Synthesis of ketoenaminoanthryl and
9,10-Bis-isoxazolylanthryl Linked Biscalix[4]arenes: Atropisomers and
Molecular Recognitions
Chia-Chen Tsai,
†I-Ting Ho,
†Jean-Ho Chu,
†Li-Ching Shen,
†Shou-Ling Huang,
‡and Wen-Sheng Chung*
,††
Department of Applied Chemistry, National Chiao Tung University, Hsinchu, Taiwan 30050, Republic of China
‡Instrumentation Center, National Taiwan University, Taipei, Taiwan 106, Republic of China
*
S Supporting InformationABSTRACT:
An efficient synthetic pathway for the synthesis
of biscalix[4]arenes 5
−10 using 1,3-dipolar cycloaddition
reactions is reported. Biscalix[4]arene 10 is capable of forming
a complex with methyl viologen because of favorable cation
−π
interactions and a proper cavity size to accommodate the
guest. Moreover, biscalix[4]arenes 8a and 8b were found to be
atropisomers at room temperature. These two conformers
were unable to exchange at room temperature because of the
restricted rotation of the C
9−C
11or C
10−C
12bonds of the
β-amino-α,β-unsaturated ketones of anthracene.
■
INTRODUCTION
Biscalixarenes
1have been studied extensively in recent years
because the structures usually contain interesting properties
including allosteric effect,
2intramolecular oscillation,
3and
conformational conversion.
4An internal cavity, formed
naturally through the linkage of two calixarenes, can be used
as a host not only for metal ions but also for neutral molecules.
For example, Gutsche and co-workers reported that 5,5
′-biscalix[5]arene can be used to selectively recognize
full-erene[70] over fullerene[60], where the biscalix[5]arene
undergoes an anti to syn conformational change upon
complexation with a fullerene to maximize the interaction
between host and guest.
4aMethyl viologen is one of the most widely used herbicides in
the world. It is shown to be toxic to humans and animals and is
linked to the development of Parkinson’s disease;
5accordingly,
it is highly desirable to have a selective and sensitive method in
the fast screening of methyl viologen. Currently, most of the
detection of methyl viologen relies on
1H NMR titration
experiments using various macrocycles such as calix[4]arenes,
6crown ethers,
7triptycenes,
8and pillar[5]arenes.
9There has
been very few reports on the fluorescent sensing of methyl
viologen. To the best of our knowledge, Wagner and Isaacs
were the first to report a fluorescent sensing of methyl viologen
using cucurbit[6]uril as the host.
10The design and synthesis of
a highly specific fluorescent sensor for viologen is still
demanding, and it would have following benefits: high
sensitivity, easy to use, low cost, and low background
interference.
We have been using a strategy to construct a variety of
functionalized isoxazoline and isoxazole unit(s) onto the
calix[4]arene skeletons through double and/or quadruple
1,3-dipolar cycloaddition reactions of alkenes/alkynes with aryl
nitrile oxides.
11,12In order to further expand the diversity of
biscalix[4]arenes, we also explored possible ring-opening
reactions of mono- and bis-isoxazole substituted
calix[4]-arenes.
12−14To our delight, Mo(CO)
6-mediated ring-opening
reactions of these isoxazole-substituted calix[4]arenes led to the
formation of various enaminone (
β-amino-α,β-unsaturated
ketone) appended calix[4]arenes efficiently.
14Using the
protocol described above, we report herein the synthesis of
biscalix[4]arenes 6
−10, which contain an ellipsoidal cavity and/
or an anthryl group as fluorophore. The ring-opening reaction
of biscalixarene 7 led to two 9,10-bis-ketoenaminoanthryl
biscalix[4]arenes, 8a and 8b, which showed interesting
atropisomeric properties.
15The application of biscalix[4]arene
10
as a fluorescent chemosensor for methyl viologen is also
studied.
■
RESULTS AND DISCUSSION
The synthetic pathways for biscalix[4]arenes 5, 7, and 10 are
depicted in Scheme 1. Our synthetic strategy for linking two
calix[4]arenes started with the double 1,3-dipolar cycloaddition
reactions between aryl dinitrile oxides (prepared in situ from 3
and 4) and propargyl ether to yield 5 and 7 in 43% and 62%
yield, respectively. In principle, the quadruple cycloaddition
reactions, of two bispropargyloxycalix[4]arenes 2 with two
anthracene-9,10-bis(carbonitrile oxide) 4, should lead to a
doubly bridged biscalix[4]arene 10; however, when 2 (5.5 mM)
was refluxed with 4 (5.5 mM) in THF for 24 h, the reaction
mixture became very messy and was difficult to be purified by
Received: December 7, 2011
Published: February 21, 2012
column chromatography. Alternatively, the doubly bridged
biscalix[4]arene 10 could be synthesized via a two-step reaction
sequence starting from 7. First, the bispropargyl ether
substituted biscalix[4]arene 9 was obtained in 86% yield
through S
N2 reaction of 7 with 2 equiv of propargyl bromide
under basic conditions.
Second, a double 1,3-dipolar cycloaddition of the
bispro-pargyl ether substituted biscalix[4]arene 9 with 4 afforded the
doubly bridged biscalix[4]arene 10 in 32% yield.
1H NMR
spectrum of the methylene bridge protons and the isoxazole
protons of biscalix[4]arene 10 showed only two singlets
implying that its structure was highly symmetrical.
The N
−O bond cleavage of the isoxazole units of
biscalix[4]arenes 5 by Mo(CO)
6-mediated ring-opening
reaction led to the formation of 1,4-bisketoenaminophenyl
biscalix[4]arene 6 and recovered calix[4]arene in 32 and 40%
yield, respectively. Under similar reaction conditions, the
ring-opening reaction of 7 gave the 9,10-bisketoenaminoanthyl
biscalix[4]arenes 8a and 8b, as a mixture of atropisomers, in
67% yield (Scheme 2).
1H NMR spectra of these compounds
showed that the amino protons of the ketoenaminos appeared
as two singlets: one around
δ10.0−10.2 (due to H-bonding
with the carbonyl groups) and the other around
δ 5.6−5.8 ppm.
The structures of all products (5−10) were fully characterized
by spectral data including
1H and
13C NMR (Figures S12
−S27,
Supporting Information), mass, and high resolution mass
spectrometry (Experimental Section). Furthermore, the
structure of biscalix[4]arene 10 was confirmed by a
single-crystal X-ray single-crystallography analysis (Figure 1). The X-ray
crystal structure of 10 clearly shows that it contains a
rectangular cone cavity. This biscalix[4]arene is a
nanometer-sized macrocycle (3.0 nm long) with two parallel anthracene
moieties, and the distance between the two anthracene planes is
4.0 Å. The two anthracenes are not in juxtaposition; they are
slightly staggered. The cavity of 10 is constructed by the walls
of two parallel anthracene moieties and two tail-to-tail
calix[4]arenes; therefore, it has a potential for
π−π interaction
and recognition of dications by the two bridged calix[4]arenes.
To this end, we envisaged that methyl viologen and its
analogues may have the potential to be snugly fit into the
rectangular cavity of biscalix[4]arene 10.
Unexpectedly, the
1H NMR spectra of the ring-opened
products 8 clearly showed two sets of signals (Figure 2b),
indicating the existence of two conformational isomers or
so-called atropisomers.
15In contrast, the ring-opened product 6,
from the reaction of 1,4-bisisoxazolylphenyl substituted
biscalix[4]arene 5, showed only one set of proton signals
(Figures 2a, S1, and S2, Supporting Information).
To determine whether steric hindrance between the anthryl
and the ketoenamino groups or steric bulkiness of the
calix[4]arene plays the crucial role in making compounds 8
atropisomers, we synthesized a control compound 12, in which
the two calix[4]arenes were replaced by two para-t-butylphenyl
ether groups (Scheme 3). The
1H NMR spectrum of 12 at
room temperature gives rise to two well-resolved sets of signals
(Figure S26, Supporting Information). The results imply that
adding a bulky substituent or not at a remote position from the
9,10-bisketoenamino substituted anthracene did not affect its
atropisomeric properties. There is no need to replace t-butyloxy
group with a bulkier substituent, such as calix[4]arene, to
achieve atropisomeric properties in the 9,10-bisketoenamino
substituted anthracene. Note that no atropisomeric properties
were found for the phenyl bridged 1,4-biscalix[4]arene 6
(Figure 2a); thus, the hindered rotation in the
9,10-bisketoenamino substituted anthracene of 8 or 12 plays a key
role in forming atropisomers.
1
H NMR spectra of the two biscalixarenes 8a and 8b show
that some of their signals are separated and allowed for area
integrations. At room temperature (298 K), the ring-opened
products 8a and 8b exist as a mixture of conformers with a ratio
of 46:54 in CDCl
3(Figures S2 and S3, Supporting
Information). However, the assignment of the cis- or
trans-atropisomers cannot be unambiguously determined yet. We
tried to separate the atropisomers 8a and 8b by HPLC using
various columns;
16however, it was unsuccessful.
Variable-temperature NMR studies at Variable-temperatures as high as 393 K
(sample started to decompose) showed that the two sets of
proton signals of 8a and 8b did not have any symptoms of
merging (Figures S4 and S5, Supporting Information), implying
a very high energy barrier for the rotations of C
9−C
11and C
10−
C
12bonds.
15e,fThe energy barriers for the restricted rotation in
Scheme 2. Syntheses of Ring-Opened Biscalix[4]arene 6 and Atopisomers 8a and 8b
9-phenylanthracenes have been predicted by DFT
calcula-tions
15and confirmed experimentally by VT NMR
15e,17to be
∼21 kcal mol
−1. On the other hand, the simplicity of the NMR
spectra of 7 even at
−50 °C (Figures S6, Supporting
Information) implies that rapid rotation occurs at this
temperature and that there is a very low energy barrier of the
rotations of C
9−C
11and C
10−C
12in 7. The rotational energy
barrier of the bis-isoxazole substituted anthracene 7 is estimated
to be smaller than 10 kcal mol
−1.
18The normalized fluorescence spectra of biscalix[4]arenes 7,
10
and control compound 11 are shown in Figure 3.
Biscalix[4]arene 10 displayed a broader emission band (
λ
maxat 443 nm) compared to those of biscalix[4]arene 7 and control
compound 11 (both showed a
λ
maxat 432 nm). The results
implied that an intramolecular
π−π interaction of the two
parallel anthracenes of biscalix[4]arene 10 should have
occurred in cosolvent MeOH/CHCl
3(v/v, 1:2), which led to
a longer emission wavelength.
Since biscalix[4]arene 10 contains anthracenes as
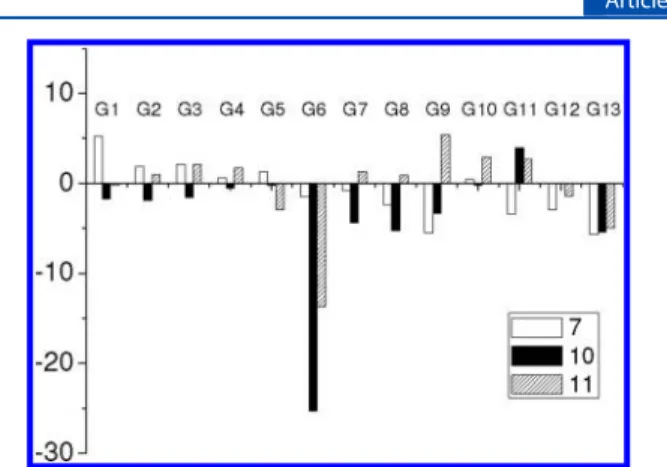
fluoro-phores, we then used it in fast screening on a series of aromatic
guests, alkyldiamines, and methyl viologen (G1−G13, Chart 1)
using fluorescence spectroscopy. The binding properties of 10
in cosolvent MeOH/CHCl
3(v/v, 1:2) were assessed by adding
200 equiv of various guests, and their relative fluorescence
intensity changes are shown in Figure 4. There was basically no
(or very small) change in the fluorescence spectra of
biscalix[4]arene 10 when it was mixed with excess aromatic
guests (G1
−G5) and alkyldiamines (G7−G13). To our
delight, only methyl viologen (G6) caused a significant
quenching on the fluorescence of biscalix[4]arene 10 (Figures
4 and 5). The fluorescence quantum yield of 10 was
determined to be 0.80
± 0.02 using 9,10-diphenylanthracene
Figure 2.1H NMR spectra of the ring-opened products (a) 6 and (b) 8, where* denotes signals from the residual of chloroform-d. In spectrum (b),
the signals labeled with a prime come from atropisomers.
as a standard.
19Upon titration with G6, the fluorescence
intensity of 10 gradually decreased, which gave a fluorescence
quantum yield of 0.57
± 0.01 (30% decrease) at 200 equiv of
G6. The association constant of complex 10
·G6 was
determined to be 137.4
± 7.6 M
−1by a Stern
−Volmer plot
20(Figure 5b). Furthermore, the excimer emission of 10 was
slightly blue-shifted at high equivalents of G6, indicating that
the
π−π interaction of the two parallel anthracenes of
biscalix[4]arene 10 was reduced. The results imply that G6
might have been embedded into the cavity of biscalix[4]arene
10, hence favoring the monomer emission compared to that of
the excimer.
1
H NMR titration experiments of biscalix[4]arene 10 with
methyl viologen (G6) were also carried out to shed light on its
binding mode (see Figures S7 and S8, Supporting
Informa-tion). The proton signals of the anthracene of the host 10 were
slightly upfield shifted by the addition of G6, which is
consistent with the inclusion of G6 in the cavity of 10.
Moreover, we also found that the proton signals of G6 were
broadened and high field shifted in the presence of 10 equiv of
10
(Figure S8, Supporting Information). Diffusion-ordered
NMR spectroscopy (DOSY) has been particularly useful in the
characterization of complex host
−guest systems in solution.
21Thus, 2D DOSY experiments were used to investigate the
complex between biscalixarene 10 and G6. When a 1:1 mixture
of 10 and G6 was measured in CD
3OD/CDCl
3(v/v, 1/2) at
295 K, the diffusion coefficients for host 10 and guest G6 were
determined to be 3.16
× 10
−10and 6.03
× 10
−10m
2/s,
respectively. However, when 100 equiv of G6 with 1 equiv of
10
were measured by 2D DOSY, a new species with a different
diffusion coefficient (4.17
× 10
−10m
2/s) appeared. This
indicates that biscalixarene 10 and methyl viologen G6 form a
complex. (Figure S9, Supporting Information)
In order to know whether the rectangular cavity of
biscalix[4]arene 10 is necessary for the recognition of methyl
viologen, we synthesized a control compound 11, in which the
two calix[4]arene units are replaced by two para-t-butylphenyl
groups. Furthermore, the fluorescence study of the
open-chained biscalix[4]arene 7 toward methyl viologen G6 was also
used for comparison (Figure 4). The fluorescence of the
open-chained biscalix[4]arene 7 showed very little change at 200
equiv of G6;
19however, the fluorescence quantum yield of the
other control compound 11 did show some quenching by G6.
The quenching effect of methyl viologen G6 on the open-chain
bis-para-t-butylphenyl 11 was smaller (
Φ
Fdecreased by 20%)
compared to that on the biscalix[4]arene 10 (
Φ
Fdecreased by
30%). The association constant of complex 11
·G6 was
determined to be 77.6
± 0.6 M
−1by a Stern
−Volmer plot.
20(Figure S10, Supporting Information). The
1H NMR titration
spectra of control compound 11 with G6 showed no change
even with 10 equiv of G6 (Figure S11, Supporting
Information). On the basis of these observations, we conclude
that not only the cation
−π interaction but also a proper cavity
size must have played important roles in the binding of methyl
viologen (G6) by biscalix[4]arene 10.
Finally, an optimized geometry of 10 with G6 was calculated
by the molecular modeling DMol
3and simulated in CHCl
3
environment (Figure 6 and Tables S4
−S5, Supporting
Information).
22,23The DMol
3method from Material Studio
5.0 is developed by Accelrys Inc., in which the wave functions
are expanded in terms of an accurate numerical basis set. We
used a double-numeric quality basis set with polarization
functions (DNP). The size of the DNP basis set is comparable
to Gaussian 6-31G
**, but DNP is more accurate than a
Figure 3.Normalized fluorescence spectra of biscalixarenes 7, 10 and control compound 11 (10μM, MeOH/CHCl3(v/v, 1:2)). Excitation
wavelength was at 393 nm for 7, 395 nm for 10, and 392 nm for 11.
Chart 1
Figure 4.Relative fluorescence intensity changes ((I− I0)/I0× 100%)
of biscalixarenes 7, 10 and the control compound 11 (each of 10μM) in MeOH/CHCl3(v/v, 1:2) at 298 K upon addition of various guests
(200 equiv). Excitation wavelength was at 393 nm for 7, 395 nm for 10, and 392 nm for 11.
Gaussian basis set of the same size.
23The tolerances of the
energy, gradient, and displacement convergences were 2
× 10
−5Ha, 4
× 10
−3Ha Å
−1, and 5
× 10
−3Å, respectively.
23bThe
optimized geometries of 10 with G6 by calculation showed a
sandwich-like structure. The distance between two anthracenes
of 10 increased from 4.0 Å (crystal) to ca. 6.5 Å when the G6
was embedded into the cavity of biscalix[4]arene 10. The
results explain why the excimer emission of 10 was slightly
blue-shifted.
■
CONCLUSION
Using a two-step reaction sequence, we have successfully
synthesized a novel fluorescent biscalix[4]arene 10 with
rectangular cavity. The biscalix[4]arene 10, with two parallel
anthracene units, was found to show some affinity to dication
molecules such as methyl viologen (G6). Although the binding
constant of biscalix[4]arene 10 with G6 is small (137.4
± 7.6
M
−1), it has the advantages of fast and easy screening by
fluorescence spectroscopy. From a comparison of the results
with two other control compounds (7 and 11), we believe that
the cation
−π interaction as well as a proper cavity size play key
roles in the complexation of biscalix[4]arene 10 with G6.
Moreover, 2D DOSY experiments provided strong evidence to
support the complex formation between 10 and G6.
We also found that not only 9,10-bisketoenaminoanthryl
biscalix[4]arenes (8a and 8b) but also
9,10-bisketoenami-noanthryl bis-t-butyl-phenol ethers (12a and 12b) are
atropisomers, where hindered rotation between the
ketoena-mino group and the nearby C
−H hydrogens of the anthracene
were the key features. The estimated energy barriers for the
restricted rotation of in the 9,10-bisketoenaminoanthryl
derivatives 8a,b are >23 kcal mol
−1from VT NMR. In sharp
contrast, the isoxazole substituted 9,10-bisisoxazolylanthryl
biscalix[4]arene 7 has a much lower energy barrier on the
rotation of C
9−C
11; therefore, even at temperatures as low as
−50 °C, it did not show any symptom of proton NMR signal
splitting between its atropisomers.
■
EXPERIMENTAL SECTION
General Methods.1H NMR spectra were measured with either a
300 or 500 MHz spectrometer. Natural abundance13C NMR spectra
were measured using pulse Fourier transform techniques, with a 300 or 500 MHz NMR spectrometer operating at 75.4 and 125.7 MHz, respectively. Mass spectra were recorded in the FAB mode with m-nitrobenzyl alcohol (NBA) as the matrix. UV−vis and fluorescence spectra were measured with spectrometer and spectrofluorimeter using HPLC-grade solvents.
1,4-Bis-isoxazolyl-phenylmethyl Linked Biscalix[4]arene, 5. Triethylamine (0.35 mmol) in ethanol (1.9 mL) was slowly added to a well-stirred solution of 1 (0.40 g, 0.69 mmol) and hydroximoyl chloride 3 (0.07 g, 0.31 mmol) in ethanol (30 mL). The reaction mixture was stirred at reflux for 24 h under N2(g). After evaporation
of the solvent, the mixture was washed with water and extracted with dichloromethane. The organic phase was dried over MgSO4, and the
solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography using ethyl acetate/n-hexane as eluent to give 0.20 g (42.7%) of 5 as a yellow solid: mp 178−180 °C; Rf= 0.45 (ethyl acetate/n-hexane = 1:4);1H NMR (CDCl3,300 MHz) δH10.00 (s, 2H), 9.16 (s, 4H), 8.03 (s, 4H), 7.12−6.99 (m, 16H), 5.39 (s, 4H), 4.33 (d, 4H, J = 13.2 Hz), 4.26 (d, 4H, J = 13.7 Hz), 3.46 (d, 4H, J = 13.2 Hz), 3.44 (d, 4H, J = 13.7 Hz), 1.22−1.20 (m, 72H) ppm; 13C NMR (CDCl 3, 75.5 MHz) δC 167.6 (Cq), 162.1 (Cq), 149.0 (Cq), 148.9 (Cq), 148.3 (Cq), 147.5 (Cq), 143.7 (Cq), 143.2 (Cq), 133.2 (Cq), 130.4 (Cq), 128.1 (Cq), 127.7 (Cq), 127.6 (CH), 127.4 (Cq), 126.7 (CH), 125.8 (CH), 125.7 (CH), 125.6 (CH), 102.8 (CH), 68.1 (CH2), 34.3 (Cq), 34.0 (Cq), 33.9 (Cq), 32.9 (CH2), 32.2
Figure 5.(a) Fluorescence emission spectra of biscalix[4]arene 10 (10μM) in the presence of various equivalents of methyl viologen (G6) and (b) its corresponding Stern−Volmer plot using the intensity at 443 nm as a parameter (Ka= 137.4± 7.6 M−1). All measurements were in a cosolvent of
MeOH/CHCl3(v/v, 1:2), and the excitation wavelength was 395 nm.
(CH2), 31.4 (CH3), 31.2 (CH3) ppm; FAB-MS m/z 1534 (M + H+),
1533 (M+); HRMS (FAB) calcd for C102H120O10N21532.8943, found
1532.8916.
1,4-Bis-ketoenamino-phenylmethyl Linked Biscalix[4]arene, 6. A mixture of 5 (0.05 g, 0.03 mmol), Mo(CO)6(0.02 g, 0.08 mmol),
and H2O (0.2 mL) in CH3CN (10 mL) was stirred and heated at
reflux for 5 h. The solvent was removed under a vacuum, and the residue was dissolved in 10 mL of dichloromethane. Then, to the solution was added 10 mL of NH4OH (aq) to remove remaining
molybdenum salts. After stirring for 1 h, the organic layer was washed with water and 1 M EDTA (aq). The organic phase was dried over MgSO4, and the solvent was removed under reduced pressure. The
residue was purified by neutral silica gel column chromatography with ethyl acetate/n-hexane (v/v, 1:5) as eluent to give 0.016 g (31.9%) of yellow solid 6 with para-tert-butyl calix[4]arene (40%) as a side product. 6: mp 182−184 °C; Rf= 0.1 in ethyl acetate/n-hexane (v/v,
1:3);1H NMR (300 MHz, CDCl 3)δH10.28 (s, 2H), 10.07 (bs, 2H), 9.55 (s, 4H), 7.83 (s, 4H), 7.09−6.98 (m, 16H), 6.04 (s, 2H), 5.56 (bs, 2H), 4.85 (s, 4H), 4.53 (d, 4H, J = 12.9 Hz), 4.29 (d, 4H, J = 13.8 Hz), 3.44 (d, 4H, J = 13.5 Hz), 3.41(d, 4H, J = 12.9 Hz), 1.50−1.36 (m, 72H) ppm;13C NMR (CDCl 3, 75.5 MHz)δC193.6 (Cq), 161.7 (Cq), 150.3 (Cq), 148.3 (Cq), 148.0 (Cq), 143.5 (Cq), 143.0 (Cq), 139.2 (Cq), 133.4 (Cq), 128.2 (Cq), 127.4 (Cq), 127.3 (CH), 126.5 (CH), 125.8 (CH), 125.6 (CH), 91.1 (CH), 79.2 (CH2), 34.2 (Cq), 34.0 (Cq), 33.9 (Cq), 33.1 (CH2), 32.3 (CH2), 31.5 (CH3), 31.2 (CH3) ppm; FAB-MS m/z 1539 (M + 2), 1538 (M + H+); HRMS (FAB) calcd for C102H124N2O101536.9256, found 1536.9266.
9,10-Bis-isoxazolylanthryl-methyl Linked Biscalix[4]arene, 7. A mixture of 1 (0.20 g, 0.30 mmol) and 4 (0.04 g, 0.15 mmol) in THF (15 mL) was heated at reflux for 24 h under N2(g). After evaporation
of the solvent, the mixture was washed with water and extracted with dichloromethane. The organic phase was dried over MgSO4, and the
solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography with ethyl acetate/n-hexane as eluent to give 0.15 g (62.2%) of 7 as a yellow solid: mp 180−182 °C; Rf = 0.35 (ethyl acetate/n-hexane (v/v, 1:5));1H NMR (300 MHz, CDCl3)δH10.13 (s, 2H), 9.34 (s, 4H), 8.11−8.08 (m, 4H), 7.61−7.58 (m, 4H), 7.28−7.07 (m, 18H), 5.63 (s, 4H), 4.55(d, 4H, J = 13.2 Hz), 4.33 (d, 4H, J = 13.8 Hz), 3.61 (d, 4H, J = 13.2 Hz), 3.51 (d, 4H, J = 13.8 Hz), 1.33−1.29 (m, 72H) ppm;13C NMR (CDCl 3, 75.5 MHz) δC167.2 (Cq), 161.4 (Cq), 149.1 (Cq), 148.8 (Cq), 148.3 (Cq), 147.7 (Cq), 143.6 (Cq), 143.3 (Cq), 133.5 (Cq), 130.3 (Cq), 128.1 (Cq), 127.7 (Cq), 127.7(Cq), 126.8 (CH), 126.0 (CH), 125.9 (CH), 125.7 (CH), 125.7 (CH), 125.5 (Cq), 108.8 (CH), 67.6 (CH2), 34.3 (Cq), 34.0 (Cq), 33.9 (Cq), 32.9 (CH2), 32.4 (CH2), 31.5 (CH3), 31.2 (CH3) ppm; FAB-MS m/z 1634 (M + H+), 1633 (M+); HRMS (FAB)
calcd for C110H124O10N21632.9256, found 1632.9275.
9,10-Bis-ketoenaminoanthryl Linked Biscalix[4]arene, 8. A mixture of 7 (0.10 g, 0.06 mmol), Mo(CO)6(0.07 g, 0.25 mmol), and
3 drops H2O in THF/CH3CN (1 mL/10 mL) was stirred and heated
at reflux for 5 h. The solvent was removed under a vacuum, and the residue was dissolved in 10 mL of dichloromethane. Then, to the solution was added 10 mL of NH4OH (aq) to remove remaining
molybdenum salts. After stirring for 1 h, the organic layer was washed with water and 1 M EDTA (aq). The organic phase was dried over MgSO4, and the solvent was removed under reduced pressure. The
residue was purified by neutral silica gel column chromatography with ethyl acetate/n-hexane (v/v, 1/5) as eluent to give 0.07 g (66.7%) of yellow solid 8a and 8b (atropisomers): mp 218−220 °C; Rf= 0.43
(ethyl acetate/n-hexane (v/v, 1:3)); 1H NMR (300 MHz, CDCl 3)
atropisomers 25°C, area ratio = 46:54, δH10.20 (bs, 1H), 10.14 (bs,
1H), 9.96 (s, 2H), 9.87 (s, 2H), 9.39 (s, 4H), 8.28−8.27 (m, 4H), 7.61−7.59 (m, 4H), 7.08−6.90 (m, 16H), 5.76 (bs, 1H), 5.68 (s, 1H), 5.51 (s, 2H), 4.94 (s, 2H), 4.90 (s, 2H), 4.60−4.44 (m, 4H), 3.99− 3.91 (m, 4H), 3.42−3.21(m, 8H)1.23−1.19 (m, 72H) ppm;13C NMR (75.5 MHz)δC207.1 (Cq), 193.5 (Cq), 193.2(Cq), 161.5 (Cq), 161.2 (Cq), 150.9 (Cq), 150.6 (Cq), 148.2 (Cq), 148.1, (Cq), 148.1 (Cq), 147.9 (Cq), 147.7 (Cq), 143.3 (Cq), 143.2 (Cq), 142.9 (Cq), 133.6 (Cq), 133.5 (Cq), 132.8 (Cq), 132.7 (Cq), 128.2 (Cq), 128.1 (Cq), 127.9 (Cq), 127.8 (Cq), 127.5 (Cq), 127.5 (Cq), 127.0 (CH), 126.9 (CH), 126.4 (CH), 126.3 (CH), 125.9 (CH), 125.8 (CH), 125.5 (CH), 95.3 (CH), 94.7 (CH), 78.9 (CH2), 78.7 (CH2), 78.2 (CH2), 34.1 (Cq), 33.9 (Cq), 33.9 (Cq), 33.9 (Cq), 33.0 (Cq), 32.7 (CH2), 32.6 (CH2), 32.4 (CH2), 31.5 (CH3), 31.4 (CH3), 31.2 (CH3), 30.9 (CH3), 29.7 (CH2) ppm; FAB-MS m/z 1638 (M + H+), 1637 (M+);
HRMS (FAB) calcd for C110H128N2O101636.9569, found 1636.9586.
Bispropargyl Ether Substituted 9,10-Bis-isoxazolylanthryl-methyl Linked Biscalix[4]arene, 9. A mixture of 7 (0.11 g, 0.07 mmol), sodium methoxide (0.01 g, 0.18 mmol), and propargyl bromide (0.04 mL, 0.34 mmol) in CHCl3/CH3CN (3 mL/30 mL)
was stirred and heated at reflux for 24 h. The solvent was removed under a vacuum, and the residue was purified by silica gel column chromatography with ethyl acetate/n-hexane as eluent to give 0.10 g (85.8%) of 9: mp 239−241 °C; Rf= 0.18 (ethyl acetate/n-hexane = 1:5);1H NMR (300 MHz, CDCl 3)δH8.00−7.96 (m, 4H), 7.51−7.48 (m, 4H), 7.25−6.74 (m, 22H), 5.40 (s, 4H), 4.54 (d, 4H, J = 2.3 Hz), 4.36 (d, 4H, J = 13.2 Hz), 4.31 (d, 4H, J = 13.4 Hz), 3.39 (d, 4H, J = 13.2 Hz), 3.32 (d, 4H, J = 13.4 Hz), 2.13 (t, 2H, J = 2.3 Hz), 1.30− 1.15 (m, 72H) ppm;13C NMR (75.5 MHz) δ C168.5 (Cq), 161.0 (Cq), 150.4 (Cq), 149.5 (Cq), 149.3 (Cq), 147.6 (Cq), 147.5 (Cq), 141.7 (Cq), 132.6 (Cq), 132.4 (Cq), 130.2 (Cq), 127.8 (Cq), 127.8 (Cq), 126.6 (CH), 126.1 (CH), 125.8 (CH), 125.7 (Cq), 125.6 (CH), 125.1(CH), 107.9 (CH), 78.2 (Cq), 76.2 (Cq), 68.2 (CH2), 63.3 (CH2), 37.1 (Cq), 33.9 (Cq), 33.8 (Cq), 32.1 (CH2), 31.9 (CH2), 31.7 (CH3), 30.9 (CH3) ppm; FABMS m/z 1709 (M + 2), 1708 (M + H+), 1707 (M+); HRMS calcd for C 116H128O10N21708.9569, found 1708.9546.
Doubly Bridged 9,10-Bis-isoxazolylanthryl Substituted Biscalix[4]arene, 10. A mixture of 9 (0.10 g, 0.05 mmol) and 4 (0.02 g, 0.06 mmol) in THF (15 mL) was stirred and heated at reflux for 24 h under N2system. The solvent was removed under a vacuum,
and the residue was purified by silica gel column chromatography with ethyl acetate/n-hexane as eluent to give 0.04 g (32.2%) of biscalix[4]arene 10 as a yellow solid: mp > 260°C (decomposed); Rf = 0.23 (ethyl acetate/n-hexane (v/v, 1:4));1H NMR (300 MHz, CDCl3)δH7.72−7.69 (m, 8H), 7.32−7.11 (m, 8H), 6.94 (s, 8H), 6.32 (s, 4H), 5.24 (s, 8H), 4.37 (d, 8H, J = 13.2 Hz), 3.47 (d, 8H, J = 13.2 Hz), 1.28 (s, 36H), 1.08 (s, 36H) ppm;13C NMR (75.5 MHz)δ C 168.2 (Cq), 160.6 (Cq), 150.5 (Cq), 149.8 (Cq), 147.9 (Cq), 141.9 (Cq), 132.6 (Cq), 129.8 (Cq), 127.5 (Cq), 126.4 (CH), 126.0 (CH), 125.8 (CH), 125.3 (CH), 125.2 (Cq), 107.1 (CH), 68.2 (CH2), 34.1 (Cq), 33.9 (Cq), 32.0 (CH2), 31.7 (CH3), 31.0 (CH3) ppm; FAB-MS
m/z 1970 (M + H+); HRMS (FAB) calcd for C
132H136O12N4
1969.0155, found 1969.0137.
X-ray Crystal Data for 10. C137H155Cl3N4O16; M = 2220.00; T =
150(2) K; triclinic; a = 11.9430(8) Å, b = 16.0684(10) Å, c = 16.8946(8) Å;α = 82.311(4)°, β = 81.842(5)°, γ = 78.038(5)°; V = 3121.4(3) Å3; space group P1̅; Z = 1; ρ
calcd= 1.181 mg m−3; crystal
dimensions 0.20× 0.15 × 0.10 mm3;λ = 1.54178 Å; 29213 reflections
collected; 11277 independent reflections [Rint = 0.0379]; absorption
coefficient 1.176 mm−1; 1184 parameter refined on F2; R
1= 0.1173,
wR2[F2] = 0.3066 (all data); GOF on F2= 2.333;Δρmax= 1.774 e Å−3.
CCDC-853852 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/ data_request/cif.
3,3 ′-Anthracene-9,10-diylbis{5-[4-tert-butylphenoxy)-methyl]isoxazole}, 11. A mixture of 1-tert-butyl-4-(prop-2-ynyloxy)-benzene (0.20 g, 1.07 mmol) and 4 (0.12 g, 0.48 mmol) in THF (25 mL) was stirred and heated at reflux for 24 h under a N2system. The
solvent was removed under a vacuum, and the residue was purified by silica gel column chromatography with ethyl acetate/n-hexane as eluent to give 0.16 g (52.1%) of 11 as a yellow solid: mp 231−233 °C; Rf = 0.45 (ethyl acetate/n-hexane (v/v, 1:4));1H NMR (300 MHz, CDCl3)δH7.88−7.85 (m, 4H), 7.49−7.45 (m, 4H), 7.38 (d, 4H, J = 8.7 Hz), 7.00 (d, 4H, J = 8.7 Hz), 6.64 (s, 2H), 5.37 (s, 4H), 1.31 (s, 18H) ppm;13C NMR (75.5 MHz, CDCl 3)δC168.9 (Cq), 160.8 (Cq), 155.5 (Cq), 144.7 (Cq), 130.1 (Cq), 126.5 (CH), 125.9 (CH), 125.5 (Cq), 114.4 (CH), 106.8 (CH), 61.7 (CH2), 34.1 (Cq), 31.5 (CH3)
ppm; FABMS m/z 637 (M + H+), 636 (M+); HRMS calcd for
C42H40O4N2636.2988, found 636.2994.
(3 Z,3′Z)-4,4′-Anthracene-9,10-diylbis[4-amino-1-(4-tert-butylphenoxy)but-3-en-2-one], 12a−b. A mixture of 11 (0.07 g, 0.11 mmol), Mo(CO)6 (0.07 g, 0.25 mmol) and H2O (0.2 mL) in
THF/CH3CN (1 mL/10 mL) was stirred and heated at reflux for 24 h.
The solvent was removed under a vacuum, and the residue was dissolved in 10 mL of dichloromethane. Then, to the solution was added 10 mL of NH4OH (aq) to remove remaining molybdenum
salts. After stirring for 1 h, the organic layer was washed with water and 1 M EDTA (aq). The organic phase was dried over MgSO4, and the
solvent was removed under reduced pressure. The residue was purified by neutral silica gel column chromatography with ethyl acetate/n-hexane (1/5) as eluent to give trace amount of yellow solid 12 (atropisomers): mp > 180°C (decomposed); Rf= 0.20 (ethyl acetate/
n-hexane (v/v, 1:3));1H NMR (300 MHz, CDCl 3)δH10.44 (bs, 2H), 8.11−8.07 (m, 4H), 7.61−7.46 (m, 4H), 7.28−7.25 (m, 2H), 6.91− 6.79 (m, 2H), 5.82 (s, 1H), 5.78 (s, 1H), 5.50 (bs, 1H) 5.43 (bs, 2H), 4.63, (s, 2H), 4.62 (s, 4H), 1.28−1.26 (m, 9H) ppm;13C NMR (75.5 MHz, CDCl3) δC 195.6 (Cq), 195.6(Cq), 161.0 (Cq), 160.9(Cq), 155.9 (Cq), 143.8 (Cq), 132.8 (Cq), 127.9(Cq), 126.8 (CH), 126.2 (CH), 125.6 (CH), 114.0 (CH), 95.8 (CH), 72.0 (CH2), 34.1 (Cq),
31.5 (CH3) ppm; HRMS (FAB) calcd for C42H40N2O4 636.2988,
found 636.2994.
■
ASSOCIATED CONTENT
*
S Supporting InformationCrystallographic data for compound 10 (CIF), data calculated
by molecular modeling using DMol
3for the optimized
geometry of complex 10
·G6,
1H and
13C NMR spectra for all
products 5
−12, and spectroscopic data. This material is
available free of charge via the Internet at http://pubs.acs.org.
■
AUTHOR INFORMATION
Corresponding Author
*E-mail: [email protected].
Notes
The authors declare no competing financial interest.
■
ACKNOWLEDGMENTS
We thank the National Science Council (NSC) and the MOE
ATU program of the Ministry of Education, Taiwan, the
Republic of China, for financial support.
■
REFERENCES
(1) (a) Marchand, A. P.; Chong, H.-S.; Takhi, M.; Power, T. D. Tetrahedron 2000, 56, 3121. (b) Webber, P. R. A.; Beer, P. D.; Chen, G. Z.; Felix, V.; Drew, M. G. B. J. Am. Chem. Soc. 2003, 125, 5774. (c) Chen, C.-F.; Lu, L.-G.; Hu, Z.-Q.; Peng, X.-X.; Huang, Z.-T. Tetrahedron 2005, 61, 3853.
(2) (a) Haino, T.; Yamanaka, Y.; Araki, H.; Fukazawa, Y. Chem. Commun. 2002, 402. (b) Nabeshima, T.; Saiki, T.; Sumitomo, K.; Akine, S. Tetrahedron Lett. 2004, 45, 4719.
(3) Ohseto, F.; Sakaki, T.; Araki, K.; Shinkai, S. Tetrahedron Lett. 1993, 34, 2149.
(4) (a) Wang, J.; Bodige, S. G.; Watson, W. H.; Gutsche, C. D. J. Org. Chem. 2000, 65, 8260. (b) Haino, T.; Fukunaga, C.; Fukazawa, Y. Org. Lett. 2006, 8, 3545.
(5) Tanner, C. M.; Kamel, F.; Ross, G. W.; Hoppin, J. A.; Goldman, S. M.; Korell, M.; et al. Environ. Health Perspect. 2011, 119 (6), A259 DOI: 10.1289/ehp.1002839.
(6) (a) Hwang, G. T.; Kim, B. H. Tetrahedron 2002, 58, 9019. (b) Hwang, G. T.; Kim, B. H. Tetrahedron Lett. 2000, 41, 5917. (c) Pierro, T.; Gaeta, C.; Troisi, F.; Neri, P. Tetrahedron Lett. 2009, 50, 350.
(7) (a) Su, Y.-S.; Chen, C.-F. Org. Lett. 2010, 12, 1888. (b) Xu, Z.; Jiang, L.; Feng, Y.; Zhang, S.; Liang, J.; Pan, S.; Yang, Y.; Yang, D.; Cai, Y. Org. Biomol. Chem. 2011, 9, 1237.
(8) (a) Zong, Q.-S; Chen, C.-F. Org. Lett. 2006, 8, 211. (b) Peng, X.-X.; Lu, H.-Y.; Han, T.; Chen, C.-F. Org. Lett. 2007, 9, 895. (c) Zhao, J.-M.; Zong, Q.-S.; Han, T; Xiang, J.-F.; Chen, C.-F. J. Org. Chem. 2008, 73, 6800. (d) Jiang, Y.; Cao, J.; Zhao, J.-M.; Xiang, J.-F.; Chen, C.-F. J. Org. Chem. 2010, 75, 1767. (e) Hu, S.-Z.; Chen, C.-F. Chem.Eur. J. 2011, 17, 5423.
(9) (a) Li, C.; Xu, Q.; Li, J.; Yaoa, F.; Jia, X. Org. Biomol. Chem. 2010, 8, 1568. (b) Ogoshi, T.; Hashizume, M.; Yamagishi., T.-A.; Nakamoto, Y. Chem. Commun. 2010, 46, 3708.
(10) Lagona, J.; Wagner, B. D.; Isaacs, L. J. Org. Chem. 2006, 71, 1181.
(11) (a) Hwang, G. T.; Kim, B. H. Tetrahedron Lett. 2000, 41, 10055. (b) Morales-Sanfrutos, J.; Ortega-Muñoz, M.; Lopez-Jaramillo, J.; Hernandez-Mateo, F.; Santoyo-Gonzalez, F. J. Org. Chem. 2008, 73, 7768.
(12) (a) Shu, C.-M.; Lee, G.-H.; Peng, S.-M.; Chung, W.-S. J. Chin. Chem. Soc. 2000, 47, 173. (b) Shiao, Y.-J.; Chiang, P.-C.; Senthilvelan, A.; Tsai, M.-T.; Lee, G.-H.; Chung, W.-S. Tetrahedron Lett. 2006, 47, 8383.
(13) (a) Nitta, M.; Kobayashi, T. J. Chem. Soc., Perkin Trans. 1985, 1, 1401. (b) Tranmer, G. K.; Tam, W. Org. Lett. 2002, 4, 4101. (c) Kociolek, M. G.; Straub, N. G.; Marton, E. J. Lett. Org. Chem. 2005, 2, 280.
(14) (a) Senthilvelan, A.; Lee, G.-H.; Chung, W.-S. Tetrahedron Lett. 2006, 47, 7179. (b) Senthilvelan, A.; Ho, I.-T.; Chang, K.-C.; Lee, G.-H.; Liu, Y.-G.-H.; Chung, W.-S. Chem.Eur. J. 2009, 15, 6152. (c) Ho, T.; Chu, J.-H.; Chung, W.-S. Eur. J. Org. Chem. 2011, 1472. (d) Ho, I.-T.; Huang, K.-C.; Chung, W.-S. Chem.Asian J. 2011, 6, 2738.
(15) (a) Port, A.; Moragas, M.; Sánchez-Ruiz, X.; Jaime, C.; Virgili, A.; Alvarez-Larena, A.; Piniella, J. F. J. Org. Chem. 1997, 62, 899. (b) Irngartinger, H.; Weber, A.; Escher, T.; Fettel, P. W.; Gassner, F. Eur. J. Org. Chem. 1999, 2087. (c) Lunazzi, L.; Mazzanti, A.; Minzoni, M.; Anderson, J. E. Org. Lett. 2005, 7, 1291. (d) Schwab, G.; Stern, D.; Stalke, D. J. Org. Chem. 2008, 73, 5242. (e) Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; Muldoon, J.; McGlinchey, M. J. Org. Lett. 2011, 13, 256. (f) Friebolin, H. Basic One- and Two-Dimensional NMR Spectroscopy; VCH: Weinheim, Germany, 2005; p 313. (g) ΔG≠was estimated to be 23 kcal mol−1 on the basis of variable temperature NMR experiments (see Figures S4 and S5, Supporting Information). The activation free energies were obtained using equations from ref 15f. kc
= 2.22Δν, ΔG≠= 4.58 Tc(10.32 + log (Tc/kc)) 10−3kcal/mol.
(16) Normal phase HPLC was performed with a Gilson 321-H1 pump system with a 506C interface, a Rheodyne 7725I injector, a Gilson 155 UV−vis detector (Gilson, Inc., Middleton, WI). For silica column (Hypersil, 4.6× 250 mm, 5 μm), the mobile phase was eluted from hexane to hexane/EA (v/v, 2:3) over 30 min at a flow rate of 1 mL/min. For CHIRALPAK AS-H and CHIRACEL OD-H columns (both are 4.6 × 250 mm, 5 μm, Daicel Chemical Industrial, Ltd., Tokyo), the mobile phase of 2-propanol/hexane (v/v, 1:9) was eluted isocratically at a flow rate of 0.5 mL/min.
(17) (a) Nowak, W.; Wierzbowska, M. THEOCHEM 1996, 368, 223. (b) Nori-shargha, D.; Asadzadeha, S.; Ghanizadehb, F.-R.; Deyhimic, F.; Aminic, M. M.; Jameh-Bozorghi, S. THEOCHEM 2005, 717, 41. (c) Nikitin, K.; Fleming, C.; Müller-Bunz, H.; Ortin, Y.; McGlinchey, M. J. Eur. J. Org. Chem. 2010, 5203.
(18) The rotation barrier was calculated on the basis of the variable temperature NMR experiment using the equation in ref 15g.
(19) The relative fluorescence quantum yields were determined by comparison of the integrated area of the emission spectra of the samples with the reference compound 9,10-diphenylanthracene (ΦF=
0.90± 0.02 in cyclohexane). For the guest-free studies, biscalixarenes 7, 10 and control compound 11 were at 10 μM concentration in MeOH/CHCl3(v/v, 1:2). For the complexation studies, 200 equiv of
G6(methyl viologen) was added to 10μM solution of biscalixarenes 7, 10 or control compound 11 in MeOH/CHCl3(v/v, 1:2). Emission
excitation at 388 nm for 7 and 384 for 11 and from 395 to 650 nm for 10with excitation at 391 nm. The relative fluorescent quantum yield for 7, 10, and 11 are 0.70 ± 0.02, 0.80 ± 0.02, and 0.96 ± 0.02, respectively. The relative quantum yields were calculated using equation:ΦF= (Aref/A) × (F/Fref) × (n2chloroform/n2cyclohxane) × Φref,
where A is the absorbance at the excitation wavelength, F is the integrated emission area, and n is the refractive index of the solvent (nchloroform = 1.4459, ncyclohexane = 1.4262 at 25 °C). See related
references: (a) Eaton, D. F. Pure Appl. Chem. 1988, 60, 1107. (b) Dawson, W. R.; Windsor, M. W. J. Phys. Chem. 1968, 72, 3251.
(20) The association constant was calculated by using a Stern− Volmer plot; see: Valeur, B. Molecular Fluorescence: Principles and Applications; Wiley-VCH: Weinheim, Germany, 2001; p 77.
(21) We thank one of the reviewers for this suggestion. (a) Morris, K. F.; Johnson, C. S. J. Am. Chem. Soc. 1992, 114, 3139. (b) Keresztes, I.; WIlliard, P. G. J. Am. Chem. Soc. 2000, 122, 10228. (c) Balayssac, S.; Gilard, V.; Delsuc, M.-A.; Malet-Martino, M. Spectrosc. Eur. 2009, 21, 9.
(22) (a) Delley, B. J. Chem. Phys. 1990, 92, 508. (b) Delley, B. J. Chem. Phys. 2000, 113, 7756.
(23) (a) Benedek, N. A.; Snook, I. K.; Latham, K.; Yarovsky, I. J. Chem. Phys. 2005, 122, 144102. (b) Kusama, H.; Orita, H.; Sugihara, H. Langmuir 2008, 24, 4411. (c) Inada, Y.; Orita, H. J. Comput. Chem. 2008, 29, 225.
![Figure 1. (a) X-ray single crystal structure of biscalix[4]arene 10 and (b) a snapshot of the structure in (a) by 90° rotation in its horizontal-axis.](https://thumb-ap.123doks.com/thumbv2/9libinfo/7939348.157483/3.938.92.847.122.761/figure-crystal-structure-biscalix-snapshot-structure-rotation-horizontal.webp)

![Figure 6. A possible binding mode of the complexation of biscalix[4]arene 10 with G6 (a) side view and (b) top view.](https://thumb-ap.123doks.com/thumbv2/9libinfo/7939348.157483/6.938.103.845.381.573/figure-possible-binding-mode-complexation-biscalix-arene-view.webp)
