Synthesis and Properties of a Novel Electrochromic Polymer
Obtained from the Electropolymerization of a
9,9’-Spirobifluorene-Bridged Donor-Acceptor (D-A)
Bichromophore System
Luis Otero,
†Leonides Sereno,
†Fernando Fungo,*
,†Yuan-Li Liao,
‡Chi-Yen Lin,
‡and
Ken-Tsung Wong*
,‡Departamento de Quı´mica, UniVersidad Nacional de Rı´o Cuarto, Agencia Postal 3 (5800), Rı´o Cuarto, Argentina, and Department of Chemistry, National Taiwan UniVersity, Taipei 106, Taiwan
ReceiVed March 3, 2006
The synthesis and photophysical, electrochemical, and spectroelectrochemical characterization of a novel donor-acceptor (D-A) bichromophore system composed of two D-A segments linking through a spiro center are reported. The electron-donating (D) moieties are triphenylamine (TPA) groups, whereas the electron-withdrawing (A) moieties are cyano groups. The particular “spiro” configuration that perpendicularly bonds the D-A chromophores by a tetrahedral carbon, impedes orbital interactions between the branches. Thus, the two TPA substituents act independently, rendering an efficient electropolymerization process feasible. The polymer film obtained showed reversible electrochemical oxidation accompanied by strong color changes with high coloration efficiency and contrast ratio, which can be switched by potential modulation. The remarkable electrochromic behavior of the film is clearly interpreted on the basis of spectroelectrochemical studies. A plausible polymerization mechanism involved with the TPA dimerization reaction is proposed for the electropolymerization process.
Introduction
Polymeric and oligomeric materials applied in thin layer electro-optical devices such as organic light emitting diodes (OLEDs), solar cells, and organic field effect transistors (OTFTs)1should exhibit reasonable electronic conductivity
and adequate band-gap energies or light absorption/emission in the desired region of the electromagnetic spectrum. For example, low-band-gap polymers with absorption bands in the near-infrared region can be obtained by maximizing the π-electron delocalization within the conjugated polymer backbone through aromatic and quinoid type structures adopting coplanar conformations between the polymer’s consecutive repeating units.2a-c,3aCommon examples are the
classical conducting polymers4such as polyaniline,5-7
poly-thiophenes,8,9 and polypyrroles.10
Another strategy employed in designing polymeric struc-tures with desired electronic properties involves the synthesis
of the donor-acceptor (D-A) type polymers in which the A and D units are strong electron-withdrawing (A) and electron-donating (D) moieties, respectively.3a-b,11,12Making
appropriate choices for the A and D units allows one to control of theπ-system’s HOMO and LUMO energy levels. Therefore, the properties such as redox behavior and pho-tophysical characteristics of the polymers can be tailored. In addition, because of the electronic nature of D-A polymers, they are able to show two different redox processes (oxidation and reduction), depending on which moiety (D or A) is involved.
In this paper, we report the synthesis and photophysical, electrochemical, and spectroelectrochemical studies of a novel D-A chromophore system composed of two D-A segments linking through a spiro center (Scheme 1). This molecule is basically a D-A type chromophore in which the electron-donating (D) moiety is a triphenylamine (TPA) group and the electron-withdrawing (A) moiety is a cyano group. Upon oxidation, the TPA can undergo well-known radical cation dimerization to produce the
tetraphenylben-* To whom correspondence should be addressed. E-mail: ffungo@
exa.unrc.edu.ar (F.F.), [email protected]. (K.-T.W.).
†Universidad Nacional de Rı´o Cuarto.
‡National Taiwan University.
(1) (a) Thelakkat, M. Macromol. Mater. Eng. 2002, 287, 442. (b) Cumpston, B. H.; Ananthavel, S. P.; Barlow, S.; Dyer, D. L.; Ehrlich, J. E.; Erskine, L. L.; Heikal, A. A.; Kuebler, S. M.; Lee, I. Y. S.; McCord-Maughon, D.; Qin, J.; Rochkel, H.; Rumi, M.; Wu, X. L.; Marder, S. R.; Perry, J. W. Nature 1999, 398, 51. (c) Drobizhev, M.; Karotki, A.; Dzenis, Rebane, A.; Suo, Z. Y.; Spangler, C. W. J. Phys. Chem. B 2003, 107, 7540.
(2) (a) Bre´das, J. L. J. Chem. Phys. 1985, 82, 3808. (b) Jenekhe, S. A. Nature 1986, 322, 345. (c) Chen, W. C.; Jenekhe, S. A. Macromol-ecules 1995, 28, 465.
(3) (a) Roncali, J. Chem. ReV. 1997, 97, 173. (b) Agrawal, A. K.; Jenekhe, S. A. Macromolecules 1993, 26, 895.
(4) Skotheim, T. A.; Elsenbaumer, R. L.; Reynolds, J. R. Handbook of Conducting Polymers, 2nd ed.; Marcel Dekker: New York, 1997.
(5) Rodrigues, M. A.; De Paoli, M. A.; Mastragostino, M. Electrochim. Acta 1991, 36, 2143.
(6) Panero, S.; Scrosati, B.; Baret, M.; Cecchini, B.; Masetti, E. Sol. Energy Mater. Sol. Cells 1995, 39, 239.
(7) Tassi, E. L.; De Paoli, M. A. Electrochim. Acta 1994, 39, 2481. (8) Dietrich, M.; Heinze, J.; Heywang, G.; Jonas, F. J. Electroanal. Chem.
1994, 369, 87.
(9) Sapp, S. A.; Sotzing, G. A.; Reynolds, J. R. Chem. Mater. 1998, 10, 2101.
(10) De Paoli, M. A.; Zanelli, A.; Mastragostino, M.; Rocco, A. M. J. Electroanal. Chem 1997, 435, 217.
(11) Zhang, Q. T.; Tour, J. M. J. Am. Chem. Soc. 1998, 120, 5355. (12) Devasagayaraj, A.; Tour, J. M. Macromolecules 1999, 32, 6425. 10.1021/cm0605264 CCC: $33.50 © 2006 American Chemical Society
Published on Web 06/24/2006
Downloaded by NATIONAL TAIWAN UNIV on September 2, 2009 | http://pubs.acs.org
zidine (TPB), which does not effectively couple to give polymers because of the high delocalization of the radical cation between the two nitrogen atoms.13-16However, it has
been reported that blocking the radical cation delocalization can be achieved by introducing electron-deficient components between two TPA units, leading to an improvement in their reactivity toward electropolymerization.17The particular spiro
configuration that perpendicularly bonds two D-A chro-mophores with a tetrahedral carbon impedes the π-orbital interaction between the different D-A chromophore branches. Thus, the two TPA substituents could act independently, rendering the electropolymerization processes feasible. In addition, these kinds of D-A structures, implanted with a rigid and coplanar fluorene ring, may lead to a good emitter with promising potential applications in OLEDs.18-20
Experimental Section
Synthetic Procedures. Synthesis of
7,7’-Dibromo-9,9’-spirobi-fluorene-2,2’-bis(carboxylic acid methyl ester) (2). To a solution
of 7,7′-dibromo-9,9′-spirobifluorene-2,2′-dicarboxylic acid (5.62 g, 10 mmol) in methanol (90 mL) was slowly added sulfuric acid (10 mL). The solution was allowed to reflux for 3 days. The white solid was collected and washed with methanol to afford white diester 2 (5.3 g, 90%). Mp: 307-309°C. IR (KBr): ν 3058 (w), 2985 (w), 2952 (w), 1719 (s), 1613 (m), 1427 (m), 1301 (s), 1255 (s) cm-1.1H NMR (CDCl 3, 400 MHz):δ 8.12 (dd, J ) 8.0, 1.4 Hz, 2H), 7.89 (d, J ) 8.0 Hz, 2H), 7.77 (d, J ) 8.0 Hz, 2H), 7.56 (dd, J ) 8.0, 1.6 Hz, 2H), 7.35 (s, 2H), 6.84 (d, J ) 1.4 Hz, 2H), 3.79 (s, 6H).13C NMR (CDCl 3, 100 MHz): δ 166.5, 150.0, 147.0, 145.2, 139.5, 131.8, 130.4, 130.1, 127.4, 125.3, 123.1, 122.5, 120.2, 50.1. MS (m/z, FAB+): 590 (12), 559 (15), 460 (10), 329 (30), 307 (100), 289 (53). Anal. Calcd: C, 59.01; H, 3.07. Found: C, 58.79; H, 2.96.
Synthesis of 2,2’-Dibromo-7,7’-bis(hydroxymethyl)-9,9’-spiro-bifluorene (3). A solution of diester 2 (5.9 g, 10 mmol) in THF
(150 mL) was stirred under N2and cooled to 0°C.
Diisobutyla-luminum hydride (45 mL, 1.0 M in hexane solution) was slowly added. The reaction was allowed to warm to room temperature and was stirred for another 4 h. The mixture was quenched with 10% sodium hydroxide solution (6 mL), and the precipitate was filtered out. The solvent was evaporated and then extracted with CH2Cl2.
The organic phase was collected and dried over anhydrous MgSO4.
After the solvent was evaporated in vacuo, the white solid was washed with hexane to afford white dihydroxy compound 3 (5.32 g, 86%). Mp: 276-278°C; IR (KBr): ν 3946 (m), 3761 (w), 3695 (w), 3045 (s), 2979 (s), 2693 (m), 2395 (m), 2302 (s), 1427 (s), 1248 (s), 897 (m), 724 (s) cm-1.1H NMR (CDCl 3, 400 MHz): δ 7.79 (d, J ) 8.0 Hz, 2H), 7.69 (d, J ) 8.0 Hz, 2H), 7.50 (dd, J ) 8.0, 2.0 Hz, 2H), 7.38 (d, J ) 7.2 Hz, 2H), 6.82 (d, J ) 1.6 Hz, 2H), 6.69 (s, 2H), 4.54 (s, 4H).13C NMR (CDCl 3, 100 MHz)δ 149.4, 147.4, 140.8, 139.9, 139.6, 130.8, 126.9, 126.7, 122.2, 121.2, 121.1, 119.9, 65.5, 65.1. MS (m/z, FAB+): 534 (40), 516 (50), 460 (10), 369 (5), 341 (10), 307 (100), 289 (60). Anal. Calcd: C, 60.70; H, 3.40. Found: C, 60.36; H, 3.30. Synthesis of 2,2’-Dibromo-7,7’-bis(bromomethyl)-9,9’-spirobi-fluorene (4). A solution of dihydroxy compound 3 (5.32 g, 10
mmol) in CH2Cl2(100 mL) was slowly added with PBr3(9.6 mL,
100 mmol). After being stirred 2 h at room temperature, the mixture was poured into water and extracted with CH2Cl2. The organic phase
was collected, dried over anhydrous MgSO4, then concentrated by
rotary evaporation. The solid was washed with hexane to afford white product tetrabromo compound 4 (6.27 g, 95%). MpL 267-268°C. IR (KBr): ν 3946 (m), 3767 (w), 3688 (w), 3058 (s), 2979 (s), 2700 (m), 2322 (s), 1732 (w), 1427 (s), 1275 (s), 903 (s), 718 (m) cm-1.1H NMR (CDCl 3, 400 MHz): δ 7.79 (d, J ) 8.4 Hz, 2H), 7.70 (d, J ) 8.0 Hz, 2H), 7.52 (dd, J ) 8.0, 1.6 Hz, 2H), 7.47 (dd, J ) 8.0, 1.6 Hz, 2H), 6.82 (d, J ) 2.0 Hz, 2H), 6.70 (d, J ) 1.2 Hz, 2H), 4.36 (s, 4H).13C NMR (CDCl 3, 100 MHz): δ 149.6, 147.6, 140.7, 139.8, 138.0, 131.4, 129.6, 127.2, 124.4, 121.9, 121.5, 120.5, 65.3, 33.3. MS (m/z, FAB+): 663 (10), 460 (15), 369 (20), 307 (100), 289 (50). Anal. Calcd: C, 49.13; H, 2.44. Found: C, 49.15; H, 2.52.
Synthesis of 2,2’-Dibromo-7,7’-bis(diethyl methylphosphonate)-9,9’-spirobifluorene (5). A mixture of tetrabromo 4 (0.66 g, 1 mmol)
and triethyl phosphite (1.0 mL) was heated under reflux for 8 h. The mixture was quenched with water and then extracted with CH2
-Cl2. The organic layer was collected and dried over anhydrous
MgSO4. After CH2Cl2was evaporated, excess triethyl phosphite
was evaporated at 140°C under a vacuum (0.05 Torr). The residue was allowed to cool to room temperature and purified by precipita-tion from CH2Cl2and hexane to afford white diphosphite compound 5 (0.66 g, 85%). Mp: 113-114°C. IR (KBr): ν 3940 (m), 3754 (w), 3695 (w), 3058 (s), 2985 (s), 2693 (m), 2402 (m), 2302 (s), 1427 (s), 1275 (s), 1029 (m), 903 (s) cm-1.1H NMR (CDCl 3, 400 MHz): δ 7.76 (d, J ) 7.6 Hz, 2H), 7.68 (d, J ) 8.4 Hz, 2H), 7.49 (d, J ) 7.6 Hz, 2H), 7.35 (d, J ) 7.6 Hz, 2H), 6.80 (s, 2H), 6.58 (s, 2H), 3.90-3.76 (m, 6H), 3.07-2.90 (m, 4H), 1.06 (q, J ) 6.8 Hz, 6H).13C NMR (CDCl 3, 100 MHz): δ 149.5, 147.6, 140.2, 139.1, 131.9, 131.1, 129.8, 127.0, 125.1, 121.3, 120.3, 65.2, 62.3, 62.0, 34.5, 33.1, 16.3. MS (m/z, FAB+): 776 (10), 775 (10), 663 (10), 549 (20), 327 (20), 281 (50), 207 (100). HRMS ((M + H+), FAB+): calcd for C35H3779Br79BrO6P2, 773.0432; found, 773.0421;
calcd for C35H3779Br81BrO6P2, 775.0412; found, 775.0400; calcd
for C35H3781Br81BrO6P2, 777.0391; found, 777.0406.
Synthesis of 2,2’-Dibromo-7,7’-bis(4’’-N,N-diphenylaminostyryl)-9,9’-spirobifluorene (6). A solution of diphosphite 5 (774 mg, 1
mmol) and 4-N,N-diphenylaminobenzaldehyde (546 mg, 2 mmol) in anhydrous THF (20 mL) was added to a solution of potassium
tert-butoxide (0.83 g, 10 mmol) in anhydrous THF (20 mL) at 0 °C for 16 h. The solution was extracted with CH2Cl2. The organic
layer was collected and dried over anhydrous MgSO4. After CH2
-Cl2was evaporated, the solid was washed with hexane to afford
light yellow product dibromo compound 6 (0.912 g, 90%). Mp: 325-327°C. IR (KBr): ν 3953 (m), 3761 (w), 3688 (w), 3058 (s), 2979 (s), 2680 (m), 2395 (w), 2309 (s), 1427 (s), 1255 (s), 897 (s), 738 (s) cm-1.1H NMR (DMSO-d 6, 400 MHz): δ 8.05 (d, J ) 8.0 Hz, 2H), 8.00 (d, J ) 8.0 Hz, 2H), 7.69 (d, J ) 8.0 Hz, 2H), 7.62 (dd, J ) 8.0, 1.6 Hz, 2H), 7.40 (d, J ) 7.6 Hz, 4H), 7.27 (t, J ) 8.0 Hz, 8H), 7.06-6.97 (m, 16H), 6.84 (d, J ) 8.4 Hz, 6H), 6.76 (d, J ) 1.8 Hz, 2H).13C NMR (CDCl 3, 100 MHz): δ 150.2, 148.2, 147.5, 147.4, 140.4, 139.7, 138.2, 131.3, 131.1, 129.2, 128.5, 127.3, 126.7, 126.4, 124.5, 123.2, 123.0, 121.7, 121.4, 121.3, 120.4, 65.4. MS (m/z, FAB+): 1014 (5), 732 (5), 663 (10), 549 (15), 460
(13) Oyama, M.; Nozaki, K.; Okazaki, S. Anal. Chem. 1991, 63, 1387. (14) Seo, E. T.; Nelson, R. F.; Fritsch, J. M.; Marcoux, L. S.; Leedy, D.
W.; Adams, R. N. J. Am. Chem. Soc. 1966, 88, 3498.
(15) Creason, S. C.; Wheeler, J.; Nelson, R. F. J. Org. Chem. 1972, 37, 4440.
(16) Zhang, T.; Toth, B. Anal. Chem. 2000, 72, 2533.
(17) Leung, M.-k.; Chou, M.-Y.; Su, Y.-O.; Chiang, C.-L.; Chen, H.-L.; Yang, C.-F. Yang, C.-C.; Lin, C.-C.; Chen, H.-T. Org. Lett. 2003, 5, 839.
(18) Fungo, F.; Wong, K.-T.; Ku, S.-Y.; Hung, Y.-Y.; Bard, A. J. J. Phys. Chem. B 2005, 109, 3984.
(19) Patra, A.; Pan, M.; Friend, C. S.; Lin, T. C.; Cartwright, A. N.; Prasad, P. N. Chem. Mater. 2002, 14, 4044.
(20) Chiang, C.-L.; Wu, M.-F.; Dai, D.-C.; Wen, Y.-S.; Wang, J.-K.; Chen, C.-T. AdV. Funct. Mater. 2005, 15, 231.
Downloaded by NATIONAL TAIWAN UNIV on September 2, 2009 | http://pubs.acs.org
(10), 369 (20), 307 (100). HRMS (M+, FAB+): calcd for C65H4479
-Br79BrN
2, 1010.1871; found, 1010.1821; calcd for C65H4479Br81
-BrN2, 1012.1851; found, 1012.1856; calcd for C65H4481Br81BrN2,
1014.1830; found, 1014.1860.
Synthesis of 2,2’-Dicyano-7,7’-bis(4’’-N,N-diphenylaminostyryl)-9,9’-spirobifluorene (1). A mixture of dibromo compound 6 (450
mg, 0.44 mmol) and copper cyanide (315 mg, 3.52 mmol) in DMF (20 mL) was heated under reflux for 48 h. The reaction was quenched with ammonium water (30 mL) and then extracted with CH2Cl2. The organic layer was collected and dried over anhydrous
MgSO4. After CH2Cl2was evaporated, the solid was purified by
column chromatography on silica gel (hexane/ethyl acetate ) 4/1) to afford the final product 1 as a yellow solid (160 mg, 40%). Mp: 218-220°C. IR (KBr): ν 3946 (m), 3761 (m), 3058 (s), 2979 (s), 2687 (w), 2309 (s), 1593 (w), 1420 (s), 1281 (s), 903 (s) cm-1.1H NMR (DMSO-d6, 400 MHz): δ 8.25 (d, J ) 8.0 Hz, 2H), 8.18 (d, J ) 8.0 Hz, 2H), 7.91 (dd, J ) 8.0, 1.2 Hz, 2H), 7.76 (d, J ) 8.0 Hz, 2H), 7.41 (d, J ) 8.0 Hz, 4H), 7.28 (t, J ) 8.0 Hz, 8H), 7.15 (s, 2H), 7.11 (s, 2H), 7.06-6.98 (m, 14H), 6.89 (s, 2H), 6.85 (d, J ) 8.4 Hz, 4H).13C NMR (CDCl 3, 100 MHz): δ 148.4, 148.2, 147.6, 147.3, 145.8, 139.8, 138.7, 132.6, 130.5, 129.6, 129.2, 127.5, 127.4, 127.1, 125.7, 124.6, 123.1, 122.9, 121.7, 121.6, 120.7, 118.7, 110.8, 65.2. MS (m/z, FAB+): 904.4 (40), 663.4 (100), 662.4 (60), 607.4 (10), 459.2 (10), 369.3 (5), 258.1 (20), 219.3 (10). HRMS (M+, FAB+): calcd for C67H44N4, 904.3566; found, 904.3586.
Electrochemical Characterization. Electrochemical experiments
were conducted with a potentiostat-galvanostat Autolab (Electro-chemical Instruments) employing a three-electrode cell at room temperature. The electrolyte solution as supporting electrolyte consisted of an acetonitrile (MeCN, anhydrous) solution with 0.1 M tetra-n-butylammonium perchlorate (TBAP). The working electrode consisted of a 2.16× 10-3cm2inlaid platinum disk that
was polished on a felt pad with 0.3µm alumina and sonicated in
water and absolute ethanol for 3 min each; it was then dried in an oven at 50°C. A platinum coil was used as the counterelectrode and a silver wire was used as a pseudo-reference electrode. All potential values in this study are expressed relative to a silver pseudo-reference electrode (Ag). The ferrocene/ferrocenium redox couple (Fc/Fc+) was added to the electrolyte as an internal standard and shows an oxidation potential value of 0.37 V vs Ag.
Spectroelectrochemical experiments were carried out in a homemade cell built from a commercial UV-visible cuvette. The ITO-coated glass (a piece that fit in the cuvette) was used as the working electrode, a platinum wire as the counter electrode, and an Ag wire as the reference electrode. The cell was placed in the optical path of the sample light beam in a Hewlett-Packard diode array (model 8453) spectrophotometer. The background correction was obtained by taking a UV-vis spectrum of a blank cell (an electrochemical cell with an ITO working electrode without the polymer film) with conditions and parameters identical to those of the polymer experiment.
Results and Discussion
Synthesis. The synthesis of spiro-configured D-A
mol-ecule 1 is shown in Scheme 1. Esterification of 2,2′ -dibromo-9,9′-spirobifluorene-7,7′-dicarboxylic acid21with methanol
in the presence of sulfuric acid afforded diester 2 in 90% yield. The ester groups of 2 were subsequently reduced to alcohol with diisobutylaluminum hydride (DIBAL-H) at low temperature to give dialcohol 3 in 86% yield. The hydroxy
(21) Pudzich, R.; Salbeck, J. Synth. Met. 2003, 138, 21.
Scheme 1
Downloaded by NATIONAL TAIWAN UNIV on September 2, 2009 | http://pubs.acs.org
groups in 3 were then converted to bromo groups by treating it with an excess amount of PBr3. Treatment of the resulting
tetrabromo compound 4 with triethyl phosphite afforded the diethyl phosphite compound 5 in 85% yield. The donor moiety (an N,N-diphenyl aminophenylene group) was then introduced by Wittig-Horner olefination of 4-N,N-diphe-nylaminobenzaldehyde with the diethyl phosphite compound
5 in the presence of KtOBu as a strong base. The acceptor
moieties (cyano groups) were introduced by treating the dibromo compound 6 with CuCN in refluxing DMF to give the final product in 40% yield after column chromatography purification. A similar spiro-configured bichromophore molecule 7 (Scheme 1) is used for comparison studies. The synthesis and characterization of molecule 7 (Scheme 1) have been described elsewhere.22
Photophysical Properties. The absorption and emission
spectra of spiro-configured D-A system 1 were studied in different organic solvents. Figures 1 and 2 depict the steady-state absorption and emission spectra of 1, respectively, in various solvents. The absorption peak wavelength is nearly solvent-polarity independent, indicating a small interaction between D and A in the ground state. In contrast, the emission spectra of 1 show a strong solvent-polarity depen-dence, exhibiting broad emission bands that undergo
sig-nificant bathochromic shifts as the solvent polarity increases. For example, 1 displays an emission band centered at 447 nm in cyclohexane, which is then red-shifted to as far as 546 nm in acetonitrile. In all solvents studied, the excitation spectra are effectively identical to the absorption profiles, excluding the possibility of trace impurities. On the basis of the absorption and emission spectroscopy, it is most likely incorrect to attribute the emission simply to a solvato-chromism process, in which a remarkable change of dipole moment takes place upon excitation. Alternatively, the results can be more plausibly attributed to a mechanism involving a fast photoinduced electron-transfer (PET) process, resulting in a large change of dipole moment in the excited state. Subsequently, because of the unstable solvation environment, a solvent relaxation process takes place, leading to the solvent-polarity-dependent emission.23,24
Electrochemical Characterization. Cyclic voltammetry
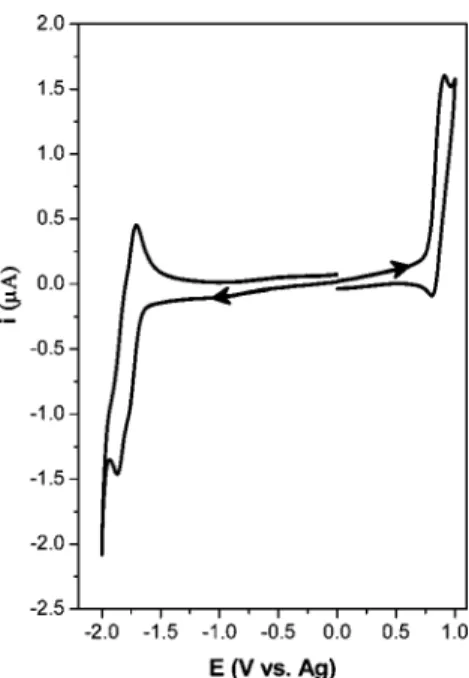
(CV) was used to obtain information about the redox behavior and the electrogenerated radical ions stability of spiro-configured D-A systems 1 and 7. The electrochemistry was studied in acetonitrile (MeCN) containing 0.1 M tetra-n-butylammonium perchlorate (TBAP) as a supporting electrolyte. The first CV of 1 obtained on a Pt electrode is shown in Figure 3. In agreement with our expectations for these kinds of molecules, the CV shows two redox responses, one of them centered around 1.0 V that is assigned to the oxidation of D units, and another centered around -1.75 V that corresponds to the reduction of A moieties. On the other hand, both cathodic and anodic processes give a very similar peak profile, and the current peaks show the same values within the experimental error. This fact suggests that equal numbers of electrons are exchanged. On the other hand,
(22) Chiang, C.-L.; Shu, C.-F.; Chen, C.-T. Org. Lett. 2005, 7, 3717.
(23) Chien, Y.-Y.; Wong, K.-T.; Chou, P.-T.; Cheng, Y.-M. Chem. Commun. 2002, 2874.
(24) Wong, K.-T.; Ku, S.-Y.; Cheng, Y.-M.; Lin, X.-Y.; Hung, Y.-Y.;
Pu, S.-C.; Chou, P.-T.; Lee, G.-H.; Peng, S.-M. J. Org. Chem. 2006, 71, 456.
Figure 1. Absorption spectra of 1 in different solvents.
Figure 2. Photoluminescence spectra of 1 in different solvents; inset shows the Lippert plots of emission peaks as a function of solvent polarities.
Figure 3. Cyclic voltammogram of 1 (7.6× 10-4M) on a Pt electrode in
MeCN with 0.1 M TBAP, V ) 0.3 V s-1, area of electrode ) 2.16× 10-3 cm2.
Downloaded by NATIONAL TAIWAN UNIV on September 2, 2009 | http://pubs.acs.org
in the reverse scan of the anodic sweep, the complementary reduction peak shows an ipbackward/ipforward relationship
less than one (ipc/ipa , 1) in the entire range of scan rate
studied (0.05-2.00 V s-1). This behavior is typical for an irreversible chemical reaction coupled to heterogeneous charge transfer.25-27 We estimated the apparent number of
electron exchanged in the overall electrode processes by comparison of the experimental current function (ψ) with model compounds.28,29The experimentalψ can be defined
asψ ) ip/(AV1/2C*), where A is the working electrode area,
in cm2, V the potential scan rate in V s-1, and C* the bulk concentration of species in M. In similar experimental conditions, ferrocene was selected as a model for one-electron exchange (ψ ) 0.73 A (V s-1)-1/2cm-2M-1), and N,N,N’,N’-tetramethyl-1,1-naphthidine as a two-electron exchange model30(ψ )1.80 A (V s-1)-1/2cm-2 M-1). We
calculated for both anodic and cathodic processes at different scan rates a value ofψ≈ 1.8, indicating that the number of electrons exchanged in the overall electrode processes is two. The electro-oxidation of TPA was extensively studied by others.31-35All those studies concluded that the radical cation
formed by oxidation is involved in a dimerization reaction that produced tetraphenylbenzidine (TPB). The electrochemi-cal processes can be described by an EC2E mechanism,36
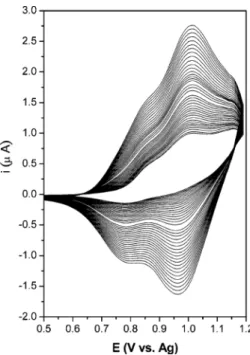
where the dimerization reaction is second order. The TPB has a lower oxidation potential than TPA because of its more-extended π-electron delocalization. However, when 1 is successively cycled between 0 and 1.2 V, the current increases, indicating the characteristic behavior of the formation of a deposited electroactive film. Typical multiple CV scans of dipolar molecule 1 on a Pt electrode are shown in Figure 4.Moreover, when a Pt electrode, which has been treated as previously described, is removed from the cell and subsequently rinsed with MeCN and transferred to a molecule
1 free-electrolyte solution, it shows the redox responses
depicted in Figure 5. It confirms that the oxidation processes of molecule 1 form an irreversibly adsorbed product on the Pt electrode surface.
Conversely, molecule 7, which has a related molecular structure, shows quite different electrochemical properties. A representative CV of 7 is shown in Figure 6, where a well-defined and reversible couple is observed at 1.1 V. The process hasψ ) 1.80 A (V s-1)-1/2cm-2M-1, corresponding
to the oxidation of the two amino moieties present in 7 at the same potential. The ipc/iparelationship is approximately
equal to 1, indicating that the intermediates produced by the oxidation processes are stable in the time scale of a CV scan. Repetitive scans between 0 and 1.2 V show the same patterns observed in the first one, being affected only by the diffusion contribution, and the formation of new peak systems was not detected. This reversibility suggests that the radical cations of molecule 7 are more stable than the ones produced from electro-oxidation of 1. We proposed that when the TPA is also a constituent of the fluorene moiety, the dimer formation (TPB) is not efficient, because of the better radical cation stabilization through the rigid coplanar fluorene rings. We observed similar stabilization behavior in related (25) Nicholson, R. S.; Shain, I. Anal. Chem. 1964, 36, 706.
(26) Nicholson, R. S.; Shain, I. Anal. Chem. 1965, 37, 178.
(27) Mastragostino, M.; Nadjo, L.; Saveant, J. M. Electrochim. Acta 1968, 13, 721.
(28) Miras, M. C.; Silber, J. J.; Sereno, L. Electrochim. Acta 1988, 33, 851.
(29) Fungo, F.; Otero L.; Durantini, E.; Thompson, W. J.; Silber, J. J.; Moore, T. A.; Moore, A. C.; Gust, D.; Sereno, L. Phys. Chem. Chem. Phys. 2003, 5, 464.
(30) Miras, M. C.; Silber, J. J.; Sereno, L. Electrochim. Acta 1988, 33, 851.
(31) Seo, E. T.; Nelson, R. R.; Fritsch, J. M.; Marcoux, L. S.; Leedy, D. W.; Adamns, R. N. J. Am. Chem. Soc. 1966, 88, 3498.
(32) Marcoux, L. S.; Adamns, R. N.; Feldberg, S. W. J. Phys. Chem. 1969, 73, 2623.
(33) Nelson, R. F.; Feldberg, S. W. J. Phys. Chem. 1969, 73, 2623. (34) Creason, S. C.; Wheeler, J.; Nelson, R. F. J. Org. Chem. 1972, 37,
4440.
(35) Nelson, R. F.; Philp, R. H. J. Phys. Chem. 1979, 83, 713. (36) Bard, A. J.; Faulkner, L. R. Electrochemical Methods: Fundamentals
and Applications; John Wiley & Sons: New York, 1980.
Figure 4. Repetitive cyclic voltammograms of the oxidation of a 1.0 mM
solution of 1 in MeCN with 0.1 M TBAP on a Pt electrode, V ) 0.1 V s-1, area of electrode ) 2.16× 10-3cm2.
Figure 5. Cyclic voltammogram of the electro-deposited film derived from
1 on a Pt electrode in MeCN with 0.1 M TBAP, V ) 0.1 V s-1, area of
electrode ) 2.16× 10-3cm2.
Downloaded by NATIONAL TAIWAN UNIV on September 2, 2009 | http://pubs.acs.org
TPA-spirobifluorene systems, for which we obtained fully reversible electro-oxidation processes.18This stability avoids
the formation of an electro-deposited film of molecule 7 when it is successively cycled.
On the other hand, it is interesting to note that in the molecules under consideration, we have two fluorene rings bonded perpendicularly through a common sp3carbon atom,
and the electron donor and acceptor are distributed in both fluorene rings (Scheme 1). This molecular geometry impedes π orbital interactions between the two rings, and the inductive effects between the rings are minimal. In consequence, the redox potentials are only slightly affected by the substituents in a neighboring fluorene ring. It is expected that the electron-transfer number observed in the CV studies is the combina-tion of the individual oxidacombina-tion and reduccombina-tion of the electron-donor and -acceptor components, respectively.18
Electrochemical Characterization of the Electrogen-erated Film. The electrochemical response of molecule 1
films shows a stable behavior for more than several hundred cycles, and the same remains unchanged after air exposure and storage. Thus, this is congruent with the fact that there are not noticeable transformations in the film. Figure 5 shows that the film has a nearly symmetrical current response. At a low scan rate, the film presents anodic and cathodic peak potential separation (∆Ep) Epa- Epc) of around 0.1 V, but at V > 0.1 V s-1,∆Epshifts to larger values, which suggests nonideal behavior.36,37 However, similar i-E profiles and
identical peak systems are obtained regardless of the CV starting potential value. When the CV experiment is initiated at 1.4 V with a cathodic scan, the CV shape is very similar to the one showed in Figure 5. This fact suggests a high reversibility in the electrochemical behavior of the film and precludes instability as an explanation for the observed nonideal behavior. Furthermore, the oxidation charge of films
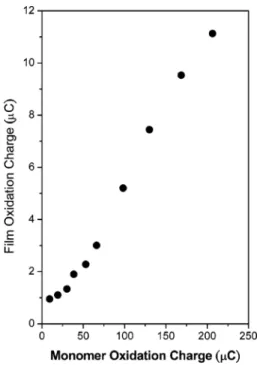
grows linearly with the monomer charge oxidations involved in the film formation, as shown in Figure 7. This observation is an indication of the good electrical conductivity of the film; we did not find a plateau or diminution in the film growing after more than 100 cycles in the film formation.
Spectroelectrochemical Characterization of the Elec-trogenerated Film. We performed a spectroelectrochemical
study on the films of molecule 1 in order to obtain information about the possible molecular structure. To do that, the film was electro-deposited on an ITO electrode, where it showed the same characteristics of those obtained over a Pt electrode. During this experiment, we found a strong electrochromic effect and clear spectral changes. Those changes occur during the redox switching between oxidized and reduced forms of the film. The results observed imply that the electro-deposited films of molecule 1 have the potential for electrochromic applications. The ITO/1 film electrode was immersed in a 0.1 M TBAP-MeCN solution and subjected to potential steps between the values where the film showed different electrochromic states. After each potential step, the current in the electrochemical system was allowed to decay to background before the optical absorption spectrum was taken. Figure 8 shows photographs of molecule
1 film in uncharged (neutral, pale yellow), half (Ox-1, red),
and fully (Ox-2, blue) oxidized states, taken under ambient (37) Brown, A. P.; Anson, F. C. Anal. Chem. 1977, 49, 1589.
Figure 6. Cyclic voltammogram of molecule 7 on a Pt electrode in MeCN
with 0.1 M TBAP. V ) 0.2 V s-1[7] ) 8.56× 10-4M, area of electrode ) 2.16 × 10-3cm2.
Figure 7. Film oxidation charge vs the anodic charge involved in the
oxidation of monomer 1.
Figure 8. Pictures of molecule 1 film on the ITO electrode at different coloration states.
Downloaded by NATIONAL TAIWAN UNIV on September 2, 2009 | http://pubs.acs.org
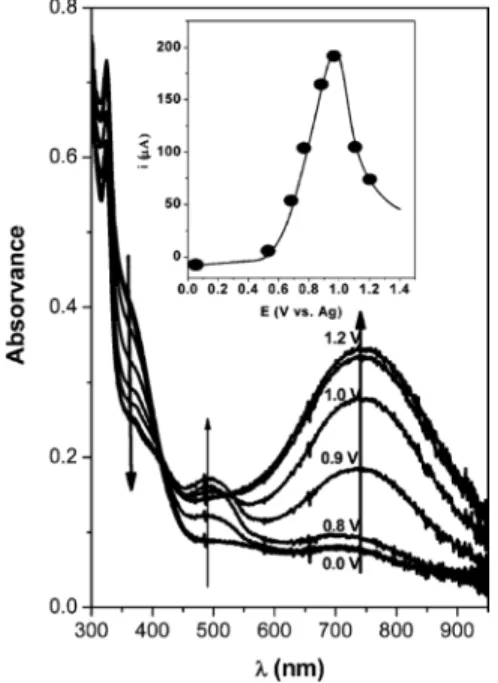
conditions. The pictures also show the experimental setup used in the spectroelectrochemical studies. The film colors are homogeneously distributed across the electrode surface, and the color changes are easily detected by the naked eye. The absorption spectra of the electro-deposited film of molecule 1 were measured as a function of applied potentials between 0.0 and 1.3 V (Figure 9). In the neutral form at 0 V, the film has a pale yellow color, with light absorption below 450 nm. As the potential increased to positive values, the intensity of the shoulder at 364 nm (that extends to the visible region) decreased. On the other hand, the growth of two new absorption bands was observed. At first, the intensity of a peak at 495 nm started to increase and reached a maximum at potential values of around 0.9 V, producing the observed red coloration of the film (Ox-1, Figure 8). The absorbance at 495 nm then started to decrease when the potentials became more anodic. Meanwhile, the intensity of the second absorption peak, with a wavelength maximum centered at 740 nm, increased as the potential was gradually turned to around 0.8 V and reached a maximum at 1.2 V. This absorption is ascribed to the fully oxidized state of the electro-deposited film of molecule 1 (Figure 8). Thus, the blue color (Ox-2, Figure 8) results from a decrease in light absorption at shorter wavelengths, associated with an increase of the broad absorption band at lower energy.
The spectroelectrochemical behavior of the electro-deposited film of molecule 1 agrees with the presence of tetraphenylbenzidine (TPB) in its molecular structure. As mentioned above, TPB is the product of the radical cation dimerization reaction of triphenylamine (TPA).13-15 The
spectroelectrochemical data suggest that the TPB moiety is the electroactive center in the film. When the TPB molecule is oxidized in solution at potentials around 0.65 V, it produces the radical cation, which has an absorption maximum at 480 nm. At more-anodic potential, the TPB dication is produced, which has an absorption band maximum at 678 nm.13The
absorption wavelength maximum of the electro-deposited
film of molecule 1 is red-shifted with respect to those of TPB in MeCN solution.13This difference could be attributed
to the different dielectric environment in film.
To obtain an electrochromic parameter of molecule 1 film, we applied potential switching steps between the reduced and oxidized states, where the absorbance changes were monitored with the time. The ITO/1 film oxidation state was switched by stepping the potential between 0.5 and 1.3 V, with time intervals of 10 s, and the current and absorbance at 740 nm was monitored versus time. As is shown in Figure 10, the current transient shapes of the oxidation and reduction current peaks are different, reaching zero in a few seconds during the potential pulses, but the charge injected in both processes are the same. Typically, in most electrochromic films, the oxidation of the neutral form (coloration step) proceeds more slowly than the reduction of the oxidized form (bleaching step) because of the difference in charge-transport rates between the two forms.38-41In general, the absorbance
switching follows the current switching, where the kinetics of the charge-transport process are reflected in the coloration response time. In the case of molecule 1 film, we estimate a response time required for 90% full-transmittance change at 740 nm of 1.7 s for the coloration step and 1.0 s for the bleaching step. On the other hand, the amount of charge (Q) in each current curve in both processes was very similar, in agreement with the stable anodic electrochemical properties of molecule 1 film. This value of the charge was used to calculate the coloration efficiency,η, using the equation42η ) ∆A(λ)/Q, where Q is the injected charge per unit electrode
area (C cm-2) and∆A is the change in absorbance during a
redox step of the device. The absorbance change at 740 nm under the same potential switching conditions as shown in Figure 10 yielded a blue coloration efficiency of
ap-(38) Panero, S.; Scrosati, B.; Baret, M.; Cecchini, B.; Masetti, E. Sol. Energy Mater. Sol. Cells 1995, 39, 239.
(39) Sapp, S. A.; Sotzing, G. A.; Reynolds, J. R. Chem. Mater. 1998, 10, 2101.
(40) De Paoli, M.-A.; Zanelli, A.; Mastragostino, M.; Rocco, A. M. J. Electroanal. Chem. 1997, 435, 217.
(41) Fungo, F.; Jenekhe, S. A.; Bard, A. J. Chem. Mater. 2003, 15, 1264. (42) Monk, P. M. S.; Mortimer, R. J.; Rosseinsky, D. R. Electro-chromism: Fundamentals and Applications; VCH: Weinheim, Ger-many, 1995.
Figure 9. Absorption spectra of the electro-deposited film of molecule 1
on an ITO electrode at various applied potentials in MeCN with 0.1 M TBAP. Inset shows the potentials at which spectra were obtained.
Figure 10. ITO/1 film electrochromic switching, current and optical
response to a potential step between 0.5 and 1.3 V applied to the electro-deposited film of 1 in MeCN with 0.1 M TBAP. Absorption was recorded at 740 nm.
Downloaded by NATIONAL TAIWAN UNIV on September 2, 2009 | http://pubs.acs.org
proximately -308 C cm-2. This value is higher than that of electrochromic cells employing polyaniline43-45 and
poly-pyrrole46 as the anodic coloring material and WO 3 as the
cathodic coloring material,42but is lower than that of dual
electrochromic cells composed with complementary elec-trochromic polymers, with total deviceη values ranging from 250 to 1413 C cm-2.39,46,47
We also evaluated the contrast ratio for both colored states from the maximum transmission change observed in Figure 10. The transmittance of molecule 1 film in the neutral state (very pale yellow) is 97% and 29.7% in the fully oxidized state (blue) at 740 nm. This gives a difference in the transmittance (∆%T) between those states of 67.3%. The
contrast achieved from the molecule 1 film is quite high when compared to other electrochromic polymer films.38-40,48-50
All these results concluded that the electropolymerized 1 films showed very good electrochromic characteristics under aerobic conditions, with a homogeneous change of color between three coloration states in a repetitive and reproduc-ible behavior.
Formation Mechanism of the Electro-Deposited Film of Molecule 1. The observed spectroelectrochemical behavior
strongly indicates the presence of a TPB moiety in the film structure and supports that the TPA radical cation dimer-ization reaction is involved in molecule 1 film formation. We propose that the oxidation of molecule 1 gives two separate triphenylamine radical cations, which dimerize rapidly to form TPB moieties. The efficient polymer forma-tion is due to the particular molecular structure of molecule
1, in which two fluorene rings holding TPA substituents are
perpendicularly bonded. This configuration precludes π-or-bital interactions between the two D-A chromophore branches,18allowing independent dimerization of both TPA
substituents to occur smoothly. A possible polymer structure is depicted in Scheme 2. This polymer structure explicates the continuous film growth and the good conductivity due to the presence of TPB, an excellent hole transporter.
Conclusions
The efficient synthetic pathways of a novel spirobifluo-rene-based donor-acceptor (D-A) system have been suc-cessfully established. The obtained bipolar molecule (1)
exhibits a fast photoinduced electron-transfer (PET) process probed by steady-state photophysical measurements. The unique molecular configuration impedes the extension of π-conjugation between two D-A branches, which allows the donors (TPA) to act independently; this leads to the formation of a stable polymer film upon electro-oxidation. The polymer film showed reversible electrochemical pro-cesses and stable color change with high coloration efficiency and contrast ratio when the polymer was switched between the neutral and oxidized states under aerobic conditions. The excellent electrochromic behavior of the film can be clearly interpreted by spectroelectrochemical studies. The spectro-electrochemical studies also agree with the TPA dimerization mechanism mainly being involved in the eletropolymeriza-tion process. Thus, a plausible polymer structure was pro-posed. The kinetics of the charge-transport process in the film have been investigated by the coloration response time, showing that the polymer films are of high interest and prom-ising potential as hole- and electron-transporting materials in organic devices. We are currently applying this polymer as an active material in organic photovoltaics and other opto-electronic devices; these results will be reported in due course.
Acknowledgment. This work is supported financially by the
National Science Council (NSC) of Taiwan (NSC94-2113-M-002-002). C.Y.L. thanks the NSC for supporting his summer research program (NSC93-2815-C-002-028M). We are grateful to Consejo Nacional de Investigaciones Cientı´ficas y Te´cnicas (CONICET-Argentina), Agencia Nacional de Promocio´n Ci-entı´fica y Tecnolo´gica (ANPCYT-Argentina), Secretarı´a de Ciencia y Te´cnica de la Universidad Nacional de Rı´o Cuarto (SECYT-UNRC), and Fundacio´n Antorchas for financial sup-port. L.O. and F.F are scientific members of CONICET. CM0605264
(43) Jelle, B. P.; Hagen, G.; Odegard, R. Electrochim. Acta 1992, 37, 1377. (44) Michalak, F.; Aldebert, P. Solid State Ionics 1996, 85, 265. (45) Panero, S.; Scrosati, B.; Baret, M.; Cecchini, B.; Masetti, E. Sol. Energy
Mater. Sol. Cells 1995, 39, 239.
(46) Arbizzani, C.; Mastragostino, M.; Zanelli, A. Sol. Energy Mater. Sol. Cells 1995, 39, 213.
(47) Rauh, R. D.; Wang, F.; Reynolds, J. R.; Meeker, D. L. Electrochim. Acta 2001, 46, 2023.
(48) Jelle, B. P.; Hagen, G.; Odegard, R. Electrochim. Acta 1992, 37, 1377. (49) Marcel, C.; Tarascon, J. M. Solid State Ionics 2001, 143, 89. (50) De Paoli, M. A.; Casalbore-Miceli, G.; Girotto, E. M.; Gazotti, W. A.
Electrochim. Acta 1999, 44, 2983.
Scheme 2
Downloaded by NATIONAL TAIWAN UNIV on September 2, 2009 | http://pubs.acs.org