國
立
交
通
大
學
生物資訊及系統生物研究所
博
士
論
文
同藥理同源途徑蛋白質對多標靶抑制劑之研發
Pharmapathlogs for Multitarget Inhibitors
研 究 生:許凱程
指導教授:楊進木 教授
同藥理同源途徑蛋白質對多標靶抑制劑之研發
Pharmapathlogs for Multitarget Inhibitors
研 究 生:許凱程 Student:Kai-Cheng Hsu
指導教授:楊進木 Advisor:Jinn-Moon Yang
國 立 交 通 大 學
生物資訊及系統生物研究所
博 士 論 文
A Thesis Submitted to Institute of Bioinformatics and Systems Biology National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Ph.D. in Bioinformatics and Systems Biology
August 2011
Hsinchu, Taiwan, Republic of China
同藥理同源途徑蛋白質對多標靶抑制劑之研發
研究生:許凱程 指導教授:楊進木博士 國立交通大學 生物資訊及系統生物研究所 博士班摘 要
「單一藥物針對單一標靶,治療單一疾病」是過去數十年藥物發展的主要概念,這個概 念促使研究者發展具有高專一性的藥物,然而這個概念逐漸變得不適用於治療疾病上, 主要原因之一是單一標靶藥物容易受到蛋白質結合位上的突變之影響,而導致抗藥性的 產生。因此,開發一個能尋找多標靶藥物之新策略,藉由抑制多個標靶來降低抗藥性之 產生並且增加療效,將對藥物開發提供重要的價值。 針對此議題,我們提出一個新的概念「同藥理同源途徑蛋白質(pharmapathlog)」來 尋找多標靶藥物。同藥理同源途徑蛋白質是一群具有下述特性的蛋白質:這些蛋白質 (1)是同反應途徑上的同源蛋白質;(2)具有相似的核心結合環境;(3)能夠被相 同的化合物所抑制。在生物體反應途徑上,因為一個蛋白質的產物是其下游蛋白質的受 質,使得這些蛋白質在結合位上具有相似的物理化學特性及構型。此外,同源蛋白質常 在結合位上具有高度保留性的區域,針對這些區域設計的藥物能降低產生抗藥性之機率。 根 據 此 概 念 , 我 們 發 展 新 的 策 略 「 以 同 藥 理 同 源 途 徑 蛋 白 質 為 基 礎 之 藥 物 篩 選 (pharmapathlog-based screening strategy)」用以尋找多物種間之同藥理同源途徑蛋白質以 及其核心結合環境,當化合物能同時符合此核心結合環境時將有潛力成為這些蛋白質之 多標靶藥物,可增加療效並且降低產生抗藥性的機率。 我們應用此策略於尋找細菌及病毒之抑制劑,包括結核桿菌、幽門螺旋桿菌和流行 性感冒病毒。在抗菌研究上,成功找出三個多標靶抑制劑(IC50 <10.0 μM)能同時抑制 幽門螺旋桿菌中的莽草酸激酶和莽草酸去氫酶,此三個抑制劑也能抑制結核桿菌中的莽 草酸激酶(IC50 <10.0 μM)。此外,我們發現三個能抑制流行性感冒病毒 H1N1 與 H5N1 神經胺酸酶之新型抑制劑(IC50 4~20 μM),實驗結果顯示此三個抑制劑能有效 抑制抗藥性之神經胺酸酶(H274Y 和 I222R),並且不引起細胞毒性,提供對抗抗藥 性病株的一個良好起始點。我們也設計了五個瑞樂沙衍生物,這些衍生物位於 150 cavity 中且有良好的抑制效果(IC50 <10.0 nM)。另外,統計結果顯示同藥理同源途徑 蛋白質中的核心結合環境為高度保留性之區域,可以用來設計不易產生抗藥性之藥物。 這些實驗結果顯示同藥理同源途徑蛋白質之藥物篩選的概念將有助於尋找多標靶抑制劑, 我們相信此策略對於藥物研究開發具有極大價值。Pharmapathlogs for Multitarget Inhibitors
Student : Kai-Cheng Hsu Adviser : Dr. Jinn-Moon Yang Institute of Bioinformatics and Systems Biology
National Chiao Tung University
ABSTRACT
The concept of "one-drug, one-target, one-disease" has been dominant to drug development strategy in the past decades. This strategy induces researchers to develop inhibitors with high specificity. However, the strategy is increasingly becoming inappropriate. One of the major reasons is that single-target inhibitors often lose potency because of even one residue mutation, leading to drug resistance. Therefore, developing a new strategy to discover multitarget inhibitors, which decrease probability of drug resistances and enhance therapeutic potency by inhibiting multiple targets, provides a great value for drug design.
To address the issue, we proposed a new concept "pharmapathlog" to discover multitarget inhibitors. Pharmapathlog are a group of proteins that satisfy the following properties: (1) they are protein orthologs in the same pathway; (2) they share comparable core binding environments; (3) they can be inhibited by the same compounds. Proteins in the same pathway may share similarities in physical-chemical properties and shapes in their binding sites because a product of one enzyme is a substrate of the next enzyme in a series of catalytic reactions. Furthermore, orthologous proteins often share conserved core binding environments during evolution, providing an opportunity to develop multitarget inhibitors to target these conserved regions for reducing the probability of drug resistance. Based on the new concept, we developed a "pharmapathlog-based screening strategy" to identify pharmapathlogs in the same pathways across multiple species and their core binding environments by using site-moiety maps. A compound highly agreeing with the core binding environments of pharmapathlogs could simultaneously inhibit the multiple proteins of pharmapathlogs.
To verify the utility of the pharmapathlog-based screening strategy, we applied this strategy to identify new inhibitors for bacteria and virus, including Helicobacter pylori,
Mycobacterium tuberculosis, and influenza virus. Based on the strategy, three multitarget
inhibitors simultaneously inhibiting shikimate dehydrogenase and shikimate kinase of
Helicobacter pylori with low IC50 values (<10.0 μM) were discovered. The three inhibitors
also showed inhibitory effects (IC50 <10 μM) for shikimate kinase of Mycobacterium
tuberculosis. Subsequently, the strategy was successfully used to discover three new inhibitors with low IC50 values (4~20 μM) for H1N1 and H5N1 neuraminidases, and design five
zanamivir derivatives located at the 150-cavity with IC50 values in the <10 nanomolar range.
Our experimental results showed that the three inhibitors may overcome the drug resistances introduced by H274Y and I222R for H1N1 neuraminidase without causing apparent
cytotoxicity, suggesting a starting point to combat drug-resistant strains. In addition, we found that core binding environments of pharmapathlogs are highly conserved, suggesting that targeting the core binding environments is useful to avoid drug-resistance. These experimental results show that the concept of pharmapathlogs is useful to discover multitarget inhibitors. We believe that the new strategy is useful to design new drugs toward human diseases.
Acknowledgement
經由很多人的努力及幫助使我能夠完成這份論文。首先我必須感謝我的指導教授楊 進木老師,他對研究的熱情感染了我,使我能在學習的路程積極向上,當我遇到瓶頸或 對問題絕望時他總是會說:『來討論一下。』,討論完之後總是會豁然開朗。除了在研 究專業上的教導與訓練之外,楊老師沒有什麼事情是解決不了的態度更令人值得學習, 這也激勵我能走得更遠且不輕言放棄。還要感謝他所提供的學習環境、研究資源以及出 國的機會,使得我在求學時能夠更加順利,也要感謝他經常提供對外合作的機會,增廣 我的見聞。 接著我要感謝我的口試委員,包含所內我的指導教授楊進木教授、黃鎮剛教授,所 外的王雯靜教授、徐祖安教授、郭盛助教授、吳永昌教授、鄭添祿教授。感謝每位教授 在百忙之中抽空擔當我的口試委員並且評鑑我的論文,以及在口試期間的對我研究所提 供的寶貴建議,有了他們的指教與建言才使得這本論文能更臻完美。 我也感謝實驗室的全體同仁與我走過這一段路。特別感謝與我同組的彥甫、伸融、 御哲、志達、淩婷、彥超、宣人,常常與我一起討論與工作到半夜,在新竹與你們吃飯 的時候總是非常開心的。感謝章維在研究與生活上給我的幫助,也感謝俊辰、其樺、一 原、宇書、峻宇、怡馨在研究上給的建議。感謝士中平日在生活上及行政上的幫助,讓 我能輕鬆很多。因為實驗室的同仁使得我的求學過程更豐富更多采多姿。 再來我要感謝合作的夥伴給予的實驗資料以及研究建議。感謝王雯靜教授與鄭文琪 博士在研究幽門螺旋桿菌之抑制劑上所提供的實驗資料,常常看到鄭文琪博士來生科實 驗館做一整天的實驗。感謝徐祖安教授、洪慧貞博士及方明瑜小姐在研究流行性感冒病 毒之抑制劑上所提供的協助,每次都必須麻煩他們花很多時間做實驗。感謝林俊成教授 與林建宏先生在合成抗流感病毒之抑制劑所提供的資料。因為這些人的努力使得我能夠 驗證我電腦預測及分析的結果,使得論文能更加完整。 最後我想特別感謝我父母與睿瑜。我的父母從小就提供了良好的環境讓我無後顧之 憂地學習,總是無怨無悔地照顧我,即使在成績不理想時也不曾責罵過我,溫暖的家是 我堅持下去的力量。睿瑜總是與我分享生活中的每一件事,我們一起度過快樂與悲傷, 甚至在研究上也能互相幫忙,在每個星期的努力後,與她在一起能讓我充完電,充滿精 力面對挑戰,希望我們未來能一直這樣走下去。僅將此論文獻給我這些敬愛的人以及幫 助我的人。Contents
摘 要 ... i
ABSTRACT ... ii
Acknowledgement ... iv
Contents... v
List of figures ... vii
List of tables ... ix
Chapter 1. Introduction ... 1
1.1 Background ... 1
1.2 Current state of computational drug design ... 5
1.3 Thesis overview... 7
Chapter 2. Site-moiety map to recognize interaction preferences between protein pockets and compound moieties ... 10
2.1 Introduction ... 11
2.2 Methods and Materials ... 12
2.2.1 Site-moiety map, anchor and pocket ... 15
2.2.2 Data sets ... 15
2.2.3 Main procedure of constructing a site-moiety map ... 16
2.2.4 Input and output of the SiMMap server ... 18
2.3 Results and Discussion ... 20
2.3.1 Thymidine kinase ... 20
2.3.2 Estrogen receptor... 22
2.3.3 Discussion ... 22
2.4 Conclusions ... 30
Chapter 3. Pharmapathlogs for discovering multitarget inhibitors of shikimate pathways using site-moiety maps ... 31
3.1 Introduction ... 32
3.2 Methods and Materials ... 33
3.2.1 Overview of pharmapathlog-based screening strategy ... 33
3.2.2 Preparations of protein structures and screening databases ... 37
3.2.3 Computational screening and establishment of site-moiety maps ... 39
3.2.4 Identification of core anchors, pharmapathlogs, and multitarget inhibitors 42 3.3 Results and Discussion ... 44
3.3.1 Site-moiety map of shikimate dehydrogenase ... 44
3.3.2 Site-moiety maps of shikimate kinases ... 48
3.3.4 New multitarget inhibitors ... 56
3.3.5 Specific site and inhibitors for shikimate dehydrogenase ... 58
3.4 Conclusions ... 59
3.5 Acknowledgments ... 60
Chapter 4. Pharmapathlogs for optimizing and identifying neuraminidase inhibitors 61 4.1 Introduction ... 62
4.2 Methods and Materials ... 64
4.2.1 Overview of pharmapathlog-based screening strategy ... 64
4.2.2 Dataset preparation ... 66
4.2.3 Main procedure for identifying new type inhibitors and lead optimization 69 4.3 Results and Discussion ... 73
4.3.1 Site-moiety maps of H1N1, H5N1, and H3N2 NAs ... 73
4.3.2 Optimization process of zanamivir derivatives ... 79
4.3.3 Identified novel inhibitors ... 86
4.3.4 Proposed binding mechanism of novel inhibitors ... 88
4.3.5 Advantages of pharmapathlog-based screening strategy ... 93
4.4 Conclusions ... 94 4.5 Acknowledgments ... 95 Chapter 5. Conclusions ... 96 5.1 Summary ... 96 5.2 Major contributions ... 97 5.3 Future works ... 98 List of publications ... 101 References ... 102
List of figures
Figure 1.1. Concept of pharmapathlogs using protein orthologs in the shikimate pathways as
the example... 3
Figure 2.1. Overview of the SiMMap server for the site-moiety map using herpes simplex virus type-1 thymidine kinase and 1000 docked compounds as the query.. ... 14
Figure 2.2. The SiMMap server analysis results using estrogen receptor and 1000 docked compounds as the query. ... 19
Figure 2.3. The relationships between the site-moiety map and 15 co-crystallized ligands of TK ... 24
Figure 2.4. The relationships between the site-moiety map and 22 co-crystallized ligands of ER. ... 25
Figure 2.5. Comparison of SiMMap with GEMDOCK. ... 26
Figure 3.1. Overview of pharmapathlog-based screening strategy for identifying multitarget inhibitors. ... 35
Figure 3.2. Discovery of multitarget inhibitors for inhibiting multiple proteins in a pathway. . 36
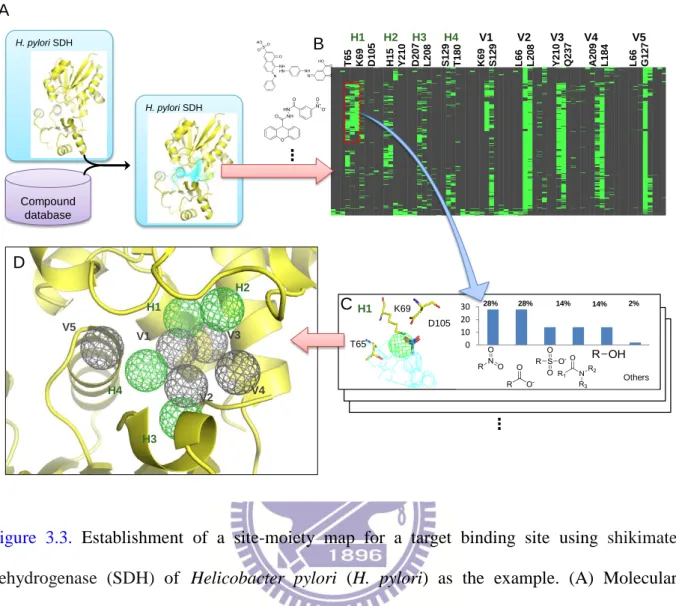
Figure 3.3. Establishment of a site-moiety map for a target binding site using shikimate dehydrogenase of Helicobacter pylori as the example. ... 41
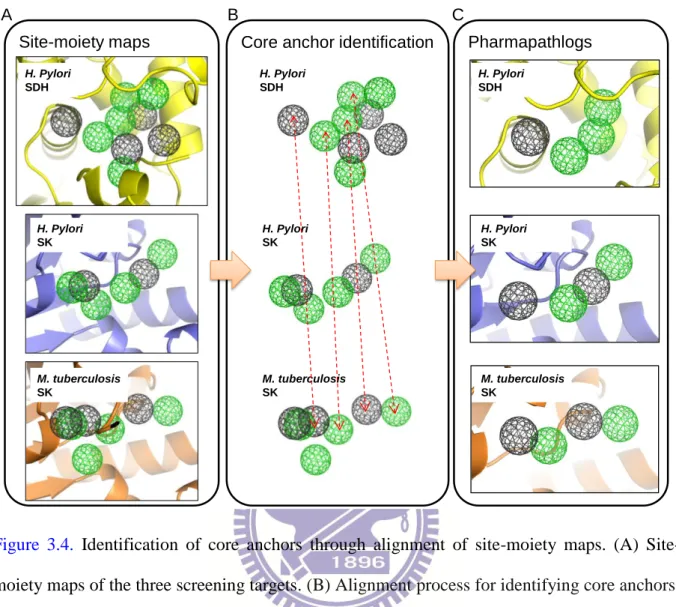
Figure 3.4. Identification of core anchors through alignment of site-moiety maps ... 44
Figure 3.5. Site-moiety map of shikimate dehydrogenase. ... 47
Figure 3.6. Importance of anchors in biological functions ... 48
Figure 3.7. Site-moiety maps of shikimate kinases of Helicobacter pylori and Mycobacterium tuberculosis ... 50
Figure 3.8. Importance of anchors in biological functions using shikimate kinase of Helicobacter pylori as the example ... 51
Figure 3.9. Core anchors of HpSDH, HpSK, and MtSK. ... 53
Figure 3.10. The division process for deciding core anchor residues, anchor residues, binding site residues, and other residues. ... 55
Figure 3.11. Conserved score distribution of core anchor residues, anchor residues, binding site residues, and other residues ... 55
Figure 3.12. New multitarget inhibitors identified by the pharmapathlog-based screening strategy ... 57
Figure 3.13. Specific site and inhibitors for shikimate dehydrogenase. ... 59
Figure 4.1. Overview of the pharmapathlog-based screening strategy for optimizing lead compounds and identifying novel inhibitors ... 65
Figure 4.2. Main steps for constructing a site-moiety map ... 66
Figure 4.4. Modeled structure of H3N2 NA with a 150-open form. ... 69
Figure 4.5. Site-moiety maps of NA pharmapathlogs ... 73
Figure 4.6. Relationship between core anchors and moieties of known inhibitors. ... 76
Figure 4.7. Conserved hydrogen-bonding interactions between water atoms and pockets of the H2 and H3 core anchors ... 78
Figure 4.8. Core anchors of NA pharmapathlogs for lead optimization processes of zanamivir, oseltamivir, and peramivir ... 80
Figure 4.9. Zanamivir, oseltamivir, and peramivir analogues for verifying core anchors of pharmapathlogs on lead optimization processes ... 81
Figure 4.10. Pearson’s correlation coefficient between moiety energies and IC50 values of compounds... 82
Figure 4.11. Lead optimization process of zanamivir derivatives for extending to the 150-cavity. ... 84
Figure 4.12. Discovered new type inhibitors ... 86
Figure 4.13. Effects of influenza virus in cytopathic effect inhibition assays of novel inhibitors. ... 88
Figure 4.14. Multiple sequence alignment of NAs. ... 89
Figure 4.15. Proposed new mechanism of H3N2 NA ... 91
Figure 4.16. Binding mode comparison of new type inhibitors. ... 92
List of tables
Table 2.1. The relationship between the anchors and moieties of 15 co-crystallized ligands for
TK ... 21
Table 2.2. Comparing SiMMap with other methods on thymidine kinase and estrogen receptor by false-positive rates ... 27
Table 2.3. The mapping between the anchors and active and typical compounds for TK ... 28
Table 2.4. The mapping between the anchors and active and typical compounds for ER ... 29
Chapter 1. Introduction
1.1 Background
The concept of "one-disease, one-target, one-drug" has been the dominating drug development strategy in the past decades 1,2. This strategy induces researchers to develop inhibitors with high specificity. For example, in anti-influenza drug development, neuraminidase is considered a valid target, and two drugs, zanamivir and oseltamivir, have been reported3-5. However, these single-target inhibitors may easily lose their effectiveness due to even one amino acid mutation in binding sites of target proteins, leading to drug resistance. For instance, some influenza strains that are resistant to oseltamivir have been reported since a single residue mutates 6. Another example is tetracycline, which is a broad spectrum antibiotic. Tetracycline loses its potency in Helicobacter pylori (H. pylori) because of a single triple-base-pair substitution7. Particularly in antibiotics, the increasing emergence of multiple-antibiotic-resistant superbugs causes a great concern in the world8-10, revealing the insufficiency of the single-target strategy. Therefore, developing a new strategy to discover multitarget inhibitors, which decrease probability of drug resistances by inhibiting multiple targets, provides a great value for drug design.
Proteins may share many similarities in physical-chemical properties and shapes in their binding sites despite low sequence or structural homology. For example, proteins in the same pathways contain comparable core binding environments because a product of one enzyme is a substrate of the next enzyme in a series of catalytic reactions. Hence, it is possible to design a multitarget inhibitor to simultaneously inhibit proteins in the same pathways by targeting their core binding environments. Furthermore, orthologous proteins often share conserved core binding environments during evolution, providing an opportunity to develop inhibitors to target
these conserved regions for reducing the probability of drug resistance and increasing hit rate. Recently, the concept of polypharmacology, which means that a drug binds multiple target proteins, has been proposed to design drugs11-13. In general, proteins with high sequence or structure similarity could be considered to be bound by the same compounds. However, designing these multitarget inhibitors is still a challenging task since these proteins often lack structural and sequence homology14,15, resulting in a difficulty for extracting core binding environments among these proteins. Therefore, a new strategy for extracting core binding environments without relying on sequences of structures will be useful for discovering multitarget inhibitors.
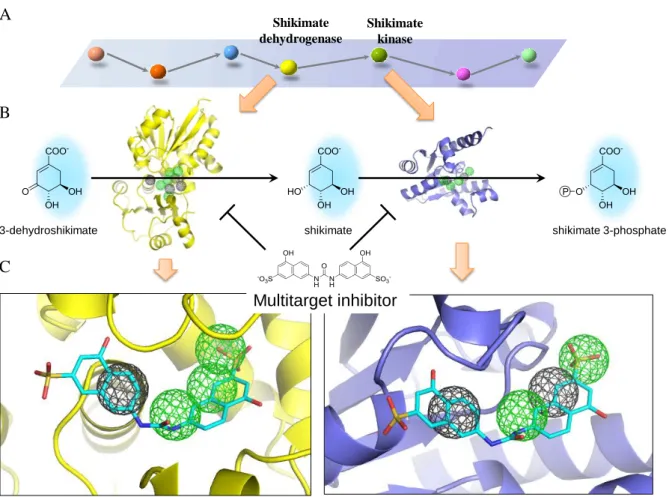
To address these issues, we propose a new strategy, called pharmapathlog-based screening strategy, to discover multitarget inhibitors (Fig. 1.1). Pharmapathlogs are a group of proteins that satisfy the following properties: (1) they are protein orthologs in the same pathway; (2) they share comparable core binding environments; (3) they can be inhibited by the same compounds. To extract core binding environments of protein binding sites, we developed the SiMMap server for generation of site-moiety maps16. A site-moiety map consists of anchors for a protein binding site. An anchor, presenting a key binding environment, includes three essential elements: (1) binding pockets, which are parts of the binding site, with conserved interacting residues; (2) moiety preferences; (3) interaction type (electrostatic, hydrogen-bonding, or van der Waals). A site-moiety map is able to present the relationship between the moiety preferences and the physico-chemical properties of the binding site through anchors. Hence, protein orthologs sharing comparable core anchors (core binding environments) in the same pathway could be considered pharmapathlogs, and the core anchors of pharmapathlogs can be used to identify multitarget inhibitors. A compound that agrees with the core anchors is often able to simultaneously inhibit the multiple targets. In addition, the moiety preferences of the core anchors can guide lead optimization processes.
Figure 1.1. Concept of pharmapathlogs using protein orthologs in the shikimate pathways as the example. Orthologous proteins sharing similar binding environments in the same pathways can be considered pharmapathlogs. For example, shikimate dehydrogenase (SDH) and shikimate kinase (SK) are adjacent proteins in the shikimate pathway. The two proteins share two key hydrogen binding environments (green sphere) and two van der Waals binding environments (grey sphere), and can be regarded as pharmapathlogs in the pathway. In addition, orthologous SKs in Helicobacter pylori (H. pylori) and Mycobacterium tuberculosis (M.
tuberculosis) are regarded as pharmapathlogs in multiple species because of their comparable
binding environments. The consensus binding environments among pharmapathlogs are core binding environments and can be used to find multitarget inhibitors agreeing with the core regions. Multitarget inhibitors have good therapeutic effectiveness and reduce probability of resistant mutations, whereas single target inhibitors often lose effectiveness when target residues mutate. Single target inhibitor SK SDH Helicobacter pylori Mycobacterium tuberculosis Other species Shikimate pathway Multitarget Inhibitor Drug
resistance Pharmapathlogs in the same pathway
… SDH SK Pharmapathlogs in multiple species H. Pylori M. tuberculosis … Pharmapathlogs
To verify the utility of the pharmapathlog-based screening strategy, at first, we applied this strategy to identify new multitarget inhibitors for shikimate pathway of H. pylori and
Mycobacterium tuberculosis (M. tuberculosis), which are human pathogens and causes peptic
ulcer disease and chronic infectious disease, respectively17-19. The shikimate pathway containing seven proteins is an attractive target pathway for drug development because the pathway is absent in human 20. By use of this strategy, we successfully discovered three multitarget inhibitors with low IC50 values (<10.0 μM) for simultaneously inhibiting shikimate
dehydrogenase and shikimate kinase by collaborating with Dr. Ching Wang and Dr. Wen-Chi Cheng of National Tsing Hua University (NTHU). Subsequently, we applied the strategy to discover three new inhibitors with low IC50 values (4~20 μM) for H1N1 and H5N1
neuraminidases, and design five zanamivir derivatives with IC50 values in the <10 nanomolar
range by collaborating with Dr. John T.A. Hsu and Dr. Hui-Chen Hung of National Health Research Institutes, and Dr. Chun-Cheng Lin and Mr. Chien-Hung Lin of NTHU. Our experimental results showed that the three inhibitors may overcome the drug resistances introduced by H274Y and I222R for H1N1 neuraminidase without causing apparent cytotoxicity, suggesting a starting point to combat drug-resistant strains. The experimental results showed that the concept of pharmapathlogs is useful to discover multitarget inhibitors.
We believe that the new strategy is useful to discover and optimize new lines of inhibitors toward human diseases.
The pharmapathlog-based screening is a general strategy for drug development, and can be extend to other human diseases and drug-resistant pathogens. A study showed that developing a drug costs 15 years and US$800 million on average21. The high cost and lengthy development time reveals the insufficiency of the traditional strategy in developing drugs for combating rapidly emerging diseases, such as malaria, tuberculosis, cholera, and avian flu. Once drug-resistant pathogens emerge, current drug treatments may be ineffective. As a result,
the pharmapathlog-based screening, which is different to currently used single-target approaches, has great potential because of the following advantages: 1) High success rate. The new strategy simultaneously considers multiple target proteins for discovering inhibitors, providing an additive opportunity to discover true hits against diseases. 2) Reduction of drug resistance. The probability of drug resistant mutations arising in all targets is extremely low. 3) High treatment efficiency. Multitarget inhibitors inhibit multiple targets; therefore, these drugs increase the efficiency of therapy and are useful to treat complexity of diseases. 4) Reduction of cost and time. Based on above reasons, we believe our research results are helpful for the drug development process.
1.2 Current state of computational drug design
Virtual screening is an efficient and promising strategy in drug discovery2,22,23. In virtual screening, thousands of compounds are ranked according to their binding affinities predicted by scoring functions, and top-ranked compounds are then tested by experiments. There are three general classes of scoring functions, including force-field-based methods24,25, empirical methods26,27, and knowledge-based methods28,29. Force-field-based scoring functions are derived from molecular mechanics force-fields such as van der Waals potentials and Columbic interactions. Empirical scoring functions measure binding affinities by summing up terms that describe physical contributions, such as hydrogen bonding, van der Waals forces, and hydrophobic contacts. Generally, empirical scoring functions have simplified energy descriptors based on physical properties and then reduce computational cost in virtual screening. Knowledge-based scoring functions are derived from energy-like functions by considering the distributions of interatomic distances in a set of crystal structures of protein– ligand complexes. Furthermore, regression techniques are often applied to scoring functions, and coefficients of descriptors are derived from a set of protein-ligand complexes with
experimental binding affinities. As a result, these scoring functions (e.g., X-SCORE30, ChemScore27, SCORE30, DrugScore29, and PLD31) usually perform well in the prediction of binding affinity.
As the number of protein structures increases rapidly, virtual screening approaches is becoming important and helpful in lead discovery2,22,23,32. Based on the various scoring functions, many popular programs (e.g., GEMDOCK 33, DOCK 34, AutoDock 35, and GOLD25) were designed for virtual screening, and were successfully applied to identify lead compounds for target proteins. However, hit rates of these programs remained intensive because of the incomplete understandings of ligand binding mechanisms in protein-ligand interactions2,22,23. For example, most of the scoring functions often lack consideration for key binding environments, such as pharmacophore spots, metal ions, and conserved residues. Pharmacophore spots are the spatial arrangement of compound moieties that are responsible for biological activity36. Metal-ligand interactions stabilize ligands to facilitate catalysis37. Conserved residues interacting with bound compounds often play important roles for biological functions. For instance, catalytic residues, which polarize substrates and thereby stabilize transition states38, are evolutionarily conserved. These key binding environments are essential for ligand binding and biological functions but ignored in most computational methods. In addition, most of these docking programs25,34,39 use energy-based scoring methods, which are often biased toward both the selection of high molecular weight compounds and charged polar compounds40,41.
Recently, some approaches have been proposed to derive key binding environments of protein-ligand interfaces (e.g., pharmacophore spots) from known compounds40,42,43. These approaches apparently increase the chance to identify active compounds. However, they are often unable to be applied for new targets, which have no known active compounds. Currently, some approaches used the scoring functions to discover multitarget inhibitors by virtual
screening 13,44. Wei et al. discovered multitarget inhibitors for the human leukotriene A4 hydrolase and the human nonpancreatic secretory phospholipase A2 by a pharmacophore-based method45,46. These approaches also contained the disadvantages as we mentioned in the previous part, leading low prediction accuracy or limiting discovery of new target inhibitors. Therefore, the more powerful techniques for identifying these key binding environments and multitarget inhibitors provide a great potential value for drug design.
In this thesis, we presented the SiMMap server to infer the key binding environments by a site-moiety map in protein-ligand interfaces. The server provides pocket-moiety interaction preferences (anchors) including binding pockets with conserved interacting residues, moiety preferences, and interaction type. We verified the site-moiety map on three therapeutic targets, including thymidine kinase, and estrogen receptors of antagonists and agonists. Experimental results showed that an anchor is often a hot spot and the site-moiety map is useful to identify active compounds for these targets. In addition, we applied site-moiety maps to extract core binding environments (core anchors) of pharmapathlogs for shikimate pathway and neuraminidases of influenza A, and several inhibitors were identified. We believe that site-moiety maps are able to provide biological insights and are useful for drug discovery and lead optimization.
1.3 Thesis overview
The thesis is organized as follows. In Chapter 2, to identify pharmapathlogs, which are ortholog proteins sharing comparable binding environments in the same pathways, we developed the SiMMap server to infer key binding environments of binding sites via site-moiety maps. A site-site-moiety map describes the relationship between the site-moiety preferences and the physico-chemical properties of the binding site through anchors. An anchor includes three
essential elements including binding pockets with conserved interacting residues; moiety preferences; and interaction type. We provided initial validation of the site-moiety map on three targets, thymidine kinase, and estrogen receptors of antagonists and agonists. Experimental results show that an anchor is often a hot spot and the site-moiety map can help to assemble potential leads by optimal steric, hydrogen-bonding, and electronic moieties. When a compound highly agrees with anchors of site-moiety map, this compound often activates or inhibits the target protein.
In Chapter 3, we presented the pharmapathlog-based screening strategy by combing site-moiety maps. The concept of strategy is to simultaneously screen multiple protein orthologs in the same pathways cross pathogens, and extract conserved binding environments (core anchors) of these proteins for discovering multitarget inhibitors. Proteins sharing similar binding environments (core anchors) are considered pharmapathlogs, and could be inhibited by the same inhibitors. In this study, we applied site-moiety maps to describe the core binding environments, called core anchors, which present conserved binding pockets with specific physico-chemical properties, similar moieties of ligands, and consensus interaction types, all of which are essential to perform biological functions during species evolution. Hence, the proteins with comparable core anchors can be regarded as pharmapathlogs, and the core anchors of pharmapathlogs can be used to identify multitarget inhibitors. Then this strategy was applied to identify multitarget inhibitors of shikimate pathways for M. tuberculosis and H.
pylori, which are human pathogens and causes peptic ulcer disease and chronic infectious
disease, respectively17-19. By use of the strategy, we successfully discovered three multitarget inhibitors with low IC50 values (<10.0 μM) for simultaneously inhibiting shikimate
dehydrogenase and shikimate kinase of in the shikimate pathway. The preliminary results show that the pharmapathlog-based screening strategy is useful to discover multitarget inhibitors.
optimize inhibitors of neuraminidases. The neuraminidase has been considered an attractive target for the treatment of influenza infection3,4. By use of the strategy, we designed five derivatives simultaneously inhibiting H1N1 and H5N1 neuraminidases with IC50 values in the
<10 nanomolar range. The derivatives could be new type inhibitors located at the 150-cavity, which is adjacent to the sialic acid binding site. Moreover, we found three novel inhibitors with IC50 values <10 μM through the strategy. Our experimental results reveals the three inhibitors
may overcome the drug resistances introduced by H274Y and I222R for H1N1 NAs without causing apparent cytotoxicity, suggesting a starting point to combat drug-resistant strains.
In the final chapter, we summarized the results of this thesis, and then discuss the future works. Currently, we propose a new concept of "pharmacologs" by extending the concept of "pharmapathlogs". Pharmacologs are a group of proteins sharing comparable binding environments in multiple pathways, and can be bound the same compounds. The major difference between pharmapathlogs and pharmacologs is that pharmacologs could be constituted by targets of multiple disease-related pathways, whereas pharmapathlogs only contain targets in a pathway. We believe that this new concept is able to identify multitarget inhibitors of multiple disease-related pathways, and then enhance therapeutic efficacy for human diseases.
Chapter 2. Site-moiety map to recognize interaction
preferences between protein pockets and compound
moieties
Identifying pharmapathlogs, which are protein orthologs sharing similar core binding environments in the same pathway and can be inhibited by the same compounds, is a challenging task. Because proteins in the same pathways often lack structural and sequence homology15,47, it is inapplicable to use sequence or structure alignment methods to find the core binding environments within these proteins. For example, in the shikimate pathway, the sequence identity between shikimate dehydrogenase (SDH) and shikimate kinase (SK) is 8.3%, and the root mean square deviation between SD and SK structures is 4.8Å . To address this issue, we developed a new server, called SiMMap16, to describe key binding environments of protein binding sites. Based on the SiMMap server, pharmapathlogs could be identified
through comparing key binding environments of proteins binding sites without considering sequence and structure similarities. In addition, consensus binding environment among these proteins are regarded as core binding environments, which can be applied to find multitarget inhibitors.
In this chapter, I cooperated with our laboratory members, including Dr. Yang, Yen-Fu Chen, Shen-Rong Lin, and Yu-Chi Huang, to develop the SiMMap server. The SiMMap server statistically derives site-moiety map with several anchors, which describe the relationship between the moiety preferences and physico-chemical properties of the binding site, from the interaction profiles between query target protein and its docked (or co-crystallized) compounds. Each anchor includes three basic elements: a binding pocket with conserved interacting
residues, the moiety composition of query compounds, and pocket-moiety interaction type (electrostatic, hydrogen-bonding, or van der Waals). We provide initial validation of the site-moiety map on three targets, thymidine kinase, and estrogen receptors of antagonists and agonists. Experimental results show that an anchor is often a hot spot and the site-moiety map can help to assemble potential leads by optimal steric, hydrogen-bonding, and electronic moieties. When a compound highly agrees with anchors of site-moiety map, this compound often activates or inhibits the target protein. We believe that the site-moiety map is useful to extract the core binding environments (core anchors) and find pharmapathlogs. The SiMMap web server is available at http://simfam.life.nctu.edu.tw/. The results were published in Nucleic
Acids Research.
2.1 Introduction
As the number of protein structures increases rapidly, structure-based drug design and virtual screening approaches are becoming important and helpful in lead discovery2,22,23,32. A number of docking and virtual screening methods25,33,34,39 have been utilized to identify lead compounds, and some success stories have been reported48-51. However, identifying lead compounds by exploiting thousands of docked protein-compound complexes is still a challenging task. The major weakness of virtual screenings is likely due to incomplete understandings of ligand binding mechanisms and the subsequently imprecise scoring algorithms2,22,23.
Most docking programs25,34,39 use energy-based scoring methods which are often biased toward both the selection of high molecular weight compounds and charged polar compounds40,41. These approaches generally cannot identify the key features (e.g., pharmacophore spots) that are essential to trigger or block the biological responses of the target
protein. Although pharmacophore techniques42 have been applied to derive the key features, these methods require a set of known active ligands that were acquired experimentally. Therefore, the more powerful techniques for post-screening analysis to identify the key features through docked compounds and to understand the binding mechanisms provide a great potential value for drug design.
To address these issues, we presented the SiMMap server to infer the key features by a site-moiety map describing the relationship between the moiety preferences and the physico-chemical properties of the binding site. According to our knowledge, SiMMap is the first public server that identifies the site-moiety map from a query protein structure and its docked (or co-crystallized) compounds. The server provides pocket-moiety interaction preferences (anchors) including binding pockets with conserved interacting residues; moiety preferences; and interaction type. We verified the site-moiety map on three targets, thymidine kinase, and estrogen receptors of antagonists and agonists. Experimental results show that an anchor is often a hot spot and the site-moiety map is useful to identify active compounds for these targets. We believe that the site-moiety map is able to provide biological insights and is useful for drug discovery and lead optimization.
2.2 Methods and Materials
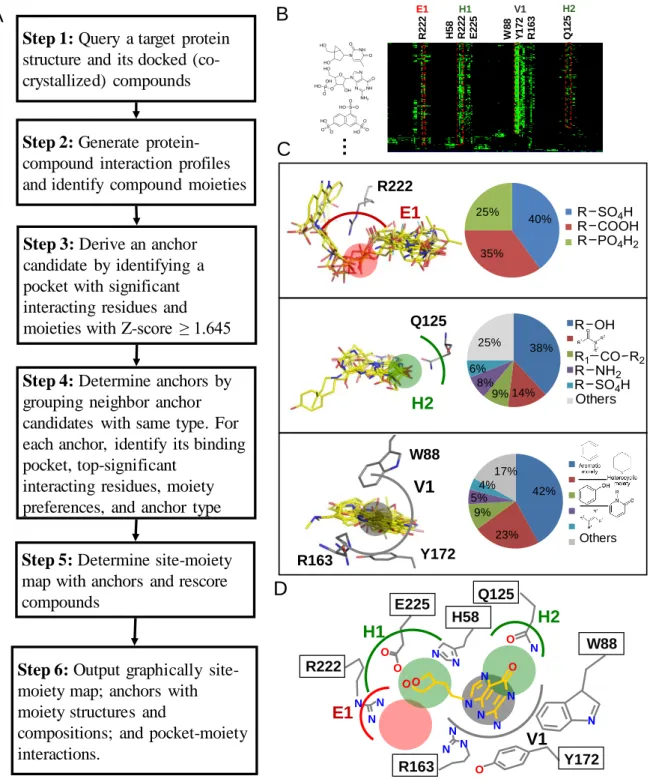
Figure 2.1 presents an overview of the SiMMap server for identifying the site-moiety map with
anchors, describing moiety preferences and physico-chemical properties of the binding site, from a query protein structure and docked compounds. The server first uses checkmol (http://merian.pch.univie.ac.at/~nhaider/cheminf/cmmm) to recognize the compound moieties and utilizes GEMDOCK33 to generate a merged protein-compound interaction profile (Fig. 2.1B), including electrostatic (E), hydrogen-bonding (H) and van der Waals (V) interactions.
According to this profile, we infer anchor candidates by identifying the pockets with significant interacting residues and moieties with Z-score ≥ 1.645. The neighbor anchor candidates, which are the same interaction type and the distances between their centers are less than 3.5Ǻ, are grouped into one anchor. These anchors form the site-moiety map describing interaction preferences between compound moieties and the binding site of the query (Figs.
2.1C and 2.1D). Finally, this server provides graphic visualization for the site-moiety map;
anchors with moiety structures and compositions; pocket-moiety interactions; and the relationship between anchors and moieties of query compounds.
Figure 2.1. Overview of the SiMMap server for the site-moiety map using herpes simplex virus type-1 thymidine kinase (TK) and 1000 docked compounds as the query. (A) Main procedure. The Z-score cutoff was set to 1.645, which was commonly used in statistics (95% confidence level). (B) The merged protein-compound interaction profile. A cell is colored by green if there is an interaction (e.g., electrostatic, hydrogen-bonding, or van der Waals interaction) between a compound and a residue (C) The pocket-moiety interaction preferences of three anchors: E1
A B … R 2 2 2 H 5 8 R 2 2 2 E 2 2 5 W 8 8 Y 1 7 2 R 1 6 3 Q 1 2 5 E1 H1 V1 H2 C D N N N O O N N N N O O N N N O N N N N R222 E225 H58 R163 E1 H1 V1 H2 W88 Q125 Y172 Others Others R222 E1 Q125 H2 Y172 R163 W88 V1 40% 35% 25% 1 2 3 40% 35% 25% 1 2 3 38% 14% 9% 8% 6% 25% 1 2 3 4 5 6 38% 14% 9% 8% 6% 25% 1 2 3 4 5 6 42% 23% 9% 5% 4% 17% 1 2 3 4 5 6 42% 23% 9% 5% 4% 17% 1 2 3 4 5 6 Step 1: Query a target protein
structure and its docked (co-crystallized) compounds
Step 2: Generate protein-compound interaction profiles and identify compound moieties
Step 3: Derive an anchor candidate by identifying a pocket with significant interacting residues and moieties with Z-score ≥ 1.645
Step 4: Determine anchors by grouping neighbor anchor candidates with same type. For each anchor, identify its binding pocket, top-significant
interacting residues, moiety preferences, and anchor type
Step 6: Output graphically site-moiety map; anchors with moiety structures and
compositions; and pocket-moiety interactions.
Step 5: Determine site-moiety map with anchors and rescore compounds
(electrostatic), H2 (hydrogen-bonding), and V1 (van der Waals). Each anchor consists of a binding pocket with conserved interacting residues, the moiety composition and anchor type; (D) The site-moiety map with four anchors.
2.2.1 Site-moiety map, anchor and pocket
The anchor (pocket-moiety interaction preference) is the core of a site-moiety map. An anchor possesses three essential elements: (1) a binding pocket with conserved interacting residues and specific physico-chemical properties; (2) moiety preferences of the pocket; (3) pocket-moiety interaction type (E, H, or V). An anchor can be considered as "key features" for representing the conserved binding environment element or a "hot spot" which involves biological functions. In addition, we regard a binding pocket, which consists of several residues significantly interacting to compound moieties, as a part of the binding site. The binding pocket often possesses specific physico-chemical properties and geometric shape to bind preferred moieties. The site-moiety map, which can help to assemble potential leads by optimal steric, hydrogen-bonding, and electronic moieties, is useful for drug discovery and understanding biological mechanisms.
2.2.2 Data sets
To describe and evaluate the utility of the SiMMap server, we tested the server on three target proteins for virtual screening. These proteins are herpes simplex virus type-1 thymidine kinase (TK, PDB code 1kim52), estrogen receptor α for antagonists (ER, PDB code 3ert53), and estrogen receptor α for agonists (ERA, PDB code 1gwr54
). Each compound set consists of 10 known active ligands and 990 compounds selected randomly from Available Chemical
Directory (ACD) proposed by Bissantz et al.55. Currently, the docked conformations of these 1000 compounds were generated by the in-house developed GEMDOCK program33 which is comparable to some docking methods (e.g., DOCK, FlexX, and GOLD) on the 100 protein-ligand complexes and some screening targets33,40. In addition, GEMDOCK has been successfully applied to identify inhibitors and binding sites for some targets51,56,57.
2.2.3 Main procedure of constructing a site-moiety map
The SiMMap server performs six main steps for a query (Fig. 2.1A). Here, we used TK as an example for describing these steps. First, users input a protein structure and its docked compounds. The server used checkmol to identify moieties of docked compounds and GEMDOCK to generate E, H and V interaction profiles. For each profile, the matrix size isN× K where N and K are the numbers of compounds and interacting residues of query protein,
respectively. An interaction profile matrix P(I) with type I (E, H, or V) is represented as
NK N N K K p p p p p p p p p I P 2 , 1 , 2 2 , 2 1 , 2 1 2 , 1 1 , 1 ) (
, where pi,j is a binary value for the compound i interacting to the residue j (Fig. 2.1B). For H
and E profiles, pi,j is set to 1 (green) if an atom pair between the compound i and the residue j
forms hydrogen-bonding or electrostatic interactions, respectively; conversely, the interaction is set to 0 (black). For van der Waals (vdW) interaction, an interaction is set to 1 when the energy is less than -4 (kcal/mol).
SiMMap identified consensus interactions between residues and compound moieties with similar physical-chemical properties through the profiles. For each interacting residue (a
column of the matrix P(I)) (Fig. 2.1B), we used Z-score value to measure the interacting conservation between this residue and moieties. Z-score values are often used to evaluate statistical significances. Here, we used to measure how significant the consensus interactions are in the protein-compound interaction profiles. High Z-score values indicate that the interactions are highly consensus. The standard deviation (σ) and mean (μ) were derived by random shuffling 1,000 times in a profile. The Z-score of the residue j is defined as
μ f Zj j
, where fj is the interaction frequency and given as
N i j i j N p f 1 .Spatially neighbor interacting residues and moieties with statistically significant Z-score ≥ 1.645 were referred as an anchor candidate. The Z-score threshold is set to 1.645 (95% confidence level) using Student's t-test. Neighbor anchor candidates, which are spatially overlapped and the same anchor type, were clustered as an anchor and the anchor center is the weighted geometric center of their interacting compound moieties. Here, two anchors were merged if the distance of two anchor centers is less than 3.5 Ǻ. In each anchor, top three residues with the highest Z-score values were regarded as key residues forming a binding pocket. For each anchor, we identified its moieties of docked compounds according to the moiety library derived from checkmol, and calculated the moiety composition (Fig. 2.1C). These anchors form the site-moiety map (Fig. 2.1D) of the query.
SiMMap can be applied to identify active compounds for structure-based virtual screening. One of weaknesses of virtual screening is likely incomplete understanding of the chemistry involved in ligand binding and the subsequently imprecise scoring algorithms. When a compound highly agrees with the anchors of the site-moiety map, this compound often activates or inhibits the target. The SiMMap server scores a compound by combining predicted binding energy of GEMDOCK and the anchor score between the map and the compound. The SiMMap score, S(i), for a compound i is defined as
n a a M i E i AS i S 1 0.5 ) ( ) 001 . 0 ( ) ( ) ( (3.1) , where ASa(i) is the anchor score of compound i in the anchor a, n is the number of anchors,E(i) is the docked energy of compound i, and M is the atom number of compound i. The anchor
score is set to 1 when the compound i agrees the moiety preference of the anchor a. Here, the anchor score and the term M0.5 are useful to reduce the deleterious effects of selecting high molecular weight compounds58. Based on SiMMap scores, we can obtain new ranks of query compounds.
2.2.4 Input and output of the SiMMap server
SiMMap is an easy-to-use web server (Fig. 2.2). Users input a protein structure without ligands in PDB format and its docked or co-crystallized compounds in MDL mol, SYBYL mol2, or PDB format (Fig. 2.2A). These docked compounds should be generated by any external docking methods (e.g., DOCK, FlexX, GOLD and GEMDOCK) before users uploaded these compounds. Typically, the SiMMap server yields a site-moiety map within 5 minutes if the number of query compounds is less than 100. This server provides the graphic visualization of the site-moiety map and anchors elements, including a binding pocket with interacting residues, moiety compositions and structures, numbers of involved compounds, and anchor types (Fig. 2.2B). For each anchor, this server shows docked conformations of compounds and the detailed atomic interactions between pocket residues and moieties (Fig. 2.2C). In addition, SiMMap shows the new rank and compound moiety structures fitting the anchors for each query compound (Fig. 2.2D). SiMMap uses two open source tools for graphic visualization: Jmol (http://www.jmol.org/) for displaying three-dimensional protein and compound structures with anchors and OASA (http://bkchem.zirael.org/oasa_en.html) for visualizing compound
structures. The server allows users to download the anchor coordinates in the PDB format; interaction profiles; new ranks and anchor scores of query compounds.
Figure 2.2. The SiMMap server analysis results using estrogen receptor (ER) and 1000 docked
compounds as the query. (A) The user interface for uploading target protein structure and docked compounds. (B) The site-moiety map has one hydrogen-bonding and three van der Waals anchors for ER. Each anchor contains the moiety structures and composition, anchor type, and key residues in the binding pocket. (C) The details of moiety structures and residue-moiety interactions in the H1 anchor. (D) The SiMMap scores, ranks and the relationships between anchors and moieties of query compounds.
A B D C
…
…
R3942.3 Results and Discussion
2.3.1 Thymidine kinase
The SiMMap server inferred the site-moiety map of TK. This map consisted of four anchors (i.e., E1, H1, H2, and V1 (Fig. 2.1D) and the moiety composition and conserved interacting residues of each anchor (Fig. 2.1C). The E1 anchor possesses a binding pocket with residue R222, and three moiety types (i.e., sulfuric acid monoester (40%), carboxylic group (35%) and phosphoric acid monoester (25%)) derived from 57 compounds. The E1 includes the phosphate moiety of ATP and its residue R222 playing a major role to interact with the substrate59,60. Furthermore, the H1 anchor is a polar pocket with three residues (H58, R222, and E225) which often form hydrogen bonds with polar moiety types among 308 compounds, for example, hydroxyl group (22%), carboxylic acid (8%), ketone (8%), ether (7%), and carboxylic amide (7%). The H2 anchor consists of the residue Q125 and 157 moieties divided into five major moiety types, including hydroxyl group (38%), carboxylic amide (14%), ketone (9%), amine (8%) and sulfuric acid monoester (6%). Finally, the V1 anchor has a binding pocket with residues W88, R163, Y172 and bulky moieties, such as aromatic ring (42%), heterocyclic group (23%), phenol (9%), and oxohetarene (5%).
The preferred moiety types of an anchor are suitable groups interacting to conserved residues of the binding pocket. The moiety preference is able to guide the suggestion of functional group substitutions for lead structures. For example, the moiety preferences of these four anchors (Fig. 2.1D) cover the moiety types derived from 15 TK co-crystal ligands (Table 2.1). In addition, these compounds contain carboxylic amide or amine groups in the H1 anchor. This result shows that the pocket-moiety interactions of these 15 complexes are highly consistent with the pocket-moiety interaction preferences.
Table 2.1. The relationship between the anchors and moieties of 15 co-crystallized ligands for TK
PDB code Compound structure
Anchor E1 H1 H2 V1 3vtk R-OH 1vtk R-OH 1p7c R-OH 1of1 R-OH 1ki6 R-OH 1ki8 R-OH 1e2k R-OH 1ki7 R-OH 1ki4 R-OH 1kim R-OH 1e2p R-OH
1ki3 R-OH Aromatic moiety
1ki2 R-OH Aromatic moiety
1qhi R-OH R-NH2 Aromatic moiety
2.3.2 Estrogen receptor
We used estrogen receptor (ER), a therapeutic target for osteoporosis and breast cancer61, as the second example. Based on 1000 docked compounds and ER, the SiMMap server identifies four anchors (H1, V1, V2, and V3) and provides moiety preferences and compositions in these anchors (Fig. 2.2B). The H1 anchor comprises three residues (E353, L387, and R394) and five main moiety types: hydroxyl group (36%), carboxylic acid (16%), amine (7%), ketone (7%), and sulfuric acid monoester (6%) summarized from 319 compounds. Furthermore, three residues (L346, T347, and L525) and 839 compounds are involved in the V1 anchor, preferring five moiety types (i.e., aromatic ring (49%), heterocyclic group (22%), alkenes (11%), phenol (8%), and oxohetarene (4%)). The anchor V2 is a hydrophobic pocket containing L346, F404, and L387, and the former two residues are highly conserved62. These hydrophobic residues interact with aromatic ring (52%), heterocyclic group (23%), phenol (12%), alkenes (5%), and oxohetarene (3%). Finally, aromatic rings (55%), heterocyclic groups (17%), alkenes (11%), and phenols (9%) summarized from 560 compounds often form vdW contacts with the long side chains of M343, M421, and L525 in the anchor V3. The ring groups of antagonists are often stabilized by the side chains of M343, L346, T347, L387, M421, and L525. In this case, most selective estrogen receptor modulators of ER (e.g., EST_01 (raloxifene), EST_06 (LY-326315,) and EST_05 (EM-343)) agree with these four anchors (Fig. 2.2D).
2.3.3 Discussion
Anchors identified by the SiMMap server often contain key pockets and moieties. To initially validate the anchors for biological mechanisms (e.g., ligand binding and catalysis mechanisms), we selected 15 TK and 22 ER co-crystallized ligands (Fig. 2.3, Fig. 2.4, and Table 2.1). The
corresponding moieties of these co-crystallized ligands were highly matched the anchors derived from 1000 docked compounds (10 known active ligands and 990 randomly selected compounds described in Data sets). The site-directed mutagenesis shows that the conserved interacting residues of the anchors are often essential for ligand binding and catalysis mechanisms. For example, the positive charged residue R222 in E1 interacts with the phosphate group of TK substrates for phosphorylation63 (Figs. 2.3A and 2.3B). The site-directed mutagenesis indicates that Q125 in H2 is essential for the substrate specificity59 and the triple mutant ,H58L/M128F/Y172F (H1 and V1), shows the drug resistance to the compound acyclovir64. In addition, the hydrogen-bonding interaction between E225 and the hydroxyl group of the substrates is able to help stabilize the LID region for the catalytic reaction64. For ER target, 22 ER co-crystallized ligands contain three consistent moieties that are hydroxyl group and aromatic rings (Fig. 2.4). The hydroxyl group forms hydrogen bonds with R394 and E353 in H1, and the aromatic ring yields vdW contacts with L346, L387, and F404 in V2. The other consistent aromatic ring forms vdW contacts with L346, T347, and L525 in V1. These results show that an anchor is often a hot spot and involved in biological functions.
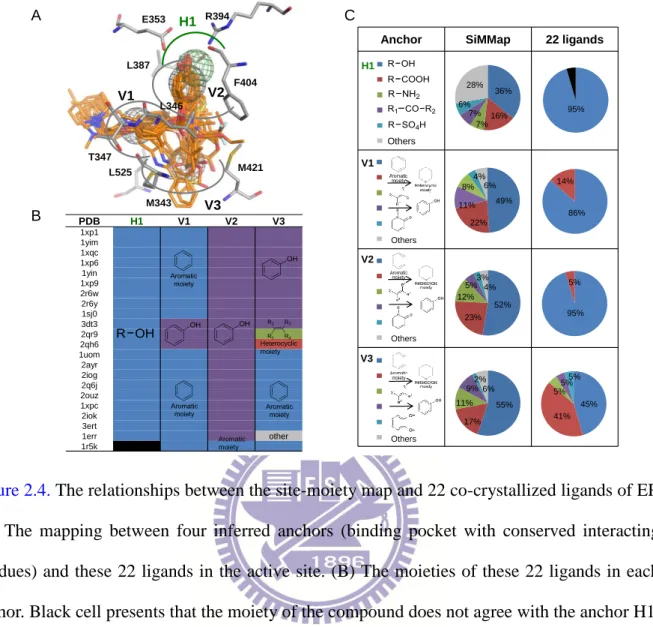
Figure 2.3. The relationships between the site-moiety map and 15 co-crystallized ligands of TK. (A) The mapping between four inferred anchors (binding pocket with conserved interacting residues) and these 15 ligands in the active site. (B) The moieties of these 15 ligands in each anchor. (C) The moiety compositions of 1000 docked compounds (SiMMap) and these 15 ligands. R222 E225 H58 R163 E1 H1 V1 H2 W88 Q125 Y172 PDB E1 H1 H2 V1 3vtk 1vtk 1p7c 1of1 1ki6 1ki8 1e2k 1ki7 1ki4 1kim 1e2p 1ki3 1ki2 1qhi 2ki5 38% 14% 9% 8% 6% 25% 1 2 3 4 5 6 38% 14% 9% 8% 6% 25% 1 2 3 4 5 6 42% 23% 9% 5% 4% 17% 1 2 3 4 5 6 40% 35% 25% 1 2 3
Anchor SiMMap 15 ligands
Others Others E1 H1 V1 40% 35% 25% 1 2 3 42% 23% 9% 5% 4% 17% 1 2 3 4 5 6 100% 27% 73% Others H2 38% 14% 9% 8% 6% 25% 1 2 3 4 5 6 87% 22% 8% 8% 7% 7% 48% 13% A B C 1 2 20%
Figure 2.4. The relationships between the site-moiety map and 22 co-crystallized ligands of ER. (A) The mapping between four inferred anchors (binding pocket with conserved interacting residues) and these 22 ligands in the active site. (B) The moieties of these 22 ligands in each anchor. Black cell presents that the moiety of the compound does not agree with the anchor H1. (C) The moiety compositions of 1000 docked compounds (SiMMap) and these 22 ligands.
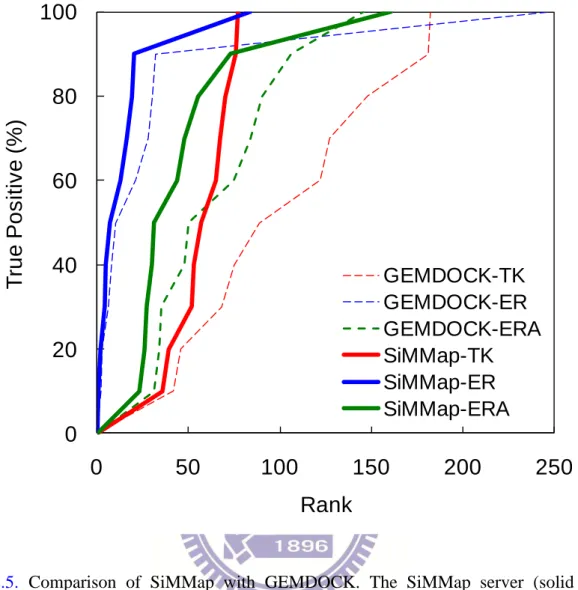
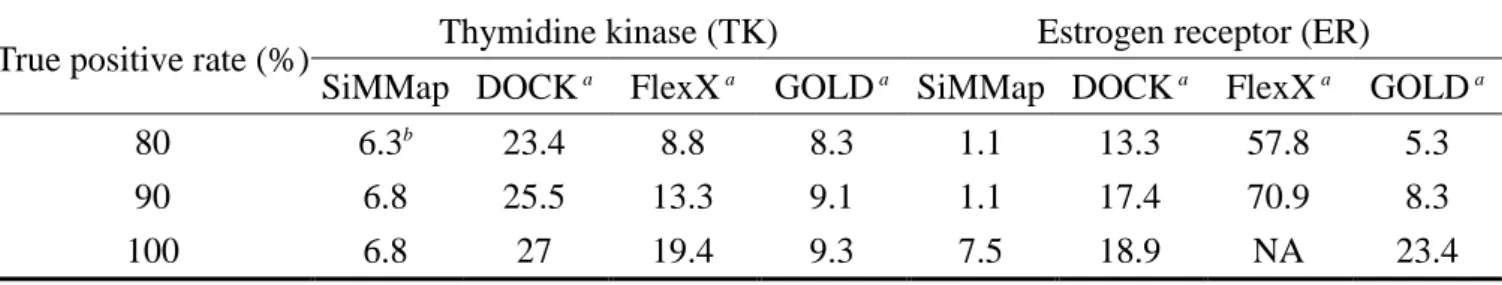
To provide initial validation of the SiMMap server for virtual screening, we selected TK, ER, and ERA with 1000 compounds as test sets. First, we compared the accuracies of SiMMap with those of GEMDOCK on these three targets based on true positive rates (Fig. 2.5). SiMMap, combining anchor scores and docking energies (Equation (3.1)), outperforms GEMDOCK on these cases. We then compared SiMMap with other three programs (DOCK, FlexX, and GOLD) on TK and ER sets. All approaches were tested using the same proteins and compound sets (Table 2.2). When the true positive rate was 90%, the false positive rates were 6.8% (SiMMap), 25.5% (DOCK), 13.3% (FlexX), and 9.1% (GOLD) for TK and were 1.1% (SiMMap), 17.4% (DOCK), 70.9% (FlexX), and 8.3% (GOLD) for ER.
1 2 3 4 5 6
Anchor SiMMap 22 ligands
A B C H1 V1 V2 R394 T347 F404 M421 M343 L525 E353 L387 L346 V3 1 2 3 4 5 6 1 2 3 4 5 6 1 2 3 4 5 6 1 2 3 4 5 6 1 2 3 4 5 6 1 2 3 4 5 6 1 2 3 4 5 6 Others Others Others Others 1 2 3 4 5 1 2 95% 5% 45% 41% 5% 5%5% 36% 16% 7% 7% 6% 28% 49% 22% 11% 8% 4% 6% 52% 23% 12% 5%3%4% 55% 17% 11% 9% 2% 6% PDB H1 V1 V2 V3 1xp1 1yim 1xqc 1xp6 1yin 1xp9 2r6w 2r6y 1sj0 3dt3 2qr9 2qh6 1uom 2ayr 2iog 2q6j 2ouz 1xpc 2iok 3ert 1err other 1r5k Aromatic moiety Heterocyclic moiety H1 V1 V2 V3 1 2 1 2 3 86% 14% 95%
Figure 2.5. Comparison of SiMMap with GEMDOCK. The SiMMap server (solid lines)
consistently outperforms GEMDOCK (dash lines) on three targets: TK (red), ER (blue) and ERA (green).
0
20
40
60
80
100
0
50
100
150
200
250
T
ru
e
Po
s
it
iv
e
(%
)
Rank
GEMDOCK-TK
GEMDOCK-ER
GEMDOCK-ERA
SiMMap-TK
SiMMap-ER
SiMMap-ERA
Table 2.2. Comparing SiMMap with other methods on thymidine kinase and estrogen receptor by false-positive rates
True positive rate (%) Thymidine kinase (TK) Estrogen receptor (ER) SiMMap DOCK a FlexX a GOLD a SiMMap DOCK a FlexX a GOLD a 80 6.3b 23.4 8.8 8.3 1.1 13.3 57.8 5.3 90 6.8 25.5 13.3 9.1 1.1 17.4 70.9 8.3 100 6.8 27 19.4 9.3 7.5 18.9 NA 23.4
a Summarized from Bissantz et al. 55
b The false-positive rate (%) is obtained based on 1000 compounds.
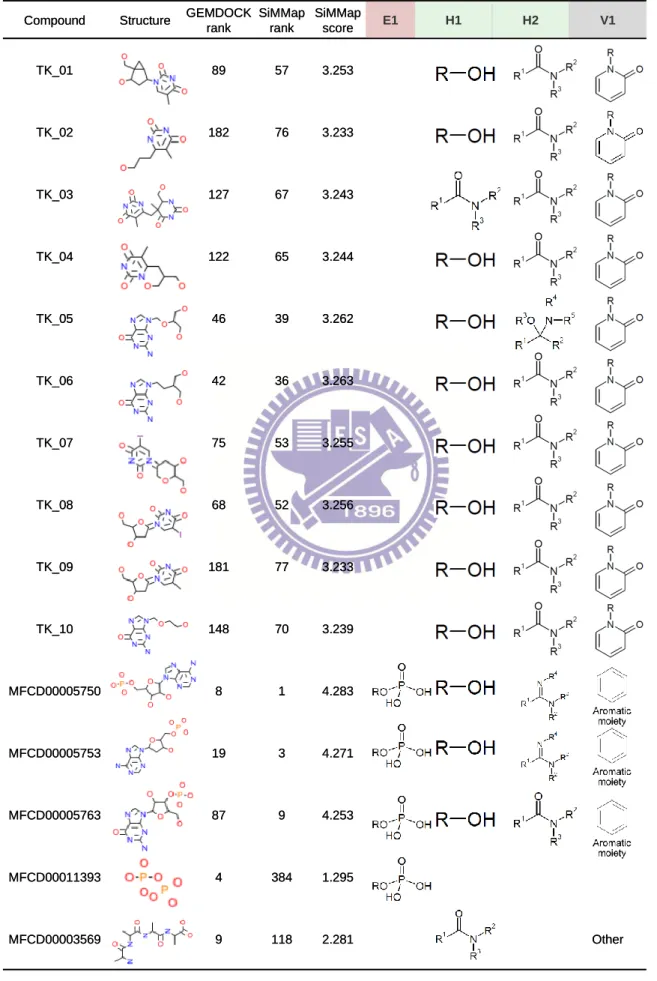
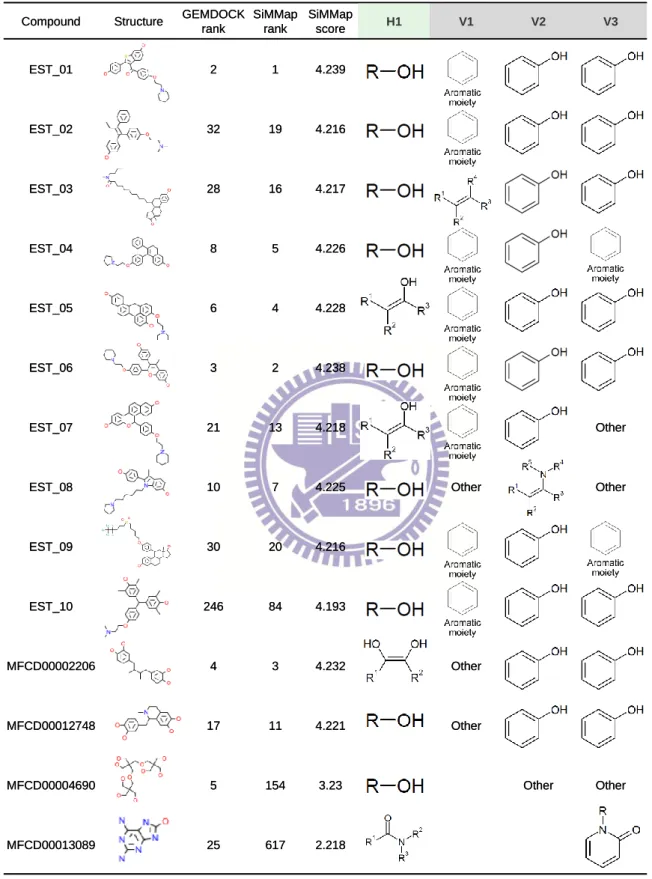
The compound, which agrees with anchors of the site-moiety map, is often able to activate or inhibit the target protein (Tables 2.1, 2.3 and 2.4). In addition, the anchor score (i.e. AS(i) defined in Equation (3.1)) of SiMMap can be used to reduce the ill-effect of the energy-based scoring methods which are often biased toward both the selection of high molecular weight compounds and charged polar compounds40,41. For example, according to the SiMMap scores (Equation (3.1)), the ranks of MFCD0005750 (adenylic acid), MFCD0005753 (deoxyadenylic acid) and MFCD0005763 (3'-guanylic acid) are 1, 3, and 9, respectively. These three compounds are thymidine analogs and agree with the four anchors of TK (Fig. 2.1 and Table 2.1). For the top ranks of ER, MFCD0002206 (masoprocol) and MFCD00012748 were also the analogs of the active compounds (Table 2.4). The anchor score of SiMMap was helpful to reduce the highly polar compounds (e.g., MFCD00011393 and MFCD00003569 in TK; MFCD00004690 and MFCD00013089 in ER) whose anchor scores are low. The anchor score of SiMMap can easily combine with other energy-based scoring functions.
Table 2.3. The mapping between the anchors and active and typical compounds for TK
Compound Structure GEMDOCK
rank SiMMap rank SiMMap score E1 H1 H2 V1 TK_01 89 57 3.253 TK_02 182 76 3.233 TK_03 127 67 3.243 TK_04 122 65 3.244 TK_05 46 39 3.262 TK_06 42 36 3.263 TK_07 75 53 3.255 TK_08 68 52 3.256 TK_09 181 77 3.233 TK_10 148 70 3.239 MFCD00005750 8 1 4.283 MFCD00005753 19 3 4.271 MFCD00005763 87 9 4.253 MFCD00011393 4 384 1.295 MFCD00003569 9 118 2.281 Other
Compound Structure GEMDOCK
rank SiMMap rank SiMMap score E1 H1 H2 V1 TK_01 89 57 3.253 TK_02 182 76 3.233 TK_03 127 67 3.243 TK_04 122 65 3.244 TK_05 46 39 3.262 TK_06 42 36 3.263 TK_07 75 53 3.255 TK_08 68 52 3.256 TK_09 181 77 3.233 TK_10 148 70 3.239 MFCD00005750 8 1 4.283 MFCD00005753 19 3 4.271 MFCD00005763 87 9 4.253 MFCD00011393 4 384 1.295 MFCD00003569 9 118 2.281 Other
Table 2.4. The mapping between the anchors and active and typical compounds for ER
Compound Structure GEMDOCK
rank SiMMap rank SiMMap score H1 V1 V2 V3 EST_01 2 1 4.239 EST_02 32 19 4.216 EST_03 28 16 4.217 EST_04 8 5 4.226 EST_05 6 4 4.228 EST_06 3 2 4.238 EST_07 21 13 4.218 Other
EST_08 10 7 4.225 Other Other
EST_09 30 20 4.216 EST_10 246 84 4.193 MFCD00002206 4 3 4.232 Other MFCD00012748 17 11 4.221 Other MFCD00004690 5 154 3.23 Other Other MFCD00013089 25 617 2.218
Compound Structure GEMDOCK
rank SiMMap rank SiMMap score H1 V1 V2 V3 EST_01 2 1 4.239 EST_02 32 19 4.216 EST_03 28 16 4.217 EST_04 8 5 4.226 EST_05 6 4 4.228 EST_06 3 2 4.238 EST_07 21 13 4.218 Other
EST_08 10 7 4.225 Other Other
EST_09 30 20 4.216 EST_10 246 84 4.193 MFCD00002206 4 3 4.232 Other MFCD00012748 17 11 4.221 Other MFCD00004690 5 154 3.23 Other Other MFCD00013089 25 617 2.218
2.4
Conclusions
This work demonstrates the utility and feasibility of the SiMMap server for statistically inferring the site-moiety map describing the relationship between the moiety preferences and physico-chemical properties of the binding site. Our experimental results show that the site-moiety map is useful to reflect biological functions and identify active compounds from thousands of compounds. In addition, the site-moiety map can guide to assemble potential leads by optimal steric, hydrogen-bonding, and electronic moieties. We believe that the SiMMap serve is able to provide the biological insights of protein-ligand binding models, enrich the screening accuracy, and guide the processes of lead optimization.
Chapter 3. Pharmapathlogs for discovering multitarget
inhibitors of shikimate pathways using site-moiety maps
Multitarget inhibitors of pharmapathlogs can enhance therapeutic treatments by inhibiting several disease-related proteins in the same pathways, and reduce probabilities of drug-resistances. However, identifying pharmapathlogs and their multitarget inhibitors are still challenging tasks since these proteins often lack structural and sequence homology. In this chapter, we used site-moiety map developed in the previous chapter to infer pharmapathlogs. Subsequently, we proposed a pharmapathlog-based screening strategy to identify multitarget inhibitors of pharmapathlogs. This strategy was initially tested in discovering multitarget inhibitors of Helicobacter pylori (H. pylori) and Mycobacterium tuberculosis (M. tuberculosis), which are human pathogens and causes peptic ulcer disease and chronic infectious disease. By uses of the strategy, three inhibitors that simultaneously showed good inhibition abilities (IC50values <10 μM) to shikimate dehydrogenase and shikimate kinase in the shikimate pathway were discovered with collaborations with Dr. Wen-Ching Wang and Dr. Wen-Chi Cheng of National Tsing Hua University. Furthermore, we found that residues of core anchors are more conserved than those of the other regions, revealing that core anchors of pharmapathlogs are often essential for catalysis or substrate binding during evolution. This suggests that designing inhibitors targeting the core anchors could decrease probabilities of drug-resistances. These experimental results reveal that the pharmapathlog-based strategy could be useful to infer pharmapathlogs and their multitarget inhibitors. We believe that the strategy can be further applied for drug design of human diseases.
3.1 Introduction
Pharmapathlogs, which are a group of protein orthologs in the same pathway that share similar binding environments and can be inhibited by the same compounds, provide clues to discover multitarget inhibitors. Therapeutics using multitarget inhibitors are more effective and less vulnerable to drug-resistances than those using single-target inhibitors. For example, cosalane, an antiretroviral drug, has potent antiviral activity by simultaneously inhibit multiple targets of HIV-1 proteins, including gp120, integrase, protease, and reverse transcriptase65-67. However, current processes of discovering multitarget inhibitors are often serendipitous, and binding mechanisms of these inhibitors may be retrospectively understood 68. One of the main reasons is that target proteins of multitarget inhibitors often lack structural and sequence homology14. In addition, the low sequence similarity of proteins in the same pathway makes discovering pharmapathlogs difficult as well15. As a result, a new strategy for inferring pharmapathlogs and their multitarget inhibitors will be valuable for drug design.
Here, we proposed the pharmapathlog-based screening strategy to infer pharmapathlogs and discover their multitarget inhibitors by applying site-moiety maps described in the previous chapter16. A site-moiety map can present and characterize key binding environments of a protein binding site by using anchors. An anchor contains three crucial elements, including conserved interacting residues constituting a binding pocket (i.e., a part of the binding site), the preference of moieties (i.e., functional groups), and pocket-moiety interaction type [electrostatic (E), hydrogen-bonding (H), or van der Waals (V)]. Therefore, we are able to infer pharmapathlogs by identifying proteins sharing consensus binding environments (core anchors) instead of using structure or sequence similarities. Furthermore, core anchors of pharmapathlogs could be used to find multitarget inhibitors by screening compounds.