國
立
交
通
大
學
生物資訊所
碩
士
論
文
正規化表示式的限制型多重序列比對之研究
On the Study of Multiple Sequence Alignment with
Regular Expression Constraints
研 究 生:李威勳
指導教授:盧錦隆 教授
正規化表示式的限制型多重序列比對之研究
On the Study of Multiple Sequence Alignment with Regular
Expression Constraints
研 究 生:李威勳 Student:Wei-Hsun Lee
指導教授:盧錦隆 教授 Advisor:Prof. Chin Lung Lu
國 立 交 通 大 學
生 物 資 訊 所
碩 士 論 文
A Thesis Submitted to Institute of Bioinformatics College of Biological Science and Technology
National Chiao Tung University in partial Fulfillment of the Requirements for the Degree of Master in
Biological Science and Technology
June 2007
Hsinchu, Taiwan, Republic of China
中文摘要
在生物資訊及計算生物領域中,多重序列比對 (Multiple Sequences Alignment) 在 發掘基因體或蛋白質序列的生物意義上是很有用的工具。通常生物學家對序列的 結構/功能/演化關係已有一些初步的認識,如活化部位的殘基、分子間的雙硫 鍵、受質結合的部位、酵素的活化性及保守性的 Motifs 等等。因此在做多重序 列比對時,生物學家希望有一個工具能讓一些結構性的/功能性的/保留性的核 甘酸或殘基可以排在一起。 2004 年 我 們 的 研 究 團 隊 已 開 發 出 一 套 限 制 型 多 重 序 列 比 對 (Multiple Sequence Alignment with Constraints) 的工具叫 MuSiC。至目前為止 MuSiC 已 被 許 多 生 物 學 家 證 實 在 生 物 的 研 究 上 是 相 當 有 用 的 。 然 而 , MuSiC 中 的 Constraint 只能是允許 Mismatches 但不能允許Gap的簡單序列片段。很多生物 重要的 Patterns 像是 PROSITE database 中的 Motifs 在 MuSiC 中是無法使用 的。因此,在此論文中我們主要的目的為研究並開發出一套能夠使用正規表示式 的限制型多重序列比對 (Multiple Sequence Alignment with Regular Expression Constraints) 的演算法與工具。
我們採用了 Progressive 的方法來解決正規表示式的限制型多重序列比對 的問題。事實上,這個方法的核心在於設計出有效率的演算法來解決正規表示式 的 限 制 型 兩 條 序 列 比 對 問 題 (Pairwise Sequence Alignment with Regular Expression Constraints Problem)。我們是將正規表示式 (Regular Expression) 轉成 有 限 狀 態 機 (Finite Automaton) , 並 使 用 Dynamic Programming 與 Divide-and-Conquer 方法來設計一個在時間與空間上都有效率的演算法來求得 最佳化的正規表示式限制型兩條序列比對。然後,我們再跟據此演算法發展出能 夠 使 用 多 個 正 規 表 示 式 的 限 制 型 多 重 序 列 比 對 工 具 : RE-MuSiC (Multiple
Sequence Alignment with Regular Expression Constraints),其網址在
ABSTRACT
Multiple sequence alignment (MSA) has received much attention in the fields of bioinformatics and computational biology because it is very useful for discovering the biological meanings of sequences. Usually, biologists may have advanced knowledge about the structural, functional, and /or evolutionary relationships about sequences of their interest, such as active site residues, intramolecular disulfide bonds, substrate binding sites, enzyme activities, conserved motifs (consensuses) and so on. They would expect an MSA program that is able to align these sequences such that the structural, functional, and/or conserved bases (i.e., nucleotides or residues) are aligned together.
In 2004, our research group has already developed a tool, called MuSiC, for multiple sequence alignment with constraint. Since then, it has been proven by many biologists to be useful in biological research. Nevertheless, the constraints allowed in MuSiC can only be simple substrings allowing mismatches but disallowing gaps in the occurrences. Many biologically important patterns such as motifs in the PROSITE database cannot be supported by MuSiC, either. Hence, in this thesis, we study and develop an algorithm and a tool for the problem of multiple sequence alignment with regular expression constraints (RECMSA).
We used a progressive approach to design an efficient program for solving the RECMSA problem. The kernel of this approach is an efficient algorithm for solving the problem of pairwise sequence alignment with regular expression constraints (RECPSA). We transform the regular expressions into a finite automaton and then use dynamic programming and divide-and-conquer approaches to develop a time and space efficient algorithm for optimally solving the RECPSA problem, which can be implemented effectively on a desktop PC with limited memory. Using this algorithm as the kernel, we developed a web-server called RE-MuSiC (Multiple Sequence Alignment with Regular Expression Constraints) that is available on-line at
誌謝
交通大學兩年忙碌的碩士生涯終於要畫下休止符了,這一路走來跌跌撞撞,嘗盡 酸甜苦辣,也因此讓我在學術研究、做人處事、產業資訊各方面能有很大的收穫 與成長。我這 EE 背景的人進入 CS 領域,並能順利完成碩士學位且找到好工 作,實在是因為威勳幸運,一路上有很多貴人的扶持與砥礪。 首先要感謝的是我最親愛的老爸、老媽、大哥和小妹,給予我幸福的家庭與 成長環境,提供我充足經濟與精神支持,讓我可以全心全力的求學而無後顧之 憂,還有每次都準備滿滿愛心的水果讓我帶回新竹。 很感謝我的指導老師盧錦隆教授,在論文的研究方向、內容、英文寫作與如 何做研究上給予我相當多的指導,並且讓我有機會與清大資工所鍾允昇學長合 作。很謝謝允昇學長在演算法、邏輯思考上給予我很大的幫助與啟發,無數次討 論和耐心溝通,讓我在這一年多來有更多的能量去克服種種難關。盧老師與允昇 學長認真、負責、嚴謹的態度,也讓 RE-MuSiC 順利發表於 NAR,與你們合 作是我的榮幸。感謝口試委員李家同、唐傳義、邱顯泰教授,從你們身上,不管 在做人處事、冷靜思考、生物知識上都讓我獲益匪淺。 感謝這兩年帶給我最多歡樂與成長的生物資訊演算法實驗室,讓我在生物資 訊所生存下來。謝謝黃彥菱學姐熱心與我分享多年程式寫作經驗與聰明工作方 法;謝謝吳家榮同學與我討論程式邏輯和讓我在很快時間就變為在地新竹人;謝 謝林光倫同學帶我寫第一支 C 程式與第一次打棒球;感謝學弟張演富在 Linux 與RE- MuSiC 的 PHP 程式上給我的協助,還有實驗室的機器管理。感謝學弟張 文勇與姜禮瑋提供研究建議與卡丁車陪伴我研究生活。另外感謝隔壁實驗室的賴 彥龍與顏士中同學,謝謝你們程式與生物上提供我諮詢,也常帶來我們實驗室許 多歡笑的種子。 此外,感謝我虎尾科技大學指導老師徐超明教授,給我資訊人應有的基本認 知,並適時給予我鼓勵與祝福。感謝財金所王淑芬教授,電信所王蒞君、陳富強教授,資工所曹孝櫟教授,清大資工所劉龍龍教授的授課;讓我在忙碌的研究生 涯中亦能接觸當前熱門產業的脈動和了解未來就業後應有的態度,這也對我在找 工作時有相當大的幫助。感謝交通大學與清華大學豐沛的學術資源和產業資訊, 讓我可以站在巨人肩上看的更遠、更清楚。 還要感謝的是我的女朋友,廖佳馨,這兩年來陪伴著我渡過每個歡笑與低 潮,更謝謝妳週末常常從台北來新竹陪我,給予我最大的動力與體諒。回憶當時, 還在中山電機所與交大生資所天人交戰,如今我已將穿碩士服畢業了!再次感謝 所有幫助過我的貴人,我會永遠記得這兩年的一切的點點滴滴。 最後,僅以最誠摯的感謝與祝福,給予威勳的所有家人、師長、實驗室的同 學、朋友們。並將取得碩士學位的喜悅與大家分享。 李威勳 2007/06 於新竹 交通大學
Contents
Chinese Abstract i Abstract ii Acknowledgement iii 1 Introduction...1 2 Preliminaries...7 2.1 Problem Formulation………...72.2 Constrained Alignment versus Weighted Finite Automaton…...8
2.3 Progressive Multiple Sequence Alignment………...……...11
3 Algorithms...14
3.1 Algorithm for RECPSA………...14
3.2 Algorithm for RECMSA………...………..………20
4 Implementation...22 4.1 RE-MuSiC...22 4.2 Usage of RE-MuSiC...22 4.2.1 Input...23 4.2.2 Output...24 4.2.3 Scoring Matrices...26 4.2.4 Gap Penalty...26
4.2.5 Syntax of Regular Expression Used in RE-MuSiC...26
5 Experiments...29
5.1 Protein Sequences with Active Site Residues...29
5.2 RNA Sequences with Phylogenetically Conserved Seudoknots...32
6 Conclusions...36
List of Figures
2.1 An illustration of a constrained alignment. Here a match has a score of 1, while all other cases are scored 0. Capital letters in the constrained alignment represent the columns responsible for the satisfactions of the constraints.. ...8 2.2 (a) Input sequences and a regular expression. (b) A is a constrained sequence
alignment. (c) A simple weighted finite automaton that can accept the
alignment A. ...10 3.1 An illustration of a constrained alignment. ...18 3.2 The matrix of weighted automata. ...18 3.3 An illustration of the weighted automaton M corresponding to the example in
Fig.3.1. State (p2, p1), etc., is denoted as p21, etc., for brevity. (a) Automaton , which is the concatenation of and . Numbers in the states are the scores , etc. Initial states of and are p
4 , 4 M M14,4 M24,4 ] [ 11 4 , 4 p W M14,4 M24,4 11 and r11,
respectively, while the final states are p22 and r22, respectively. The initial and final states of M4,4 are p11 and r22, respectively. (b) The alignment underlying
is the optimal alignment of S
] [ 22 4 , 4 r
W 1[1..4] and S2[1..4] such that state r22 is reached. ...19 4.1 The web interface of RE-MuSiC. ...24
4.2 An example of the output of RE-MuSiC for protein sequences, where the residues in the first block of columns shaded in yellow match the first regular expression of "[ST]-x-[RK]", and those in the second block of columns shaded in yellow match the second regular expression of
"G-{EDRKHPFYW}-x(2)-[STAGCN]-{P}". ……….……...25 4.3 An example of the output of RE-MuSiC for RNA sequences. Notice that a gap
appears in the block of matching the 1st regular expression constraint, which is not allowed to happen in MuSiC. ………...25 5.1 (a) Alpha class GST structure to which SjGST from non-mammalian S.
japonicum (flat worm) belongs, (b) Phi class GST structure to which AtGST
from plant A. thaliana belongs, (c) Pi class GST structure to which SsGST from mammalian S. scrofa (pig)
belongs. ………...30 5.2 The active site residues shared by all three GST proteins are marked in boxes.
The active site residues in green boxes are aligned together, but the others in red boxes are not. ……….31 5.3 The constrained sequence alignment produced by RE-MuSiC, using the pattern
of "[ST]-x(2)-[DE]" (PS00006) as the constraint, in which the residues shaded in yellow match the pattern. In addition, the residues in green boxes that correspond to the active sites shared by these three GST proteins are aligned together. ...31 5.4 Phylogenetically conserved pseudoknots in the 3'-UTRs of four coronavirus. (a)
HCoV-229E (human 229E coronavirus), (b) PEDV (porcine epidemic diarrhea virus), (c) BCoV (bovine coronavirus), (d) MHV (murine hepatitis
5.5 A partial view of the alignment produced by ClustalW, where the fragments shaded in light blue corresponds to the phylogenetically conserved pseudoknots in the 3'-UTRs of the four coronaviruses. Notably, these four shaded fragments were not aligned together. ...34 5.6 A partial view of the alignment produced by RE-MuSiC using the constraint
of "x(5)-C-U-x(4)-C-x(15,16)-U-G-x(2)-A-x(5,7)-G-x(4)-A-G-x(7,10)-U-x(3)- A-x(5)", where the fragments shaded in yellow, corresponding to the
phylogenetically conserved pseudoknots in the 3'-UTRs of the four
coronaviruses, are aligned together. ...34 5.7 The consensuses adapted from [28], which was derived by Williams et al.
from the 3'-UTRs of various coronaviruses, including HCoV-229E, PEDV, BCoV and MHV. ...35
Chapter 1
Introduction
Multiple sequence alignment (MSA) is one of the fundamental problems in bioinformatics and computational biology that have been studied extensively, because it is a useful tool in the phylogenetic analyses among various organisms, identification of conserved motifs and domains in a group of related proteins, secondary and tertiary structure prediction of a protein/RNA and so on [8, 9, 19, 36, 37]. The sum-of-pairs score is widely used for selecting an optimal MSA. This kind of MSA problem, called sum-of-pairs MSA (SPMSA) problem, can be solved by extending the dynamic programming algorithm of Needleman and Wunsch for aligning two sequence [38]. In the worst case, it needs to take time to align k sequences of length n. This exponential time limits the dynamic programming technique to align only a small number of short sequences. Actually, the SPMSA problem has been shown to be NP-complete [7, 53], which means that it seems to be impossible to design an efficient algorithm to find the mathematically optimal alignment. Hence, some approximate and heuristic methods are introduced to overcome this problem. For the approximate methods, Gusfield [18] first proposed a polynomial time approximation algorithm with performance ratio of
) 2 ( knk O k 2
performance ratio to
k
3
2− . Recently, Bafna, Lawler and Pevzner [6] further
improved the performance ratio to
k l
−
2 for any fixed l. It is worth mentioning that Li, Ma and Wang [26] have given a polynomial time approximation scheme for finding a multiple sequence alignment within a constant band, which is often useful in many practical cases. For the heuristic methods, the most widely used heuristic methods are so-called progressive strategies [13, 17, 21, 49, 50].
Standard multiple sequence alignment is based solely on the information about the residues/nucleotides constituting the sequences. In addition to merely the residues/nucleotides, however, biologists often possess more knowledge regarding function, structure or conserved patterns of the sequences to be analyzed. It is generally desirable to have such information incorporated into an alignment procedure, so that the alignment result can be more biologically meaningful. For example, functionally important sites are generally expected to be aligned together, but a typical alignment tool often fails to achieve this if the sequence similarity is low. Imposing constraints representing such information turns out to be an effective manner to incorporate biological knowledge into an alignment tool.
Motivated by such demand, Tang et al. [48] formulated the constrained multiple sequence alignment problem, where each constraint is a single residue/nucleotide. They considered alignment of RNase sequences, which are known to have a sequence of conserved residues His (H), Lys (K), and His. Using H, K, H as constraints, in the resulting constrained alignment each of these three residues can be found aligned together in a column of the alignment, appearing in the order as specified. Chin et al. [11] then proposed an improved algorithm for pairwise alignment and an approximation algorithm for multiple alignment. It is also noted that there have been other formulations regarding alignment with constraints proposed from different
perspectives with various approaches [15, 29-35, 44, 45, 51].
Conserved sites of a protein/RNA/DNA family are often of several residues/nucleotides long. For these patterns, the original formulation in [48] is not expressive enough. In addition, such patterns may not appear in the exact form in general. Consequently, Tsai et al. [52] proposed a generalized formulation and algorithm, where each constraint is a (usually short) string pattern allowing mismatches. Lu and Huang [28] then proposed a space efficient algorithm for this formulation. Web-based systems, MuSiC [52] (available at http://genome.life.nctu.edu.tw/MUSIC) and MuSiC-ME [28] (available at http://genome.life.nctu.edu.tw/MUSICME), were also developed; from now on these two systems will be referred to as MuSiC jointly. With the aid of MuSiC, Tsai et al. [52] and Lu and Huang [28] successfully identified a fragment in the 3’ untranslated region (3’-UTR) of a SARS (severe acute respiratory syndrome) coronavirus sequence that can fold into a pseudoknot, which is potentially responsible for self-replication of the virus. Indeed, since its release, MuSiC has been found useful in, e.g., detection of functionally and/or structurally important residues/motifs in sequences [10, 48], prediction of RNA pseudoknotted structures [23, 41, 52], prediction of protein structures [16], and so on.
There are, however, formulations of many biologically significant patterns beyond the capability of MuSiC. For example, many function-related protein sites as those collected in the PROSITE database [24] are expressed in regular expressions, which cannot be modeled using the substring-with-mismatch formulation of constraints implemented in MuSiC. An example of regular expression patterns is the EGF-like domain signature 2 (EGF_2, PS01186 in PROSITE): C-x-C-x(2)-[GP]-[FYW]-x(4,8)-C, which is related to the initiation of a signal transduction that results in DNA synthesis and cell proliferation. The meaning of this
pattern is that, the first residue is Cys, followed by one residue of any kind, then a Cys, followed by two residues of any kind, then a Gly or Pro, etc. Regular expressions are also convenient in describing variable ranges between patterns or between blocks within a pattern, which is necessary for some single patterns themselves, and useful in applications where different patterns are expected to exhibit proximity in their occurrences. In the above example of EGF_2, the “x(4,8)” symbol preceding the last Cys indicates a range of length varying from 4 to 8 between a residue of [F, Y or W] (Phe, Tyr or Trp) and that last Cys.
Due to the usefulness of regular expressions in describing biological patterns, Arslan formulated the problem of Regular Expression Constrained Sequence Alignment (RECSA for short) [1]. A feasible solution of RECSA is an alignment containing a run of contiguous columns such that both of the two substrings corresponding to these columns match the regular expression. The following example, constructed by Arslan, clearly indicates the difference between RECSA and a standard unconstrained alignment:
where a match of identical symbols is scored 1 and all other cases are scored 0. The alignment shown left is an optimal alignment without constraint, while that shown in the right is an optimal constrained alignment. The constraint R is [GA]-x-x-x-G-K- [ST], the P-loop motif. The starred columns “support” the satisfaction of constraint R: both GFPSVGKT and AKDDDGKS match R. Later, Chung et al. [12] proposed more time and space efficient algorithms for this problem, which, unlike Arslan’s algorithm, is capable of reconstructing the optimal alignment.
The above mentioned formulation of RECSA is for pairwise alignment with single regular expression constraints. In practice, however, it is more useful to be able to support multiple alignment with multiple constraints. In [2] Arslan extended the algorithm in [1] to support multiple alignment with multiple constraints. The algorithm proposed in [2] computes mathematically optimal constrained alignments. Unfortunately, the time complexity is extremely high, involving an exponential multiplicative factor in addition to the exponential time complexity for optimal (unconstrained) MSA computations. Even for pairwise alignment with multiple constraints, its worst case time and space requirements are intensive. In addition, the algorithms in [2] cannot find in the resulting alignment the regions responsible for the satisfactions of the constraints, either; only the alignment score, without the alignment itself, is reported. But being able to report alignments is important for practical purposes. It is therefore necessary to propose a solution more suitable for practical applications.
In this thesis, we extend the algorithm in [12] to support multiple constraints and multiple sequences. The resulting algorithm is more efficient than the one in [2] for pairwise alignment with multiple constraints. To deal with multiple sequences, a progressive method is implemented, using our improved pairwise algorithm as the kernel. This extended algorithm turns out to be more appropriate for applications than the one in [2], and we implemented a web server, RE-MuSiC (Multiple Sequence Alignment with Regular Expression Constraints), based on this algorithm. Experiments on GST proteins and on coronaviruses with phylogenetically conserved pseudoknots demonstrate that, with additional knowledge incorporated, RE-MuSiC is able to produce meaningful alignments in which important residues or structural elements can be aligned properly, even if the similarity among input sequences is low. Such ability is also useful for prediction purposes.
The rest of this thesis is organized as follows. In Chapter 2, we give the formal definition of the RECMSA (Multiple Sequence Alignment with Regular Expression Constraints) problem we study in this thesis, and also introduce weighted finite automata. In Chapter 3, we first use the dynamic programming technique to design a time-efficient algorithm for optimally solving the RECPSA (Pairwise Sequence Alignment with Regular Expression Constraints) problem. In addition, we show how to find in the resulting alignment the regions responsible for the satisfactions of the constraints, and then reconstruct the constrained sequences alignment in a space efficient manner using the divide-and-conquer approach. Based on this algorithm, we developed a program able to support multiple constraints and multiple sequences, called RE-MuSiC, using the progressive approach. In Chapter 4, we introduce the RE-MuSiC implementation and user interface. In Chapter 5, we demonstrate the applicability of our developed programs by testing them on a data set of protein and RNA sequences. Finally, we make some conclusions in Chapter 6.
Chapter 2
Preliminaries
In this chapter, we shall first formulate the problem of multiple sequence alignment with regular expression constraints. We shall then introduce the concept and basics of weighted finite automaton we use to design an efficient algorithm for the pairwise sequence alignment with regular expression constraints. Finally, we shall describe the so-called progressive approach, we use to design a heuristic algorithm for the constrained multiple sequence alignment.
2.1 Problem Formulation
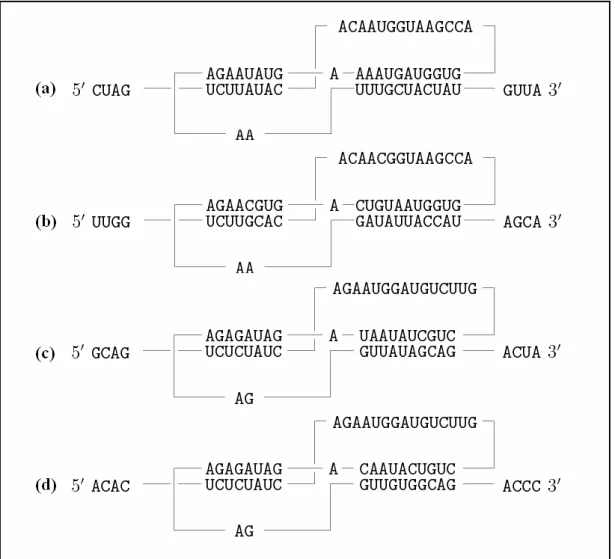
Given σ sequences S1, S2,. . . , Sσ over alphabet Σ and a sequence of m regular expression constraints R1, R2 . . . , Rm, is a regular expression, the problem of the so-called multiple sequence alignment with regular expression constraints (RECMSA) is to find an alignment with the highest possible score such that all the constraints are satisfied. An alignment A of S1, S2,. . . , Sσ is said to satisfy all the constraints if in A there exist m regions with the following property. Let the jth region, corresponding to the jth constraint, be composed of consecutive columns kj, kj+1, . . . , kj′. It is required that the jth region precedes the (j+1)st region without overlapping and the substring of each Si in the jth region matches the regular expression Rj. For example, suppose we
are given two sequences S1=cgacgta, S2=acgcgta, as well as two regular expression constraints R1=a and R2=t. Then, Figure 2.1(c) shows an optimal constraint alignment in which there are two regions (i.e., 3rd and 8th columns, respectively) whose corresponding substrings of S1 and S2 match R1=a and R2=t, respectively.
(a)
INPUT
S1
= cgacgta, S2
= acgcgta
R1
= a, R2
= t
(b) OPTIMAL UNCONSTRAINED ALUGNMENT
- c g a c g t a
a c g – c g t a
(c) OPTIMAL CONSTRAINED ALUGNMENT
c g A c g -- T a
-- A c g c g T a
Figure 2.1: An illustration of a constrained alignment. Here a match has a score of 1, while all other cases are scored 0. Capital letters in the constrained alignment represent the columns responsible for the satisfactions of the constraints.
2.2 Constrained Alignment versus Weighted Finite
Automaton
Currently, it is still a challenge to design a polynomial-time algorithm for solving the RECMSA problem, because the problem of multiple sequence alignment that can be considered a special case of RECMSA without any given constraint has been proven to be NP-hard [7, 53]. However, the pairwise version of RECMSA, simply denoted by RECPSA (Pairwise Sequence Alignment with Regular Expression Constraints), is a tractable problem, because Arslan [1] first designed a polynomial-time algorithm whose time and space complexities were further improved by Chung et al. [12].
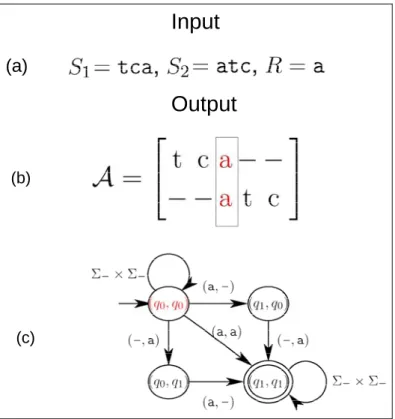
The basic idea of Arslan’s algorithm is to construct a weighted automaton that can be utilized to recognize any pairwise alignment of satisfying a required constraint.
For example, the automaton M in Figure 2.2(c) is able to recognize the pairwise alignment, as shown in Figure 2.2(b), which satisfies the given regular expression by the following steps. Initially, the automaton M is in the state (q0, q0). After reading the first two columns of A that are deletion pairs (t, -) and (c, -), M still stays in the state (q0, q0). When M reads the third column of A that is a match pair (a, a), it enters the state of (q1, q1) that is a final state of M. After reading the last two columns that are insertion pairs (-, t) and (-, c), M remains in the final state. In other words, M can enter and stay a final state from its initial state if the input pairwise alignment satisfies the given regular expression. Moreover, any non-initial state in M implies that the given regular expression is partially or completely satisfied by the input alignment. Clearly, there should be many satisfied alignments that can be accepted by M. Recall that the objective of the RECPSA problem is to find a satisfied alignment with maximum alignment score. For this purpose, a weight is assigned to each state in M for remembering the alignment that best satisfies the partial or complete constraint of regular expression. In the following, we shall formally describe how to reconstruct such a weighted automaton for recognizing the best pairwise alignment of satisfying a regular expression constraint.
Give a regular expression R, let N = (Q, Σ, δ, q0, F) be an ε-free NFA (nondeterministic finite automaton) equivalent to R, which can be constructed manually or by any established algorithm [22]. We also define δ(Q,'a) to be
, where and
U
p∈Q'δ(p,a) Q'⊆Q a∈Σ . In this thesis we use Q and Vinterchangeably. Following the notations in [23], we define a weighted automaton N
× N as the finite automaton M = (QM, WM, ΣM, δM, q0M, FM) which we construct as
Input
Output
(a)(b)
(c)
Figure 2.2: (a) Input sequences and a regular expression. (b) A is a constrained sequence alignment. (c) A simple weighted finite automaton that can accept the alignment A.
– QM = Q × Q is the set of states. Each state of M corresponds to a pair of states
in N. M remembers in each state what part of the regular expression has been seen in S1 and S2.
– WM : QM →ℜis a function that assigns real weights to each state in QM, and
initially all weights are -∞. We determine the active set of states of M by examining their weights. The active states of M have weights different than -∞.
– ΣM = (Σ × Σ) - {ε→ε}. The alphabet ΣM for M is the set of edit operations
which does not include ε→ε.
– q0M = (q0, q0)is the start state whose initial weight is 0.
– FM = F × F is the set of final states. If M is in a final state then M has processed
an alignment that satisfies the regular expression constraint. That is, there are substrings s1 of S1 and s2 of S2 that are aligned together in an alignment, and
both s1 and s2 take N to final states.
– δM is a transition function of M that
( ) ( )
(
)
( ) ( )
( ) ( ) ( )
{
}
⎩ ⎨ ⎧ ∪ × × = q p b q a p b q a p b a q p M , , , , , , , , δ δ δ δ δ( )
({
}
. , , 0 0 otherwise q q F q p if M U ∉)
2 1Function δM can be naturally extended to be defined on the cross product of QM
and the set of all possible alignments. A state (p,q) of is active iff there exists
some alignment A of S
2 1,i
i
M
1[1..i1] and S2[1..i2] such that . Each state (p,q) in has a score (p,q) assigned to it. If (p,q) is active, then (p,q)
is the score of an optimal alignment A of S
) , ( ) , (p q ∈δM q0M A 2 1,i i M 2 1,i i W 2 1,i i W
1[1..i1] and S2[1..i2] such that(p,q)∈δM(q0M,A). Otherwise no such alignment exists and (p, q) = -∞.
,i i
W
2.3 Progressive Multiple Sequence Alignment
The progressive approach is one of the widely used heuristics for efficiently finding a good MSA of several sequences. The ideas behind it are as follows [13, 17, 21, 49, 50].
1. Compute the distance matrix by aligning all pairs of sequences: Usually, this distance matrix is obtained by applying FASTA [27, 39] or the dynamic programming algorithm of Needleman and Wunsch [38] to each pair of sequences.
2. Construct the guide tree from the distance matrix: For the existing progressive methods, they mainly differ in the method used to build the guide tree for directing the order of alignment of sequence. To build the guide tree, for example, PILEUP (a program of GCG packages) uses UPGMA (Unweighted
Pair-Group Method using Arithmetic mean) method [46] and CLUSTAL W [50] uses NJ (Neighbor-Joining) method [43].
3. Progressively align the sequences according to the branching order in the guide tree: Initially, the closest two sequences in the tree are aligned using the normal dynamic programming algorithm. After aligning, this pair of sequences is fixed and any introduced gaps cannot be shifted later (i,e., once a gap, always a gap). Then the next two closest pre-aligned groups of sequences are joined in the same way until all sequences have been aligned. (Here, we may consider a sequence as an aligned group of a sequence.) To align two groups of the pre-aligned sequences, the score between any two positions in these two groups is usually the arithmetic average of the scores for all possible character comparisons at those positions. We call this kind of scoring methods as a set-to-set scoring.
In fact, MST has been used as a significant tool for data classification in the fields of biological data analysis. In [48], Tang et al. proposed a variant of progressive method by using the Kruskal MST to construct the guide tree, called Kruskal merging order tree. The Kruskal merging order tree of k sequences is constructed as follows. First, we create a complete graph G = (V,E) of k sequences in a way that each vertex of V represents a sequence and each edge e of E is associated with a weight d(e) to represent the distance between the corresponding sequences of its end-vertices. Then we use the Kruskal’s algorithm [25] to construct the Kruskal MST of G, denoted by
T .For completeness, we describe the Kruskal method for constructing T as follows.
2. Initially, T is empty. Then we repeatedly add the edges of E in non-decreasing order to T in a way that if the currently adding edge e to T dose not create a cycle in T , then we add e to T ; otherwise, we discard e.
Next, according to the Kruskal MST T , we build the Kruskal merging order tree
TK as follows.
1. Let V = {v1, v2, . . . , vk} and e1, e2, . . . , ek-1 be the edges of T with d(e1) ≦
d(e2) ≦ . . . ≦ d(ek-1).
2. For each vertex vi∈V , we create a tree Ti such that Ti contains only a node vi. For the purpose of merging trees, we consider Ti as a rooted tree by designating vi as its root, and define the merge of two tree Ti and Tj respectively rooted at vi and vj to be a new tree rooted at a new vertex u such that vi and vj become the children of u.
3. For each ek = (vi, vj), where K increases from 1 to k - 1, we find the trees Ti and
Tj containing vi and vj respectively and then merge them into a new tree. This process is continued until the remaining is only one tree. Then this final tree is the so-called Kruskal merging order tree TK.
The construction of G for k sequences can be done in O(k2) time and the computation of the Kruskal’s MST T of G can be done in O(k2 log k) time. Then the construction of TK from T can be implemented by the disjoint set union and find algorithm proposed by Gabow and Tarjan [33] in O(m + k) time, where m denotes the number of union and find operations. It is not hard to see that m = O(k) and hence the construction of TK takes O(k) time. Therefore, the total time complexity of constructing TK is O(k2log k).
Chapter 3
Algorithm
In this chapter, we shall propose an algorithm to solve the problem of pairwise sequence alignment with several regular expression constraints (RECPSA). Then, we shall present how to extend this algorithm for dealing with multiple sequences with several regular expression constraints (RECMSA).
3.1 Algorithm for RECPSA (Pairwise Sequence Alignment
with Regular Expression Constraints)
First consider the case of pairwise alignment. For simplicity let the two input sequences have equal lengths of n. Let Ah =(Qh,Σ,δh,qh,Fh) be an ε-free NFA equivalent to Rh (qh is the initial state of Ah), and
be the weighted automaton corresponding to
A ) , , , , , ( h h h h 0 h h M M M M M M h Q W q F M = Σ δ h, where M h h , Q Q Q h = × ΣMh =(ΣU
{ }
−)×(ΣU{ } ( )
− )\{
−,−}
, ,, and, for (p,q)∈ and (a,b)
(
h h)
M q q q h = × 0(
h h M F F F h = ×)
QMh ∈Σ , Mh( ) ( )
(
)
( )
( )
( )
( ) ( )
{
}
⎩ ⎨ ⎧ ∪ × × = q p b q a p b q a p b a q p h h h h Mh , , , , , , , , δ δ δ δ δ(
)
{ }
otherwise q F q p if Mh Mh 0 , ∉ UOn each pair (i1, i2) of indices of the sequences, denote as the corresponding weighted automaton. Also let M be an NFA obtained by “concatenating” M
2 1,i i h M 1, . . . , Mm
by adding an “empty move” from each final state of Mh to the initial state of Mh+1, 1 ≤
h < m. The initial state of M is set to be the initial state of M1, and the final states of M are the final states of Mm. Hence M accepts all feasible constrained alignments (see Fig. 3.3 for an example). The score held in state (p, q) of (resp. ) is denoted
as (resp. ). The score represents the score of a
best alignment A of S 2 1,i i h M Mi1,i2 ) , ( 2 1, p q Whi i Wi1,i2(p,q) Wi1,i2(p,q) h
1[1..i1] and S2[1..i2] such that all of R1, . . . ,Rh-1 are satisfied and that state (p, q) of Mh is reached if A is given to Mh as input. It is equal to , the score of optimally aligning S
) , ( 2 1, p q Wi i
1[1..i1] and S2[1..i2] such that the alignment can lead us from the initial state of to state (p,q). The goal of the algorithm is to
compute , since is the optimal score of aligning S ) , ( 2 1, p q Mi i n n m W , max(p,q) F F Wmn,n(p,q) m m× ∈ 1 and
S2 with all m regular expression constraints satisfied.
The algorithm iterates over all pairs (i1, i2) of indices of sequences S1 and S2, row by row. It computes i1,i2for all 1 ≤ i
h
M 1, i2 ≤ n and 1 ≤ h ≤ m throughout its execution. Let Vh and Eh be the set of states and set of arcs in automaton Ah, respectively. Denote as i1,i2 a |V
h
L h| × |Vh| table used to hold i1,i2. On each sequence index pair (i
h
W 1, i2), the
algorithm computes i1,i2, . . . , , in the order. For all (p, q) h
L i1,i2
m
L ∈ , [p,q] is
computed using the (first) algorithm Chung et al. proposed in [12]. The same is performed for , h > 1, except that [q
1 M Q 1,2 1 i i L 2 1,i i h L i1,i2 h
L h,qh] is initialized to the maximum of [p, q] rather than -∞ as the other states are, the maximum taken over all final states (p, q) of M 2 1, 1 i i h L − h-1.
The above procedure takes ⎟
⎠ ⎞ ⎜ ⎝ ⎛
∑
= m h h h V n E O 12 time, since each i1,i2 takes
h L
(
EhVhalignment in ⎟ ⎠ ⎞ ⎜ ⎝ ⎛
∑
= m h h n V h O 1 2space without affecting the time complexity, by a generalization of the method Chung et al. proposed in [12].
Consider an optimal alignment A satisfying all constraints. Intuitively, if we know the substrings of S1 and S2 responsible for A’s satisfaction of the constraints, then A (or another optimal solution) can be reconstructed efficiently. Suppose that the substrings of S1 and S2 responsible for A’s satisfying the lth regular expression are
[
l l]
e b
S1 1L 1 and S2
[
b2lLe2l]
, 1≤l≤m . Then we can align S1[
b1lLe1l]
and[
l l]
e b
S2 2L 2 for all l = 1,…,h, align S1
[
1Lb11−1]
and S2[
1Lb12−1]
,align
[
1 1 1]
1 1 1 + + − l l b eS L and S2
[
e2l +1Lb2l+1−1]
for l = 1, 2, . . . , m-1, andalignS
[
em n]
L 1 1 1 + and S[
e n]
m L 1 22 + . These alignments can be concatenated in the proper order to obtain A. Each alignment can be computed in linear space by Hirschberg’s celebrated divide-and-conquer algorithm [14]. In the example of Fig.3.1,
, , , , , , , .
1 1 1 =
b e11 =1 b12 =1 e12 =1 b12 =3 e12 =3 b22 =2 e22 =2
Two types of |Vh|×|Vh| tables, 1,2 and , , i i h l β 1,2 , i i h l
η 1≤l ≤h, are maintained for this
purpose. Let A be the alignment underlying the score . Then keeps the indices of S
) , ( 2 1, p q Whi i li1h,i2
[ ]
p,q , β1 and S2 corresponding to the column immediately before the first column of leaving the initial state of Ml. If l= h (resp. l<h), then keeps the indices of S1 and S2 corresponding to the first column of A which leads us to arrive at state (p, q)(resp. a final state) of Ml. It can be seen that, if (p, q) is the final state of
with the best value, then n
n m
M , Wmn,n(p,q) βln,m,n
[ ]
p,q stores and ,and stores and , where the meanings of these b and e values are as discussed in the last paragraph.
1 1 − l b b2l −1
[
p q n n m l , , , η]
l e1 el2 11 When (p, q) is not a final state, the values of [ ]
q p i i h h , 2 1, ,
η are are actually not relevant (but the values are still critical).
[
p q i i h h , 2 1, , β]
The computation of and , h=1,…, m, proceeds in the same manner as in [12]. 2 1, , i i h h β 1,2 , i i h h η 2 As to
[
and h h i i h l q ,q 2 1, , β]
i i[
h h]
h l q ,q 2 1, , η , l ≠ , we initialize them to hand , respectively, where (p, q) is the final state of with the best
score among all final states. In general, when
[ ]
p q i i h l , 2 1, 1 , − β[
p q i i h l , 2 1, 1 , − η]
1,2 1 i i h M − ) , ( 2 1, 1 p q Whi−i l≠ , for (initial or hnon-initial) state (p,q), each time when Lih1,i2
[ ]
p,q is updated to, say,where
[
p q]
(
x y Lih1',i2' ,' ' +γ ,)
( )
,{
(
1, 1) (
, 1,) (
, , 1)
}
2 1 2 1 2 1 ' 2 ' 1 i ∈ i − i − i − i i i −i and (x,y) is the
corresponding edit operation (e.g.,(x, y) = (S1[i1],-) if and ),
and are also updated to
1 1 ' 1 = i − i i2' =i2
[
p q i i h l , 2 1, , β]
li1h,i2[
p,q]
, η 1','2[
,' ']
, p q i i h l β and respectively.[
,' ']
' 2 ' 1, , p q i i h l ηWhen row i1 is being considered, all tables for rows less than i1 are not necessary and can be discarded. Therefore the overall space requirement, including the reconstruction of the optimal alignment, is ⎟
⎠ ⎞ ⎜ ⎝ ⎛
∑
= m h h n V h O 1 2 .In [2], an algorithm extending the one in [1] to support multiple constraints and multiple sequences, is proposed. In the pairwise case, the algorithm in [2] has time
complexity ⎟ ⎠ ⎞ ⎜ ⎝ ⎛
∑
= m h h n E O 1 2 2and space complexity ⎟
⎠ ⎞ ⎜ ⎝ ⎛
∑
= m h h n E O 1 2 . It is clear that(
h h O EV =
)
, hence the algorithm presented here never takes more time than the onein [2]. In the worst case, Eh can be proportional to Vh2, and the algorithm in [2]
takes ⎟ ⎠ ⎞ ⎜ ⎝ ⎛
∑
= m h h n V O 1 2 4time, while the one presented here takes ⎟ ⎠ ⎞ ⎜ ⎝ ⎛
∑
= m h h n V O 1 2 3 time.Hence the time complexity of the algorithm presented here compares favorably with
2If (p, q) is not a final state, then the value held in [ ]
q p i i h h , 2 1, ,
η may not be as described in the last paragraph. But as just mentioned, this does not affect the correctness.
the one in [2]. As to the space complexity, if Eh =O
( )
Vh , then the algorithm heremay take more space than the one in [2]. However, if ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ Ω =
∑
∑
= = m h h m h h hV E 1 1 2 , then thealgorithm here is more space efficient. More importantly, the stated space complexity for the algorithm in [2] does not include the reconstruction of the alignment; only the alignment score can be computed. It is clearly an important issue for a web-server to report the alignment. If a naive backtracking method is used to augment the algorithm in [2] to reconstruct the alignment, the space requirement would be ⎟
⎠ ⎞ ⎜ ⎝ ⎛
∑
= m h h n E O 1 2 2 ,which is too high for practical use.
Now, we apply this algorithm to solve the following example.
Input Scoring matrix
S1 = acgt, S2 = atcg Match:3
R1 = a, R2 = t Mismatch:-1 Gap=-2 Optimal unconstrained alignment
a – c g t a t c g –
Optimal constrained alignment A c g T
-A -- T c g
Figure 3.1:An illustration of a constrained alignment.
t
g
c
a
S1
g
c
t
a
S2
0 , 0M
M
0,1M
0,2M
0,3 0 , 1M
M
1,1M
1,2M
1,3 0 , 3M
M
3,1M
3,2M
3,3 0 , 2M
M
2,1M
2,2M
2,3 0 , 4M
M
4,1M
4,2M
4,3 4 , 1M
4 , 1M
4 , 3M
4 , 2M
4 , 4M
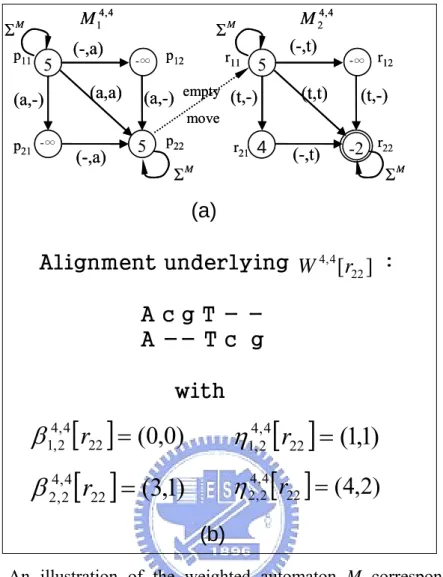
Figure 3.2:The matrix of weighted automata.5 5 -∞ -∞ 5 -2 -∞ 4 r12 r22 r21 r11 p12 p22 p21 p11 (a,-) (a,-) (-,a) (-,a) (a,a) (t,-) (t,-) (-,t) (-,t) (t,t) M Σ M Σ ΣM M Σ 4 , 4 2 M 4 , 4 1 M empty move 5 5 -∞ -∞ 5 -2 -∞ 4 r12 r22 r21 r11 p12 p22 p21 p11 (a,-) (a,-) (-,a) (-,a) (a,a) (t,-) (t,-) (-,t) (-,t) (t,t) M Σ M Σ ΣM M Σ 4 , 4 2 M 4 , 4 1 M empty move
(a)
Alignment underlying :
[
]
22 4 , 4r
W
A c g T
-A -- T c g
with
[ ]
22(
0
,
0
)
4 , 4 2 , 1r
=
β
[ ]
22(
3
,
1
)
4 , 4 2 , 2r
=
β
[ ]
22(
1
,
1
)
4 , 4 2 , 1r
=
η
[ ]
22(
4
,
2
)
4 , 4 2 , 2r
=
η
(b)
Figure 3.3: An illustration of the weighted automaton M corresponding to the
example in Fig.3.1. State (p2, p1), etc., is denoted as p21, etc., for brevity. (a) Automaton , which is the concatenation of and . Numbers in the
states are the scores , etc. Initial states of and are p 4 , 4 M M14,4 M24,4 ] [ 11 4 , 4 p W M14,4 M24,4 11 and r11,
respectively, while the final states are p22 and r22, respectively. The initial and final states of 4,4 are p
M 11 and r22, respectively. (b) The alignment underlying is the optimal alignment of S
] [ 22 4 , 4 r W
3.2 Algorithm for RECMSA (Multiple Sequence Alignment
with Regular Expression Constraints)
In this section, we use Algorithm RECPSA in previous section as kernels to design an RECMSA algorithm, for progressively aligning the input sequences into a RECMSA according to the branching order of a guide tree, where the guide tree we use here is the so-called Kruskal merging order tree. We refer the reader to Section 2 for the details of constructing the Kruskal merging order tree. Except for the adopted RECPSA kernel, the execution steps, which are described as follows.
1. Compute the distance matrix D by globally aligning all pairs of sequences using Algorithm RECPSA, where D(i, j) denotes the distance between sequences Si and Sj .
2. Create a complete graph G from the distance matrix D and then compute the Kruskal merging order tree Tk from G to serve as the guide tree.
3. Progressively align the sequences according to the branching order of the guide tree Tk in a way that the currently two closest pre-aligned groups of sequences are joined by applying Algorithm RECPSA to these two groups of sequences, where the score between any two positions in these two groups is the arithmetic average of the scores for all possible character comparisons at those positions.
In the following, we analyze the time complexity of Algorithm RECPSA. It is not hard to see that step 1 costs O(k2n2) time, where n is the maximum of the lengths of k
sequences. According to the paper of Tang et al. [48] , step 2 can be done in O(k2log k)
time. In step 3, there are at most O(k) iterations for calling Algorithm RECPSA, whose time complexity is ⎟
⎠ ⎞ ⎜ ⎝ ⎛
∑
= m h h n V O 1 2 3Hence, the time complexity of step 3 is ⎟ ⎠ ⎞ ⎜ ⎝ ⎛
∑
= m h h kn V O 1 2 3 . It is usually that ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ =∑
= m h h V O k 1 3, hence the cost of Algorithm RECMSA is dominated by step 3 and
hence its time complexity is ⎟ ⎠ ⎞ ⎜ ⎝ ⎛
∑
= m h h kn V O 1 2 3 .Chapter 4
Implementation of RE-MuSiC
In this chapter, we shall introduce the implementation of RE-MuSiC, as well as its web interface, and then describe how to use it in details. Besides, we shall introduce the syntax of regular expression utilized in RE-MuSiC.
4.1 RE-MuSiC
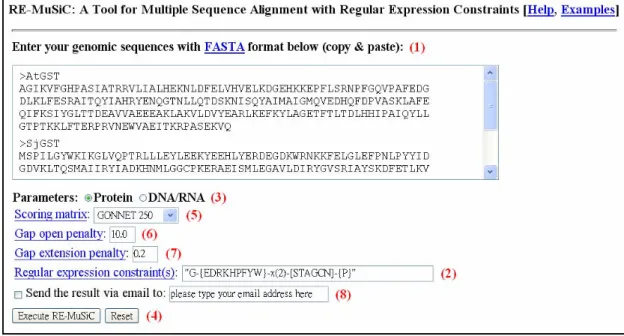
The kernel of RE-MuSiC (short of Multiple Sequence Alignment with Regular Expression Constraints) was implemented by C and its web interface by PHP and HTML. RE-MuSiC (http://140.113.239.131/RE-MUSIC) can be easily accessed via a simple web interface (see Figure 4.1). The input of the RE-MuSiC web server consists of a set of Protein/DNA/RNA sequences and a set of user-specified regular expression constraints. The output of RE-MuSiC is a multiple sequence alignment with regular expression constraints.
4.2 Usage of RE-MuSiC
In this section, we shall describe the usage of RE-MuSiC step by step, the output of RE-MuSiC and other information including scoring matrices, gap-penalty, and syntax of regular expressions currently used in RE-MuSiC.
4.2.1 Input of RE-MuSiC
1. Input a set of genomic sequences in the FASTA format in the top blank field
(1).
2. Enter one or more regular expressions that are separated by spaces in the "Regular expression constraints" field (2). Note that each constraint of regular expression should be put in quotes first. For example, if users have two regular expressions, say [ST]-x(2)-[DE] and
G-{EDRKHPFYW}-x(2)-[STAGCN]-{P}, then they may key in the following line in the "Regular expression constraints" field:
"[ST]-x(2)-[DE]""G-{EDRKHPFYW}-x(2)-[STAGCN]-{P}"
If no constraint is specified, then RE-MuSiC produces an unconstrained alignment.
3. Select the type of input sequences that can be either protein or DNA/RNA (3). 4. Just click "Execute RE-MuSiC" button (4) if users would like to run
RE-MuSiC with default parameters; otherwise, they continue with the following parameter settings.
5. Select a suitable scoring matrix for protein or DNA/RNA sequences from a list of predefined matrices (5).
6. Key in two real values for gap open penalty (6) and gap extension penalty (7), respectively, since the RE-MuSiC web server penalizes the gaps using the affine gap penalty function.
7. Check the checkbox and enter an email address (8) if the user would also like to receive an email that contains a hyperlink to the RE-MuSiC result from the server. Note that the RE-MuSiC result will be kept on the server only for 24 hours.
Figure 4.1: The web interface of RE-MuSiC.
4.2.2 Output of RE-MuSiC
In the first part of the output page, RE-MuSiC shows the user-specified parameters, including scoring matrix, gap open and extension penalties, regular expression constraints and so on. Next, RE-MuSiC outputs its result of the constrained sequence alignment, in which the columns whose residues/nucleotides match regular expression constraints are shaded in yellow (refer to Figures 4.2 and 4.3 for examples). On addition, RE-MuSiC allows users to download the RE-MuSiC alignments in FASTA format or ClustalW format.
Figure 4.2: An example of the output of RE-MuSiC for protein sequences, where the
residues in the first block of columns shaded in yellow match the first regular expression of "[ST]-x-[RK]", and those in the second block of columns shaded in yellow match the second regular expression of
"G-{EDRKHPFYW}-x(2)-[STAGCN]-{P}".
Figure 4.3: An example of the output of RE-MuSiC for RNA sequences. Notice that a
gap appears in the block of matching the 1st regular expression constraint, which is not allowed to happen in MuSiC.
4.2.3 Scoring Matrices
For protein sequences, three inbuilt series of scoring matrices are used in RE-MuSiC system: (1) GONNET 250 (default), (2) BLOSUM 30, 45, 62, and 80, and (3) PAM 30, 70, 120, 250, and 350. For DNA/RNA sequences, RE-MuSiC provides identity matrix only.
4.2.4 Gap Penalty
The RE-MuSiC web server penalizes the gaps with the so-called affine gap penalty function, which charges the score of "Gap open penalty" for the existence of a gap and the score of "Gap extension penalty" for each residue/nucleotide in the gap. The default values of "Gap open penalty" for protein and DNA/RNA sequences are 10.0 and 15.0, respectively, and those of "Gap extension penalty" are 0.2 and 6.66, respectively. All these default values can be modified by the user, depending on the evolutionary distance between the input sequences of interest.
4.2.5 Syntax of Regular Expression Used in RE-MuSiC
Regular expression is a pattern-defining notation that describes a string (or, equivalently, sequence here) or a set of strings. In RE-MuSiC, the conventions of describing a pattern of regular expression are the same as those used in PROSITE.
• The standard IUPAC one-letter codes for the amino acids and nucleotides are used in the regular expression of RE-MuSiC. Notice here that the symbol 'x'/'X' is used for a position where any amino acid or nucleotide is accepted.
IUPAC Codes for Amino Acids
Letter Meaning Letter Meaning
A A (Alanine) N N (Asparagine)
B D, N P P (Proline)
C C (Cystine) Q Q (Glutamine) D D (Aspartic Acid) R R (Arginine) E E (Glutamic Acid) S S (Serine) F F (Phenylalanine) T T (Threonine) G G (Glycine) V V (Valine) H H (Histidine) W W (Tryptophan)
I I (Isoleucine) X X (Unknown or Other Amino Acid) K K (Lysine) Y Y (Tyrosine)
L L (Leucine) Z E, Q M M (Methionine)
IUPAC Codes for Nucleotides
Letter Meaning Letter Meaning
A A (Adenine) X/N A, C, G, T B C, G, T R A, G (Purine) C C (Cytosine) S C, G D A, G, T T T (Thymine) G G (Guanine) U U (Uracil) H A, C, T V A, C, G K G, T W A, T M A, C Y C, T (Pyrimidine)
• The amino acids (or nucleotides) that are allowed to appear at a given position are indicated by listing them in a pair of square brackets '[ ]'.
o For example, [ALT] stands for Ala (A), Leu (L) or Thr (T).
• The amino acids (or nucleotides) that are not accepted at a given position are indicated by listing them in a pair of braces '{ }'.
o For example, {AM} stands for any amino acid except Ala (A) and Met (M).
• Each element in a pattern of regular expression is separated from its neighbors by a dash '-'.
o For example, [GA]-G-K-[ST] means that the first position of the pattern can be occupied by either Gly (G) or Ala (A), the second and third positions must be Gly (G) and Lys (K), respectively, and the last position can be either Ser (S) or Thr (T).
• Repetition of an element in a pattern is indicated by appending, immediately following that element, an integer or a pair of integers (meaning the allowed range of the number of repetitions) in parentheses. For example,
o x(3) equals to x-x-x, a meaning pattern of any three amino acids (or nucleotides).
o A(3) equals to A-A-A, a meaning pattern of three amino acids of Ala (A).
o x(2,4) equals to x-x, x-x-x, or x-x-x-x.
For example, based on the above conventions, [AC]-x-V-x(4)-{ED} is translated as [Ala or Cys]-any-Val-any-any-any-any-{any but Glu or Asp}. A subsequence matches a given regular expression, if it can be described by this regular expression. For example, "AgVdefgB" matches the above regular expression.
Chapter 5
Experiments
In this chapter, we shall demonstrate the applicability of our RE-MuSiC by testing it on two data sets, one with protein sequences and the other with RNA sequences.
5.1 Protein Sequences with Active Site Residues
In this experiment, we analyzed three GST (Glutathione S-Transferase) proteins as follows.
1. AtGST: a phi class GST from plant Arabidopsis thaliana whose PDBID is
1GNW:A.
2. SjGST: an alpha class GST from non-mammalian Schistosoma japonicum (flat worm) whose PDBID is 1M99:A.
3. SsGST: a pi class GST from mammalian Sus scrofa (pig) whose PDBID is
2GSR:A.
Notice that the structural similarity between these three proteins is very high (see Figure 5.1), although their pairwise sequence identities are extremely low. In particular, the glutathione binding sites (G-sites) of these GST proteins have been found to have a conserved architecture, and the glutathione backbone conformations adopted in these GSTs from different species are quite similar [42]. Also, the cheimical natures of their residues acting as G-site ligands and interactions facilitated with glutathione exhibit analogy [42]. Due to their low sequence identity, it is hard to use a typical alignment tool, like ClustalW, to produce an accurate alignment (see
Figure 5.2 for example). In this ClustalW alignment as shown in Figure 5.3, one of the active site residues shared by these three GST proteins was not aligned well. They actually should be aligned together, however, if we superpose the crystal structures of these three GST proteins [42]. By querying PROSITE with these three GST protein sequences, we noticed that they all share a pattern of PS00006 ("[ST]-x(2)-[DE]") in common. Using this pattern as the constraint, we aligned again the three GST protein sequences mentioned above with RE-MuSiC, resulting in an alignment as shown in Figure 5.3 that indeed satisfies the requested constraint (i.e., those residues matching "[ST]-x(2)-[DE]" were lined up). In addition, it is worth mentioning here that all the active site residues shared among these GSTs were also aligned together. This suggests that, with additional information regarding some common patterns, RE-MuSiC is more reliable in aligning together biologically important residues from a set of closely related proteins, even when their sequence similarities are low. Demonstrated by this experiment, therefore, our RE-MuSiC web server is helpful in the detection of active site residues in a given set of protein sequences.
Figure 5.1: (a) Alpha class GST structure to which SjGST from non-mammalian S.
japonicum (flat worm) belongs, (b) Phi class GST structure to which AtGST from
plant A. thaliana belongs, (c) Pi class GST structure to which SsGST from mammalian S. scrofa (pig) belongs.
Figure 5.2: The active site residues shared by all three GST proteins are marked in
boxes. The active site residues in green boxes are aligned together, but the others in red boxes are not.
Figure 5.3: The constrained sequence alignment produced by RE-MuSiC, using the
pattern of "[ST]-x(2)-[DE]" (PS00006) as the constraint, in which the residues shaded in yellow match the pattern. In addition, the residues in green boxes that correspond to the active sites shared by these three GST proteins are aligned together.
5.2 RNA Sequences with Phylogenetically Conserved
Pseudoknots
In this experiment, we aligned the 3' untranslated region (3'-UTR) sequences of the following four coronaviruses, where HCoV-229E and PEDV are group 1 coronaviruses, while BCoV and MHV belong to group 2.
1. HCoV-229E: human 229E coronavirus whose accession number in GenBank is af304460.
2. PEDV: porcine epidemic diarrhea virus whose accession number in GenBank is af353511.
3. BCoV: bovine coronavirus whose accession number in GenBank is af220295. 4. MHV: murine hepatitis virus whose accession number in GenBank is
af201929.
It has been reported that phylogenetically conserved pseudoknots found in the 3'-UTRs of these four coronaviruses (refer to Figure 5.4) have been postulated to be involved in their RNA replication [54]. Notice that the pairwise sequence identities between sequences in the different groups are extremely low. Hence, it was difficult for ClustalW to have the sequence fragments corresponding to the conserved pseudoknots aligned together (see Figure 5.5 for example). However, as shown in Figure 5.6, this goal was achieved by RE-MuSiC when the pseudoknot consensus (as shown in Figure 5.7), derived by Williams et al., [54] from the 3'-UTRs of various coronaviruses, was used as the constraint. In general, loops in pseudoknots are less conserved. To enhance the flexibility of the consensus, hence, we treat the nucleotides involved in the loop regions as "don't care" symbols ("x") and describe the consensus as
"x(5)-C-U-x(4)-C-x(15,16)-U-G-x(2)-A-x(5,7)-G-x(4)-A-G-x(7,10)-U-x(3)-A-x(5)". This experiment suggests that RE-MuSiC is able to help locate those sequence fragments that are conserved from the structural point of view.
Figure 5.4: Phylogenetically conserved pseudoknots in the 3'-UTRs of four
coronavirus. (a) HCoV-229E (human 229E coronavirus), (b) PEDV (porcine epidemic diarrhea virus), (c) BCoV (bovine coronavirus), (d) MHV (murine hepatitis virus).
Figure 5.5: A partial view of the alignment produced by ClustalW, where the
fragments shaded in light blue corresponds to the phylogenetically conserved pseudoknots in the 3'-UTRs of the four coronaviruses. Notably, these four shaded fragments were not aligned together.
Figure 5.6: A partial view of the alignment produced by RE-MuSiC using the
constraint of "x(5)-C-U-x(4)-C-x(15,16)-U-G-x(2)-A-x(5,7)-G-x(4)-A-G-x(7,10)-U- x(3)-A-x(5)", where the fragments shaded in yellow, corresponding to the phylogenetically conserved pseudoknots in the 3'-UTRs of the four coronaviruses, are aligned together.
Figure 5.7: The consensuses adapted from [54], which was derived by Williams et al.
from the 3'-UTRs of various coronaviruses, including HCoV-229E, PEDV, BCoV and MHV.
Chapter 6
Conclusions
In this thesis, we studied the RECMSA problem, whose aim is to find an RECMSA for the input sequences with several user-specified regular expression constraints such that substrings of the input sequences whose bases match regular expression constraint are aligned together. In this model, each of the user-specified constraints is a regular expression, which is useful in expressing biologically important sites such as those stored in PROSITE, as well as structural elements which often involve variable ranges in them. In contrast, the plain-strings-with-mismatches model adopted in previously available tools, MuSiC and MuSiC-ME, is not flexible enough to express such patterns.
We adopted the dynamic programming and divide-and-conquer techniques to design a time and memory efficient algorithm for optimally solving the RECPSA problem. In addition, we designed a method to find in the resulting alignment the regions responsible for the satisfactions of the constraints. Based on the algorithm, we developed a web Server RE-MuSiC for the RECMSA problem using the progressive approach. The algorithm underlying RE-MuSiC represents an improvement over the previously proposed algorithm [2], and is more appropriate for implementation in a web-server.
conserved pseudoknots demonstrated that, with additional knowledge incorporated, RE-MuSiC is able to produce meaningful alignments in which important residues or structural elements can be aligned properly, even if the similarity among input sequences is low. Such ability is also useful for prediction purposes.


![Figure 4.2: An example of the output of RE-MuSiC for protein sequences, where the residues in the first block of columns shaded in yellow match the first regular expression of "[ST]-x-[RK]", and those in the second block of columns shaded in yel](https://thumb-ap.123doks.com/thumbv2/9libinfo/8549761.188068/35.892.138.763.111.447/figure-example-protein-sequences-residues-columns-regular-expression.webp)