T. J. Phys. Chem. C 2007, 111, 10806. (26) Lee, I.; Han, S. W.; Kim, K. Chem. Commun. 2001, 1782. (27) Yin, Y.; Erdonmez, C.; Aloni, S.; Alivisatos, A. P. J. Am. Chem. Soc.
2006, 128, 12671.
(28) Cobley, C. M.; Campbell, D. J.; Xia, Y. AdV. Mater. 2008, 20, 748. (29) Liu, J. H.; Wang, A. Q.; Chi, Y. S.; Lin, H. P.; Mou, C. Y. J. Phys.
Chem. B 2005, 109, 40.
(30) Wang, A. Q.; Liu, J. H.; Lin, S. D.; Lin, T. S.; Mou, C. Y. J. Catal.
2005, 233, 186.
(31) Wang, A.; Hsieh, Y. P.; Chen, Y. F.; Mou, C. Y. J. Catal. 2006, 237, 197.
(32) Wang, A. Q.; Chang, C. M.; Mou, C. Y. J. Phys. Chem. B 2005, 109, 18860.
(33) Mills, G.; Gordon, M. S.; Metiu, H. J. Chem. Phys. 2003, 118, 4198. (34) Deng, X.; Min, B. K.; Guloy, A.; Friend, C. M. J. Am. Chem. Soc.
2005, 127, 9267.
(35) Guzman, J.; Carrettin, S.; Corma, A. J. Am. Chem. Soc. 2005, 127, 3286.
(36) Qu, Z.; Huang, W.; Cheng, M.; Bao, X. J. Phys. Chem. B 2005, 109, 15842.
(37) Qu, Z.; Cheng, M.; Huang, W.; Bao, X. J. Catal. 2005, 229, 446. (38) Chaki, N. K.; Tsunoyama, H.; Negishi, Y.; Sakurai, H.; Tsukuda, T.
J. Phys. Chem. C 2007, 111, 4885.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
adding 1 g of a commercial silica support (Qingdao Ocean Chemical Plant, SBET) 467 m2g-1) to a 50 mL ethanol solution containing 2.5 g of APTES (H2N(CH2)3Si(OEt)3, Acros). The mixture was refluxed for 24 h to graft APTES on the silica surface.39,40After being washed with ethanol and dried at 60 °C, the solid was redispersed in 16 g of H2O at room temperature, to which 4 mL of 1.65 wt % HAuCl4solution was added under stirring. After filtration and washing, the recovered solid was added into 10 g of water, to which 10 mL of 0.2 M NaBH4solution was added under vigorous stirring for reduction of AuCl4
-. After 20 min, the solid was recovered by filtration and thoroughly washed with water to remove Cl-for the subsequent Ag deposition, which was the same as the above Au deposition except that AgNO3was used as the precursor. Finally, the solid was calcined at 500°C in air for 6 h and reduced at 550°C in H2for 1 h to obtain the Au-Ag/SiO2catalyst. The same procedure was also applied to other supports including alumina (SBET) 198 m2g-1), nano silica (SBET) 201 m2g-1), and SBA-15 (SBET) 903 m2g-1), to obtain Au-Ag nanoparticles with different supports. In all cases, the nominal total metal loading was 6 wt %.
2.2. Characterization. The actual total metal loadings and the Au/Ag atomic ratios of various Au-Ag/SiO2 samples were determined by inductively coupled plasma spectrometer (ICP-AES) on an IRIS Intrepid II XSP instrument (Thermo Electron Corpora-tion). The surface Au/Ag atomic ratios, along with the binding energies of gold and silver, were determined by XPS on an AMICUS X-ray photoelectron spectrometer equipped with a Mg KR radiation (1253.6 eV, 10 kV, 20 mA, 200 W) under a residual pressure of 1× 10-6Pa. The raw data were corrected for substrate charging with the binding energy of the Si2p(103.4 eV) on a silica support as a reference. The recorded spectra were fit by a least-squares procedure to a product of Gaussian-Lorentzian functions after subtraction of background noise. The concentration of each element was calculated from the area of the corresponding peak, calibrated with the atomic sensitivity factor. HRTEM analyses were performed with a Tecnai G2 F30 S-Twin Transmission Electron Microscope operating at 300 kV. A few droplets of a suspension of the sample in ethanol were put on a microgrid carbon polymer supported on a copper grid and allowed to dry at room temperature for HRTEM observations. The particle size distribution was obtained by measuring more than 200 particles from randomly selected areas. Chemical compositions of individual particles or different areas were determined by energy-dispersive X-ray spec-troscopy (EDS) on a DX4 analyzer system which is attached to a Philips CM200 FEG electron microscope. X-ray Diffraction (XRD) patterns were recorded on a PW3040/60 X’ Pert PRO (PANalytical) diffractometer equipped with a Cu KR radiation source (λ )
0.15432 nm), operating at 40 kV and 40 mA. A continuous mode was used for collecting data in the 2θ range from 20°to 80°at a scanning speed of 5°min-1. UV-vis spectra were recorded at room temperature on a Cintra (GBC) apparatus with BaSO4as a reference.
2.3. Catalytic Reaction Tests. Catalytic activity was measured using a continuous flow fixed-bed reactor system. For CO oxidation, a feed stream containing 1.0 vol % CO and 1.0 vol % O2balanced with He was allowed to pass through 60 mg (20-40 mesh) of a catalyst sample at a flow rate of 20 mL min-1, corresponding to a space velocity of 20,000 mL h-1 gcat-1. For preferential CO oxidation in rich H2, the feed gas was composed of 1.0 vol % CO, 0.5 vol % O2, and 50 vol % H2balanced with He, and the total
flow rate was 40 mL min-1corresponding to a space velocity of 40 000 mL h-1 gcat-1. Prior to the reaction, the catalyst was pretreated with H2at 550°C for 1 h and then cooled to the reaction temperature under He. The inlet and outlet gas compositions were online analyzed by a gas chromatograph (HP 6890, TDX-01 column).
2.4. Density Functional Calculations. Our previous theoretical investigations indicate that with proper treatment of the relativistic effects and carefully selected exchange-correlation functional DFT methods can provide accurate information about geometries, electronic structures and properties of gold nanoclusters.41Density functional calculations were performed with the program package DMol3in the Materials Studio of Accelrys Inc.42,43The localized double-numerical basis sets with polarization functions (DNP) were used, which are more accurate than but comparable in size to the Gaussian basis set 6-31G**. Metal atom core electrons were treated using the quasi-relativistic pseudopotentials developed by Bergner et al., in which the mass-velocity and Darwin relativistic corrections were introduced.44Generalized gradient approximation (GGA) using exchange-correlation functional developed by Perdew, Burke, and Ernzerhof (PBE) was employed.45A real-space cutoff of 4.5 Å was used, with convergence criteria of 2× 10-5 hartree (1 hatree ) 27.2 eV), 4× 10-3hartree/Å, and 5× 10-3Å for energy, energy gradient, and geometry, respectively. The self-consistent field (SCF) calculations were carried out with the spin-polarization Kohn-Sham formalism and molecular symmetry was not enforced to allow for geometry relaxation. Although the actual Au-Ag nanoparticles are too large for the first-principle calculations, a series of model clusters Au38-xAgx(x ) 1, 2) were constructed based on a
face-centered cubic (FCC) octahedral Au38cluster to model the bimetallic Au-Ag nanoparticles with different compositions and structures.
3. Results and Discussion
3.1. Synthesis of Au-Ag Nanoparticles on Silica and Alumina Supports. The loading of gold and silver on the
commercial silica is illustrated in Figure 1. In this procedure, the silica surface was first functionalized with amine to create anchoring sites for gold precursor. The negatively charged AuCl4
-ions were then strongly adsorbed on the positively charged aminium ions in an acidic solution. The interaction between amine and gold precursor has been well documented in the literature,46,47and this interaction was so strong that a
relatively high amount of gold could be adsorbed on the amino-functionalized silica surface.40 After reduction with
NaBH4, small-sized gold particles were produced on the
functionalized silica surface and some of the amine groups were likely restored during the removal of Cl-ions in the washing process. Following that, Ag+cations were allowed to adsorb either on the free amine groups via complexation, or preferentially on the negatively charged gold particle surface via electrostatic attractions and metallic bonding. Our
(39) Tu, C. H.; Wang, A. Q.; Zheng, M. Y.; Wang, X. D.; Zhang, T. Appl.
Catal., A 2006, 297, 40.
(40) Chiang, C. W.; Wang, A.; Wan, B. Z.; Mou, C. Y. J. Phys. Chem. B
2005, 109, 18042.
(41) (a) Li, J.; Li, X.; Zhai, H. J.; Wang, L. S. Science 2003, 299, 864. (b) Ji, M.; Gu, X.; Li, X.; Gong, X. G.; Li, J.; Wang, L. S. Angew. Chem.,
Int. Ed. 2005, 44, 7119. (c) Gu, X.; Bulusu, S.; Li, X.; Zeng, X. C.;
Li, J.; Gong, X. G.; Wang, L. S. J. Phys. Chem. C 2007, 111, 8228. (42) Delley, B. J. Chem. Phys. 1990, 92, 508.
(43) Delley, B. J. Chem. Phys. 2000, 113, 7756.
(44) Bergner, A.; Dolg, M.; Ku¨chle, W.; Stoll, H.; Preuss, H. Mol. Phys.
1993, 80, 1431.
(45) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. ReV. Lett. 1996, 77, 3865.
(46) Ghosh, A.; Patra, C. R.; Mukherjee, P.; Sastry, M.; Kumar, R. Microp.
Mesop. Mater. 2003, 58, 201.
(47) Chi, Y. S.; Lin, H. P.; Mou, C. Y. Appl. Catal., A 2005, 284, 199.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
DFT calculations show that the latter case is predominant because of strong bimetallic interaction. In addition, we have discovered that loading gold prior to silver is pivotal for the control of the size and composition of the bimetallic particles. With a different loading sequence, either simultaneously or reversely, galvanic replacement reaction between Ag and AuCl4
-will occur,28,48 which in addition to the formation
of AgCl precipitate significantly induces the coalescence of particles upon heating treatment. The final calcination and reduction steps are necessary for removing the amine functional groups and for the formation of gold-silver particles on the support.
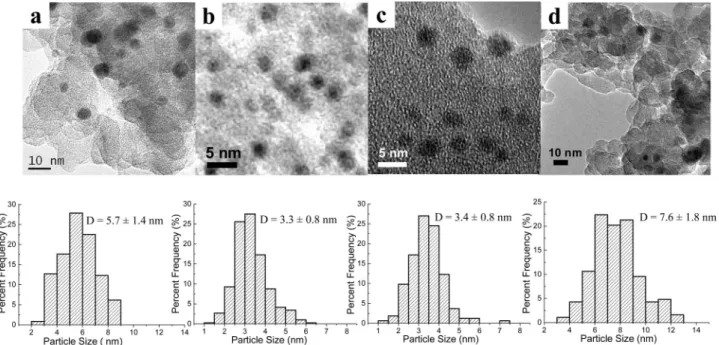
Figure 2 presents TEM images of the Au-Ag/SiO2
catalysts with different Au/Ag atomic ratios. For comparison, monometallic Au/SiO2 and Ag/SiO2 were also examined.
While Au/SiO2has an average particle size of 5.7 nm and
Ag/SiO2of 7.6 nm, the particle sizes become much smaller
upon alloying Au with Ag, 3.3 nm for Au/Ag ) 3/1 and 3.4 nm for Au/Ag ) 1/1. Note this result is in contrary to the one-pot synthesized Au-Ag@MCM-41 system,32 where
alloying gold with silver led to a remarkable increase of particle sizes. In that one-pot synthesis, Br- from the surfactant could combine with Ag+to form AgBr, which in
turn caused the enlargement of Au-Ag particles because of the much lower melting point of AgBr than that of metallic Ag. However, in our present approach, the silica surface was prefunctionalized with amine to create anchoring sites for AuCl4
-. After reduction with NaBH4and a thorough
wash-ing, the small gold particles were formed while Cl- was removed, which circumvented the formation of AgCl when Ag+was subsequently adsorbed on the surface. This is partly responsible for the small particle size of Au-Ag/SiO2.
Consistent with the TEM observations, very broad XRD peaks (Figure 3) of the Au-Ag/SiO2samples also indicate
rather small particle sizes. It should be mentioned that, different from the case of Au-Cu,23the XRD patterns of
gold and silver are totally overlapped so that one cannot distinguish Au-Ag bimetallic phase from either monome-tallic phase based on the XRD patterns.30However, from
EDS analyses (see Figure S1 in the Supporting Information), one can clearly see that each individual particle on the Au-Ag/SiO2sample is composed of both Au and Ag. This
result provides unequivocal evidence for the formation of bimetallic Au-Ag particles. It is also noted that although the Au/Ag ratio varies from one particle to another, the average Au/Ag atomic ratio, which was obtained by EDS analyses on randomly selected areas (see Figure S2 in the Supporting Information), agrees well with the nominal value. It is intriguing to note that the particle size of gold on the silica surface here is essentially identical to that on the SBA-15 obtained in our previous work,23 indicating that the
particle size is not determined by the pore size of the support. In our previous work, Au-Cu alloy particles with an average size of 2.9 nm were formed in the nanochannels of SBA-15. However, 2.9 nm is much smaller than the pore size of SBA-15 (5.7 nm), implying that the particle size is deter-mined by the thermodynamics associated with the special nanostructure of the particles, rather than by the pore size of the support. Actually, Au-Cu alloy particles with an average size of 3.4 nm can also be obtained on the commercial silica (see Figure S3 in the Supporting Informa-tion) in our present work, although this support has no orderly arranged regular pores. To further confirm that the particle size has little to do with the pore size of the support, we here synthesized Au-Ag alloy particles with the same procedure on a variety of different supports including SBA-15, nano silica, and alumina. In spite of the differences in the pore structures and chemical compositions of the supports employed (see Figure S4 in the Supporting Information), very small and uniform Au-Ag nanoparticles were produced in each case (see Figure S5 in the Supporting Information). All the above results clearly demonstrate that the present two-step approach provides a general strategy for synthesizing small, uniform, and thermally stable gold-based bimetallic particles on a variety of different supports.
To probe the key factors responsible for the size of bimetallic gold particles, we conducted several control exper-iments. First, we performed the same synthesis on the nonfunctionalized SiO2 support and obtained a large and
broad-size distribution of the Au-Ag particles (see Figure S6 in the Supporting Information). This result clearly shows that the surface functionalization with amine groups is a
(48) Sun, Y.; Xia, Y. J. Am. Chem. Soc. 2004, 126, 3892.
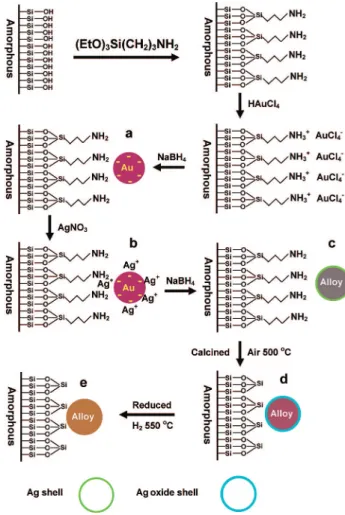
Figure 1. Schematic illustration of the procedure for synthesis of SiO2
-supported Au-Ag alloy nanoparticles. (a) Initially formed Au particles; (b) AgNO3adsorbed on the Au particles; (c) Au-Ag nanostructure with
alloy core surrounded by silver shell; (d) Au-Ag nanostructure with more gold-rich alloy core covered by Ag oxide shell; (e) random alloy of Au-Ag nanoparticles on SiO2support.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
prerequisite for avoiding agglomeration of the particles. Second, we find that the reduction of gold precursor with NaBH4 is also indispensable for obtaining the very small
bimetallic gold particles. Other reducing agents, including formaldehyde, hydrazine, and H2, all led to large particle
size. Possibly, some amine-borane complexes could have been formed during the reduction process,49 which would
act as a special reducing agent for obtaining very small bimetallic gold particles. Third and most importantly, it is found that the presence of Ag greatly limits the growth of gold particles in our two-step approach. Based on the XRD peak width of Au/Ag (111) of the samples that had been calcined at different temperatures, we calculated the average particle size by Scherrer’s equation. As shown in Figure 4, one can see that although the as-synthesized (dried at 80 °C) gold particles are smaller than the gold-silver bimetallic particles, significant sintering occurred on pure gold particles upon calcination at 500°C, which led to a sharp increase of average particle size from 1.8 to 4.1 nm. By contrast, the
gold-silver bimetallic particles are highly resistant to high-temperature calcinations, with the average particle size being essentially unchanged after 500°C calcination. It should be pointed out that the average particle size estimated from XRD is smaller than that obtained from TEM measurements, possibly because some particles are too small to be observed by TEM.
3.2. Structural Evolution during Preparation and Thermal Treatment. To uncover the underlying reason for
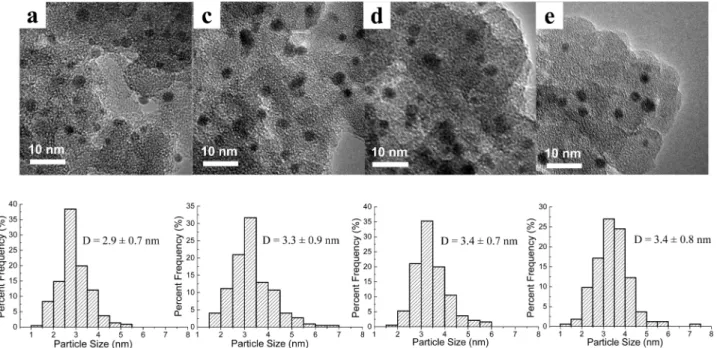
the high thermal stability of the Au-Ag nanoparticles, in particular the key role of Ag in stabilizing the gold particles, we investigated in detail the structural evolution during each step of the preparation. As shown in Figure 5a, when AuCl4
-was adsorbed on the APTS-functionalized silica surface and reduced by NaBH4, very small and uniform gold particles (2.9
( 0.7 nm) were produced. These gold particles were deposited on the silica surface possibly via interacting with the amino groups (Figure 1a). The UV-vis spectrum of the sample at this stage (Figure 6a) also shows a very broad peak centered at 525 nm, which is characteristic of surface plasma resonance
(49) Zheng, N.; Fan, J.; Stucky, G. D. J. Am. Chem. Soc. 2006, 128, 6550.
Figure 2. HRTEM images and the corresponding particle size distributions of Au-Ag/SiO2with various Au/Ag atomic ratios: (a) 1/0, (b) 3/1, (c) 1/1, (d)
0/1.
Figure 3. XRD patterns of Au-Ag/SiO2with various Au/Ag atomic ratios.
Figure 4. Changes in the average particle size with the calcination
temperature. The average particle size was determined by Scherrer’s equation according to the corresponding XRD spectra. The vertical line denotes the calcination temperature at 500°C.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
(SPR) of gold nanoparticles.50Prior to adsorbing Ag+in the
subsequent step, the sample was thoroughly washed with deionized water until no Cl-in the filtrate could be detected by Ag+. This washing step is proved to be very important because even trace amount of Cl-residue would result in the formation of AgCl precipitate on the sample surface upon adsorption of Ag+in the subsequent step, which in turn induces significantly the coalescence of particles during the final step of heat treatment due to the low melting point (455°C) of AgCl. Subsequently, Ag+cations were adsorbed on the preformed gold particle surface, or adsorbed on the amino groups that had been released. Considering that the amino groups tend to be positively charged in acidic AgNO3solution, they are difficult to bind
Ag+ cations because of electrostatic repulsion. This is also supported by the relatively low loading efficiency of Ag compared with gold on the amino-functionalized silica. For example, when a nominal amount of 6 wt % metal (Au or Ag)
was attempted to be loaded on the amino-functionalized silica, the actual Ag loading was only 0.90 wt %, whereas Au was 5.72 wt %. On the other hand, the loading efficiency of Ag could be increased significantly when gold particles were preformed on the functionalized silica surface (see Table 2 in section 3.4). Our DFT calculations also show that the gold cluster surface carries slightly negative charges, which allows the Ag+cations to be adsorbed on its surface via electrostatic and metallic interactions. Therefore, we believe that at this stage, Ag+ions were predominantly adsorbed on the preformed gold particle surface, and the UV-vis spectrum at this time still reflects the characteristic SPR of gold nanoparticles (Figure 6b). Following the reduction by NaBH4, a silver nanoshell was
produced on the gold core. According to the literature,48,51
whereas the silver atoms in the outer shell still remain as a pure silver phase, the silver atoms that are in close contact with the gold surface will diffuse into the gold core to form a gold-silver alloy phase due to the stronger bimetallic bonding, which is confirmed by our DFT calculations (see below). Thus, the UV-vis spectrum at this stage presents two distinct SPR bands (Figure 6c): one is centered at 493 nm and has a blue shift of 32 nm comparing to pure gold, which can therefore be assigned to the SPR of Au-Ag alloy phase. The other is centered at
(50) Link, S.; EI-Sayed, M. A. J. Phys. Chem. B 1999, 103, 4212.
(51) Shibata, T.; Bunker, B. A.; Zhang, Z.; Meisel, D.; Vardeman II, C. F.; Gezelter, J. D. J. Am. Chem. Soc. 2002, 124, 11989.
Figure 5. HRTEM images and the corresponding particle size distributions of Au-Ag/SiO2(Au/Ag ) 1/1) at different preparation stages. (a), (c), (d), and
(e) correspond to those in Figure 1.
Figure 6. UV-vis spectra of Au-Ag/SiO2(Au/Ag ) 1/1) at different
preparation stages. The samples of (a-e) correspond to those in Figure 1. (f) Ag/SiO2obtained by the same method as sample a. The maximum of
the absorption band is as follows: (a) 525, (b) 520, (c) 493 and 406, (d) 500, (e) 436, and (f) 406 nm.
Table 1. Calculated Average Bonding Energy (Eavg) and Impurity
Solution Energy (∆Eimp) for Pure or Impure Nanoparticles
clusters Eavga(eV) ∆Eimpb(eV)
Au38 2.231
Au37Agc face 2.214 -0.199
edge 2.213 -0.165 center 2.237 -0.477 Au36Ag2 2.242
aAverage bonding energy on each metal atom on clusters.bImpurity
solution energy, which was calcualted according to∆Eimp) E(AuN-1M)
- E(AuN) + (1/N)[E(AuN) - E(MN)]. cDifferent location of impure
metal atom was considered.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
406 nm and is characteristic of pure silver nanoparticles. Inasmuch as the penetration depth of incident light (29 nm for Ag and 37 nm for Au) is much greater than the sizes of the particles studied here,52we can observe the absorption bands
from both the surface silver nanoshell and the underlying Au-Ag alloy core. TEM image in Figure 5c also demonstrates that after deposition of silver on gold, the average particle size was slightly enlarged from 2.9 to 3.3 nm, which is in accordance with our proposed model of a thin nanoshell surrounding the Au-Ag alloy core (as shown in Figure 1c). Calcination in air at 500°C is necessary for removal of the amine ligand because any residue amine on the surface will significantly deactivate the catalyst. Unfortunately, this high-temperature treatment will induce, in most cases, severe sintering of nanoparticles for thermodynamic reasons. Comparing Figure 5a with Figure 2a, one can see that indeed gold nanoparticles without alloying with silver grow significantly from the original 2.9 nm to the after-calcination 5.7 nm. In remarkable contrast, the gold-silver bimetallic nanoparticles, with Au-Ag alloy core and an Ag nanoshell, can endure such harsh treatment, with the particle size remaining essentially unchanged (3.3 nm in Figure 5c vs 3.4 nm in Figure 5d) after calcination at 500 °C. From the UV-vis spectrum in Figure 6d, it is clear that after the calcination treatment, the SPR band from the silver nanoshell disappeared while the SPR band from the Au-Ag alloy red-shifted from 493 to 500 nm. The disappearance of the SPR band at 406 nm is most likely due to the oxidation of silver nanoshell by air to form silver oxide, which is transparent to the incident light. On the other hand, the red-shift of the SPR band of the gold-silver alloy core may be due to the segregation ofsilvertothesurfaceinducedbyhigh-temperaturecalcination,30,53
which makes the alloy core more gold-rich. This can also be visualized by the color change from yellow brown to red brown after the calcination treatment. Since significant sintering took place on pure gold nanoparticles during such high-temperature calcination while gold-silver bimetallic nanostructure was prettily sintering-resistant, we speculate that it is the silver oxide layer formed on the gold-silver alloy core that prevents the particles from sintering.
Our catalytic test shows that such nanoparticles with a silver oxide layer covering the gold-silver alloy core are totally inactive for CO oxidation at room temperature. To further activate the catalyst, a high-temperature reduction in H2 atmosphere proved to be necessary. Such a reduction
treatment did not induce the sintering of particles, but indeed
restructured the particles. As shown in Figure 6e, after the reduction treatment, the SPR band of the sample had a large blue-shift from 500 to 436 nm. The existence of only one peak indicates the formation of gold-silver alloy while its broadness may be due to the nonuniform Au/Ag ratio from one particle to another. Our EDS analyses (see Figure S1 in the Supporting Information) of different particles also confirmed this. The nonuniform composition may result from the varied amount of Ag+which was adsorbed on each gold core. The large blue-shift of the SPR band after the H2
reduction suggests that more silver atoms are alloyed with gold during that process. This can be easily understood considering that the silver oxide is reduced to metallic silver and the interdifussion between Au and Ag atoms drives completely random alloying of gold and silver. The structure changes of the nanoparticles during each step are also schematically illustrated in Figure 1.
The above proposed model was further supported by the changes of surface Au/Ag atomic ratios which were deter-mined by XPS. It should be pointed out that the binding energies (B.E.) of gold and silver for all the Au-Ag bimetallic samples we detected are characteristic of metallic gold and oxidized silver54(B.E. of Au
4f7/2varied from 83.3
to 83.5 eV, whereas that of Ag3d5/2varied from 367.4 to 367.6
eV for different samples, see Figure S7 and Table S1 in the Supporting Information). The absence of metallic Ag may be due to exposure to air during sample handling. Therefore, we just focus on the surface Au/Ag ratios. The surface Au/ Ag atomic ratio of the sample in Figure 1c is 1.60, which is significantly lower than the bulk value (determined by ICP) of 1.91 (see Table 2 in section 3.4). Clearly, the surface of the bimetallic nanoparticle at this stage is enriched with Ag, which is in accordance with our proposed model of a thin silver layer covering on the Au-Ag alloy core. When the sample in Figure 1c was calcined in air at 500°C (producing the sample in Figure 1d), the surface enrichment of silver became more pronounced, as indicated by the surface Au/ Ag atomic ratio of only 1.48. Evidently, the high-temperature calcination in air induces further segregation of Ag to the surface, leaving a more gold-rich Au-Ag alloy core. This result agrees well with our UV-vis spectra. After the final reduction by H2, the surface Au/Ag atomic ratio (1.89)
becomes very close to the bulk value, confirming the realloying of gold and silver to form a completely random
(52) Shi, H.; Zhang, L.; Cai, W. J. Appl. Phys. 2000, 87, 1572. (53) Gonza´lez, S.; Neyman, K. M.; Shaikhutdinov, S.; Freund, H. J.; Illas,
F. J. Phys. Chem. C 2007, 111, 6852.
(54) Chimentaˇo, R. J.; Kirm, I.; Medina, F.; Rodrı´guez, X.; Cesteros, Y.; Salagre, P.; Sueiras, J. E.; Fierro, J. L. G. Appl. Surf. Sci. 2005, 252, 793.
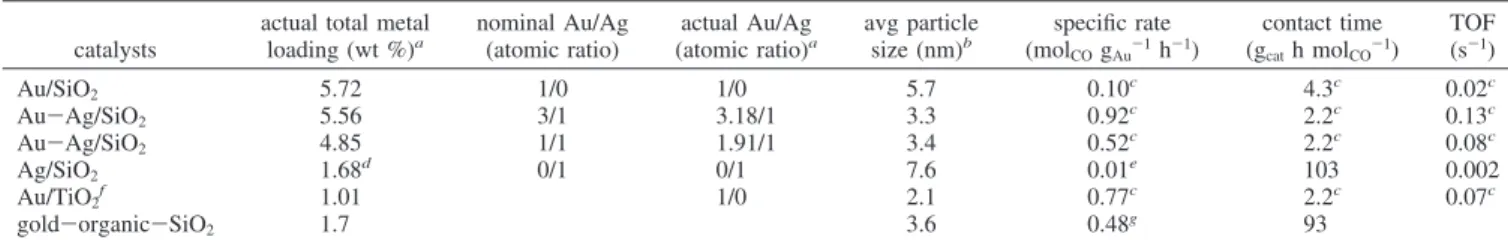
Table 2. Chemical Compositions of Au-Ag/SiO2Catalysts determined by ICP and Their Turnover Frequencies (TOFs) in CO Oxidation
catalysts
actual total metal loading (wt %)a nominal Au/Ag (atomic ratio) actual Au/Ag (atomic ratio)a avg particle size (nm)b specific rate (molCOgAu-1h-1) contact time (gcath molCO-1) TOF (s-1) Au/SiO2 5.72 1/0 1/0 5.7 0.10c 4.3c 0.02c Au-Ag/SiO2 5.56 3/1 3.18/1 3.3 0.92c 2.2c 0.13c Au-Ag/SiO2 4.85 1/1 1.91/1 3.4 0.52c 2.2c 0.08c Ag/SiO2 1.68d 0/1 0/1 7.6 0.01e 103 0.002 Au/TiO2f 1.01 1/0 2.1 0.77c 2.2c 0.07c gold-organic-SiO2 1.7 3.6 0.48g 93
aDetermined by ICP.bDetermined by HRTEM.cObtained at 20°C by controlling the CO conversion at below 10% via diluting with SiC.dThe
loading was obtained by twice deposition.eThe unit of the specific rate is mol
COgAg-1h-1.fProvided by the World Gold Council.gObtained at 30°C
in ref 18.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
alloy during the reduction process. Upon complete alloying, the particles became very active for CO oxidation.
3.3. Understanding the Exceptional Thermal Stability by DFT Calculation. From the above experimental results
one can see that Ag plays a key role in stabilizing the gold alloy particles on inert silica and alumina support. For a better understanding of such a stabilizing role, we performed quasi-relativistic density functional calculations on a series of model clusters Au38-xAgx(x ) 0, 1, 2) to elucidate the nature of the
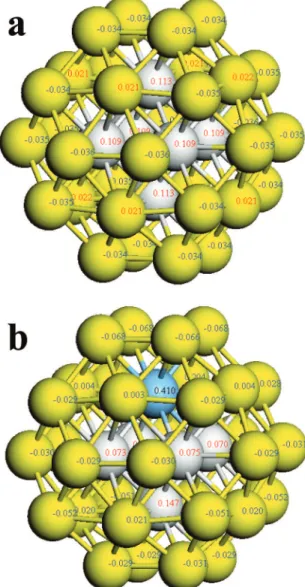
interactions between the two components of the bimetallic alloy. All these nanoclusters were constructed based on a face-centered cubic (FCC) octahedral Au38cluster.55,56As shown in Figure
7, the low-coordinated surface atoms of the pure Au38cluster
carry slightly negative charges, which will allow the Ag+ions to be preferentially adsorbed on the gold nanoparticle surface rather than on those free amine ligands. Moreover, it was found that when one Ag atom is incorporated in the Au38cluster to
substitute one Au atom, the Ag atom tends to go into the cluster center rather than stay on the surface layer (edge or face), as
indicated by the increased average bonding energies (Table 1, 2.231 eV for Au38and 2.237 eV for Au37Ag). The calculated
impurity solution energy of Au37Ag is -0.477 eV, suggesting
that alloying gold with silver is drastically exothermic and thermodynamically favorable. These calculation results on the model clusters are in good agreement with our experimental observations. When silver atoms are deposited on the preformed gold nanoparticles, those subsurface Ag atoms in close contact with the gold particle surface will spontaneously diffuse into the gold cluster to form the gold-silver alloy. The increased bonding energies of Au37Ag and Au36Ag2also suggest that the
alloy phase should have a higher melting point than the pure gold clusters. Because of the large heteronuclear Au-Ag bonding, when more silver atoms are incorporated, as in the actual Au-Ag bimetallic nanoparticles with∼3 nm size, the average bonding energies will become even higher, and thereby the rise of the melting temperature will become more pro-nounced. In agreement with our calculation results, Cheng et al.57reported that doping of Au
55with a single Cu atom can
sharply raise the melting point of the cluster. Similarly, through molecular dynamics simulations of the thermal behavior of 55-atom Au-Ag nanoalloys it is demonstrated that the melting temperature progressively goes up with increasing the Ag content.58Thus, the increased melting point by alloying gold
with silver should be partially responsible for the thermal stability of the Au-Ag nanoparticles. On the other hand, the most important factor for the thermal stability of the small-size Au-Ag particles, we believe, is due to the outmost Ag nanoshell covering the Au-Ag alloy core. As the Ag atoms are attached to the Au particles preformed in the first step, these “surface” Ag atoms are more easily oxidized during the calcination. Consistent with this notion, our DFT calculations show that the Ag atoms on the surface layer of the model cluster carry significant positive charges because of the different electronegativities for Ag and Au. These partially positively charged Ag atoms can therefore be easily oxidized during calcination in the air. The thus formed oxides serve as passivated agents that prevent the coalescence of the Au-Ag particles to form larger particles. From a more general viewpoint, the surface Ag oxide layer is just like a nanoglue that adheres strongly the inside nanoparticles to the silica support via Ag-O-Si linkages because oxides generally have a stronger interaction than the corresponding metals with the support. This effect is reminiscent of traditional metal-support interaction, which is often tuned by some promoters just like a nanoglue.59
The uniqueness of our present Au-Ag bimetallic nanostructure is that the nanoparticles are prevented from sintering under high-temperature oxidizing atmosphere because of formation of passivated layer while these nanoparticles can be activated by reducing the passivated oxide layer to reform the Au-Ag alloy phase.
3.4. Reactivity of Au-Ag Nanoparticles toward CO Oxidation. Inasmuch as the Au-Ag particles are rather small
and highly dispersed, they exhibit high activities toward CO
(55) Li, T. X.; Yin, S. Y.; Ji, Y. L.; Wang, B. L.; Wang, G. H.; Zhao, J. J.
Phys. Lett. A 2000, 267, 403.
(56) Garzo´n, I. L.; Michaelian, K.; Beltra´n, M. R.; Posada-Amarillas, A.; Ordejo´n, P.; Artacho, E.; Sa´nchez-Portal, D.; Soler, J. M. Phys. ReV.
Lett. 1998, 81, 1600.
(57) Cheng, D.; Huang, S.; Wang, W. Phys. ReV. B 2006, 74, 064117-1. (58) Chen, F.; Curley, B. C.; Rossi, G.; Johnston, R. L. J. Phys. Chem. C
2007, 111, 9157.
(59) Schubert, M. M.; Kahlich, M. J.; Feldmeyer, G.; Hu¨ttner, M.; Hackenberg, S.; Gasteiger, H. A.; Behm, R. J. Phys. Chem. Chem.
Phys. 2001, 3, 1123.
Figure 7. Mulliken net charges on (a) Au38cluster; (b) Au37Ag cluster.
Gray, inner gold atoms; yellow, outer gold atoms; blue, silver atom.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
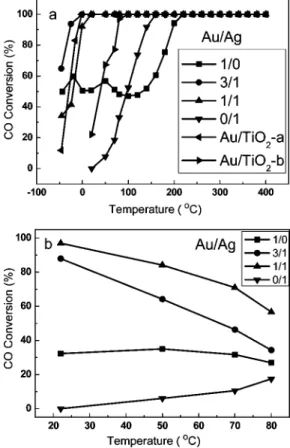
oxidation. As shown in Figure 8a, the Au-Ag alloy catalysts are active for CO oxidation even at a reaction temperature as low as -50°C, and they all greatly outperform either of the monometallic ones. Table 2 lists the turnover frequencies (TOFs) of the catalysts, which are calculated based on the relationship between the particle size and the dispersion.60
It is found that the TOF of the Au-Ag/SiO2(3/1) is nearly
5 times larger than that of the Au/SiO2. The activities follow
the order of Au-Ag (3/1)/SiO2> Au-Ag (1/1)/SiO2> Au/
TiO2 > Au/SiO2. Ag/SiO2. Compared with
Au-organic-SiO2(the last row in Table 2), which was claimed to be as
active as Au/TiO2,18 our Au-Ag/SiO2 catalysts exhibit a
much higher reaction rate at a very short contact time, at least 1 order of magnitude shorter than that used for the gold-organic-SiO2catalyst. To further show the superior
sintering-resistant property of the Au-Ag bimetallic system, we calcined Au/TiO2at 500 °C for 6 h and then reduced it in
hydrogen at 550°C for 1 h. The activity test result showed that after such high-temperature treatment, the activity of the Au/TiO2was considerably decreased (as shown by Au/
TiO2-b in Figure 8a), which is in contrast with our Au-Ag/
SiO2. It is also noted that, in spite of the comparable particle
size, Au-Ag (3/1) possesses a higher activity than Au-Ag (1/1). This implies that in addition to the particle size effect, there may be synergetic electronic effect between Au and Ag affecting the catalytic performance of Au-Ag alloy. When using different supports (nano silica, alumina, SBA-15), the Au-Ag particles also exhibited a high catalytic activity for CO oxidation (Figure S8). In the case of excess H2 present in the reacting stream (so-called preferential
oxidation of CO in H2), our Au-Ag catalysts also show
superior performances. As shown in Figure 8b, the CO conversion decreases with increasing the reaction temperature over all the gold-containing catalysts due to the competitive H2oxidation. However, at room temperature, both the CO
conversion and the selectivity to CO2over the Au-Ag (Au/
Ag ) 1/1) catalyst are nearly 100%. In comparison with our earlier reported Au-Ag@MCM-41 system,30 which was
completely inactive for the preferential oxidation of CO under excess condition, the present Au-Ag nanoparticles display exceptional performance with or without the presence of H2,
reemphasizing the importance of the small particle sizes of the nanocatalysts.
4. Conclusions
In summary, we have developed a general strategy to obtain small-sized, thermally stable, and highly active Au-Ag nano-particles on commerically available silica and alumina supports. We believe this method can be extended to a variety of other supports with rich hydroxyl groups on their surfaces. The resultant nanostructure composed of Au-Ag alloy core and a silver nanoshell can greatly limit the aggregation of nanopar-ticles during the high-temperature calcinations, thanks to the formation of a thin protective silver oxide layer that can act as a nanoglue between the silica substrate and the gold-silver particles. The final H2-reduction treatment not only reduced the
thin oxide layer but also induced realloying of gold and silver, thereby activated the catalyst. Attributed to a remarkably decreased particle size and a possible promoting role of Ag in activating oxygen, thus-prepared gold-silver alloy particles give rise to excellent performance in catalyzing CO oxidation, even in a H2-rich atmosphere. This strategy of stabilizing the gold
particles by alloying with a second metal paves a novel way for controlling the particle sizes and for enhancing the thermal stabilities of gold particles, in particular on those inert supports. Acknowledgment. Support from the National Natural Science Foundation of China (NNSFC 20673116, 20773124, 20325620) (T.Z. and A.Wang), NKBRSF (2006CB932305), and NNSFC (20525104) (J.L.) are gratefully acknowledged.
Supporting Information Available: EDS spectra, TEM images, N2 sorption isotherms of different supports, XPS spectra, and activity profiles (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
CM8027725 (60) Bond, G. C.; Thompson, D. T. Catal. ReV. Sci. Eng. 1999, 41, 319.
Figure 8. CO conversions with reaction temperature over Au-Ag/SiO2
with varying Au/Ag ratios. In each experiment, 60 mg of the catalyst was used. (a) Gas mixture, CO/O2/He ) 1/1/98 (vol.); flow rate, 20 mL min-1.
Au/TiO2is provided by the World Gold Council: Au/TiO2-a was used as
received and Au/TiO2-b was calcined at 500°C for 6 h followed by reducing
in H2at 550°C for 1 h. (b) Gas mixture, CO/O2/H2/He ) 1/0.5/50/48.5
(vol.); flow rate, 40 mL min-1.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009