Precursors of the Charge-Transfer-to-Solvent States in I
-(H
2O)
nClusters
Hsing-Yin Chen and Wen-Shyan Sheu*
Contribution from the Department of Chemistry, Fu-Jen Catholic UniVersity, Taipei, Taiwan 242, Republic of China
ReceiVed January 19, 2000
Abstract: In this paper an ab initio theoretical study of the precursors of the charge-transfer-to-solvent (CTTS) states in I-(H2O)nclusters is presented. While there is no bound excited state in monohydrated iodide I-(H2O),
the CTTS precursor states, denoted as I-(H2O)n*, emerge at cluster size n g 2, which confirms a recent
experimental observation [Serxner et al. J. Chem. Phys. 1996, 105, 7231.]. In addition, two or more bound excited states are found for larger clusters. The absorption maximum of the interior structure of I-(H2O)6is
found to be 5.02 eV, comparable to the experimental value of 5.48 eV found in the bulk, indicating that the first hydration shell of the aqueous halide makes a very significant contribution to the solvation energy of the lowest CTTS state and that the molecular details of solvent molecules play an important role in forming the CTTS states. Comparing the CTTS precursor states I-(H2O)n* with the electronic states of the corresponding
water cluster anions, e-(H2O)n, shows that the excited electron distributions are excluded from the region
occupied by the electrons of the iodine atom, which in turn results in higher energies for I-(H2O)n* compared
with e-(H2O)n. Moreover, it is shown that the cluster size dependence and isomer specificity of the excitation
energies and absorption intensities of the I-(H2O)nclusters may provide a diagnostic tool in determining the
predominate structure, surface or interior, of I-(H2O)6.
I. Introduction
For many simple inorganic anions such as OH-, SO42-, and
halide anions, there are no electronically bound excited states in the gas phase. Absorption of photons for these isolated species leads to the photodetachment of the excess electron (i.e., A-+ hν f A + e-) and displays an unstructured spectrum. However, in solutions of polar solvents (e.g., water, acetonitrile, ammonia), these anions show strong, broad absorption in the ultraviolet region, implying the presence of electronically bound excited states (i.e., A-aq+ hν f A-aq*).1Since the polar solvents do
not possess unoccupied bound orbitals to accommodate the excess electron, these excited states appear to be supported by the concerted action of the solvent molecules. Due to the crucial role played by the solvents for supporting these excited states, these spectra are known as charge-transfer-to-solvent (CTTS) spectra.2Because the CTTS states are thought to be distributed
over the solvent molecules surrounding the anion, they are particularly sensitive to the local solvent environment. Thus, CTTS spectroscopy is considered an excellent diagnostic tool for exploring the local solvation structure around an anion. This is in contrast to many other techniques used to probe solvent effects, such as measurements of solvation energies and ionic activity coefficients, which are often bedeviled with problems of assigning separate contributions to the anion and cation in solution. Because most chemical reactions occur in solution and because information about the local solvent structure surround-ing a ssurround-ingle ionic species can be obtained from CTTS spectra, CTTS spectra have attracted considerable interest.1
Earlier work on this subject mainly focused on the effect of environmental changes on the CTTS band (e.g., temperature
dependence, pressure dependence, spectra in mixed solvents, the effect of added solutes, etc.). Most of these results have been reviewed by Blandamer and Fox1. Interested readers are
referred to the reference for details. Here we just summarize some rudimental theories concerning CTTS states. In 1954, Platzman and Franck proposed a model that was conceptually important for the later theoretical treatments of the CTTS spectra3. In this model, the solvent was treated as a dielectric
continuum characterized by static Dsand optical Dopdielectric
constants. The excited electron was trapped by the electrostatic potential field created by the electric polarization of the medium
induced by the ground-state anion. Here q and r are the electronic charge and the distance from the center of the anion, respectively. In this picture, the CTTS states are hydrogen-like with an effective atomic charge of -q[(1/Dop) - (1/Ds)]. The
absorption maximum hνmaxwas evaluated from a
thermody-namic cycle proposed by the authors. Although the theory predicted values of hνmax for the anions very close to the
experimental values, it was incapable of explaining the depen-dence of hνmaxon temperature or environmental changes. This
shortcoming was later overcome by Stein and Treinin.4,5They
retained the physical picture underlying the theory of Platzman and Franck, but adopted a different thermodynamic cycle and introduced an adjustable parameter to describe the radius of the cavity occupied by the anion. This modified theory is usually named as the “diffuse model”. In nearly the same period, there
(1) Blandamer, M. J.; Fox, M. F. Chem. ReV. 1970, 70, 59.
(2) Smith, M.; Symons, M. C. R. Trans. Faraday Soc. 1958, 54, 338.
(3) Platzman, R.; Franck, J. Z. Phys. 1954, 138, 411. (4) Stein, G.; Treinin, A. Trans. Faraday Soc. 1959, 55, 1086. (5) Stein, G.; Treinin, A. Trans. Faraday Soc. 1959, 55, 1091.
V(r) ) -q r
(
1 Dop- 1Ds
)
10.1021/ja000207s CCC: $19.00 © 2000 American Chemical Society Published on Web 07/21/2000
was another theory called the “confined model” proposed by Smith and Symons.6-8 In this treatment, the CTTS states are
thought to resemble the ground state of a solvated electron. The excited electron is confined in a spherical infinitely deep well with an adjustable radius of the primary solvent shell around the anion. In all of these earlier theoretical treatments, the effect of the neutral parent solute molecule on the CTTS state was ignored and the structure of solvent molecules was not considered explicitly.
To understand the explicit molecular role that the neutral parent solute molecule and the solvent molecules play in the CTTS states, Sheu and Rossky have recently studied the CTTS spectra of an aqueous halide via semiclassical adiabatic mo-lecular dynamics (MD) simulations.9While these simulations
treated the water molecules and halogen atom classically, the excess electron was described quantum mechanically, interacting with the halogen atom and water molecules with prescribed pseudopotentials. Hence, this is still a very simplified one-electron model. They found approximately nine bound CTTS states in their simulation.9The lower six states manifest mixed
s/d symmetry, while the higher three states are approximately of p characteristic. The CTTS band was regarded as consisting of the six subbands associated with the transitions from the halide p ground state to the six s/d CTTS states. They also pointed out that the halogen atom played an important role in determining the energies of the CTTS states, since these states have considerable charge (∼55%) distributed within the attrac-tive potential of the halogen atom. Recently, with the progress of the pulsed laser technique, the ultrafast dynamics of the photodetachment process following the excitation of the CTTS state were also studied by the femtosecond time-resolved spectra of CTTS excitations10-14 and semiclassical nonadiabatic
mo-lecular dynamics simulations.15-21
Another interesting question regarding the CTTS states is how many solvent molecules are needed to support these states. This question has been experimentally addressed by Johnson and co-workers using cluster ion techniques.22They recorded absorption
spectra of the gas-phase clusters, I-(H2O)n, n ) 1-4, via the
action spectra of the photoexcited complexes, in which the electron loss was the only decomposition channel. By comparing the absorption spectra with the vertical electron binding energies (VBE) of I-(H2O)nmeasured by Markovich et al.,23they found
that the photodetachment maxima lay below the VBE at the
cluster size n g 2, indicating the existence of electronically bound excited states. The authors speculated that these excited states were dipole-bound and could be regarded as the CTTS precursor states. On the other hand, Combariza et al. tried to find these CTTS precursor states for I-(H2O)nusing the ab initio
CIS method.24However, their results were negative. This is not
surprising because these dipole-bound excited states are weakly bound and beyond the reach of the accuracy of the CIS method. In addition, the dipole-bound excited states are expected to be very diffuse. Therefore, standard basis sets should be augmented with diffuse functions, as the cases in the study of dipole-bound anions.25Nevertheless, these diffuse basis sets were not used
in their treatment due to the authors’ primary interest in the ground-state structures of the systems.
In this paper we report the ab initio results for the CTTS precursor states in I-(H2O)nclusters. We developed a scheme
for calculating diffuse excited states, such as the CTTS states or the excited states of a solvated electron. The nature of the CTTS precursor states and the effect of the iodine atom on these states are investigated. In addition, the VBE and the excitation energies of the CTTS precursor states are calculated and the implications for the determination of the predominate structure, surface and interior, of I-(H2O)6will be discussed.
II. Computational Methods
The equilibrium structures of I-(H2O)n, n ) 1-6, have been
optimized at the HF level by Combariza and co-workers.24 These
structures were directly used in this work when searching for the CTTS precursor states. This is because, according to the Born Oppenheimer approximation, the nuclear configuration is unchanged when the electron is excited from its ground state upon light absorption. Two important ingredients in finding the CTTS precursor states, the basis sets used and calculation strategies, are described in the following paragraphs. A. Basis Sets. The standard 6-31++G* basis sets were adopted for describing the water molecules. For the iodide anion, Christiansen’s pseudopotentials and basis sets,26modified by Combariza et al.24to
reproduce the experimental ionization potential (IP) in the gas phase, were still used in this work. The MP2 result of the IP for I-is 3.02 eV, which is in good agreement with the experimental value of 3.06 eV, presuming the lower2P
3/2spin-orbit state of the iodine atom is
considered. Hence, the calculated energetic results in this work, such as the VBE and excitation energies of the CTTS precursor states, should be directly compared with the lower peaks observed in the experiments, without further spin-orbit corrections.
To describe the diffuse nature of the CTTS precursor states, denoted as I-(H2O)n*, additional diffuse sets consisting of six sp shells were
added onto the iodide. The exponents form a geometric series from 7.36× 10-3to 2.3552× 10-6with a progression factor of 5. On finding that the contributions from the outmost diffuse sp functions to the highest occupied molecular orbital (HOMO) of I-(H2O)n* are
insig-nificant, these supplemental 6sp diffuse sets were determined to be sufficient for the present calculations.
B. Calculation of the CTTS Precursor States I-(H2O)n*. In this
work, the CTTS precursor states I-(H2O)n* were located by the
“initial-guess” method. For the method to work, the initial-guess wave function for the Hartree-Fock (HF) calculation has to be close enough to a specific excited state, so that using the self-consistent field (SCF) procedure, the initial-guess wave function may converge to the excited state. Therefore, it is very important to make a reasonable estimate of the initial wave function for the initial-guess method to be successful. (6) Smith, M.; Symons, M. C. R. Discuss. Faraday Soc. 1957, 24, 206.
(7) Smith, M.; Symons, M. C. R. Trans. Faraday Soc. 1958, 54, 346. (8) Griffiths, T. R.; Symons, M. C. R. Trans. Faraday Soc. 1960, 56, 1125.
(9) Sheu, W. S.; Rossky, P. J. J. Am. Chem. Soc. 1993, 115, 7729. (10) Long, F. H.; Lu, H.; Shi, X.; Eisenthal, K. B. Chem. Phys. Lett.
1990, 169, 165.
(11) Long, F. H.; Lu, H.; Shi, X.; Eisenthal, K. B. J. Phys. Chem. 1994, 98, 7252.
(12) Assel, M.; Laenen, R.; Laubereau, A. Chem. Phys. Lett. 1998, 289, 267.
(13) Kloepfer, J. A.; Vilchiz, V. H.; Lenchenkov, V. A.; Bradforth, S. E. Chem. Phys. Lett. 1998, 298, 120.
(14) Lehr, L.; Zanni, M. T.; Frischkorn, C.; Weinkauf, R.; Neumark, D. M. Science 1999, 284, 635.
(15) Sheu, W. S.; Rossky, P. J. Chem. Phys. Lett. 1993, 202, 186. (16) Sheu, W. S.; Rossky, P. J. Chem. Phys. Lett. 1993, 213, 233. (17) Sheu, W. S.; Rossky, P. J. J. Phys. Chem. 1996, 100, 1295. (18) Borgis, D.; Staib, A. Chem. Phys. Lett. 1994, 230, 405. (19) Borgis, D.; Staib, A. J. Chem. Phys. 1995, 103, 2642. (20) Borgis, D.; Staib, A. J. Chem. Phys. 1996, 104, 4776. (21) Borgis, D.; Staib, A. J. Chem. Phys. 1996, 104, 9027.
(22) Serxner, D.; Dessent, C. E. H.; Johnson, M. A. J. Chem. Phys. 1996, 105, 7231.
(23) (a) Markovich, G.; Giniger, R.; Levin, M.; Cheshnovsky, O. J. Chem. Phys. 1991, 95, 9416. (b) Markovich, G.; Pollack, S.; Giniger, R.; Cheshnovsky, O. ibid. 1994, 101, 9344.
(24) Combariza, J. E.; Kestner, N. R.; Jortner, J. J. Chem. Phys. 1994, 100, 2851.
(25) Gutsev, G. L.; Adamowicz, L. J. Phys. Chem. 1995, 99, 13412. (26) (a) Pacios, L. F.; Christiansen, P. A. J. Chem. Phys. 1985, 82, 2664. (b) Hurley, M. M.; Pacios, L. F.; Christiansen, P. A.; Ross, R. B.; Ermler, W. C. ibid. 1986, 84, 6840. (c) LaJohn, L. A.; Christiansen, P. A.; Ross, R. B.; Atashroo, T.; Ermler, W. C. ibid. 1987, 87, 2812.
This is especially important for finding the CTTS precursor states I-(H2O)n* since these states are presumably very diffuse.
To find a good initial-guess wave function, it is useful to analyze the properties of the CTTS precursor states I-(H2O)n* in detail. The
excited electron in I-(H2O)n* is reasonably regarded as being trapped
in the electrostatic potential created by two types of polarization of the solvent molecules. One is the atomic polarization induced by the anion, which is the consequence of the geometric distortion of the solvent molecules. The other is the electronic polarization, which results from the distortion of the electron distributions of the solvent molecules induced by the excited electron. The atomic polarization has already been taken into account once the equilibrium structure of the ground-state I-(H2O)nis used in the calculation of the CTTS state I-(H2O)n*.
On the other hand, due to the diffuse nature of the CTTS excited electron in I-(H2O)n*, the electronic polarization should be small and
can be neglected in the first approximation. On the basis of these considerations, the CTTS precursor states I-(H2O)n* can be
ap-proximately pictured as an excess electron attached to the corresponding neutral frame I(H2O)nin which the electrons of the solvent molecules
and iodine atom are frozen in the ground-state configurations. This picture appears to have the same physical meaning as the Koopmans’ theorem for the virtual orbital of the corresponding neutral frame I(H2O)n.27Hence, the bound virtual orbitals of I(H2O)nthat are higher
than the empty iodine 5p orbital (i.e., LUMO of I(H2O)n) can be
regarded as good first approximations for the CTTS orbitals. Therefore, they are good candidates as the initial-guess wave functions for the HF calculation of I-(H2O)n*.
Based on the statements above, the scheme for calculating the CTTS precursor states I-(H2O)n* used in this work is designed as follows:
(1) An unrestricted Hartree-Fock (HF) calculation of the corre-sponding neutral frame I(H2O)nis first performed. The existence of
CTTS states is judged by examining if there are bound virtual states (i.e., LUMO + 1, LUMO + 2,... etc.) higher than the empty iodine 5p state (LUMO).
(2) If these bound virtual states of I(H2O)n do exist, the excess
electron is put into each bound LUMO + n (n > 0) orbital of I(H2O)n,
to form the initial guess wave function for the unrestricted HF calculations of the CTTS states I-(H2O)n*. This overall spin for the
cluster is zero.
(3) Once the HF wave functions of I-(H2O)n* are obtained, the MP2
calculation is performed to improve the accuracy of the energies of these states.
To assess the effect of the iodine core on the CTTS precursor states, this scheme is also adopted for calculating the electronic states of the corresponding water cluster anions e-(H2O)n.
The quadratically convergent SCF procedure (QC-SCF)28is adopted
throughout this work. It is noted that the QC-SCF28and direct energy
minimization method29are both suitable for locating the excited states
in the present calculations. This is because these methods select the occupied orbitals by the criterion of a maximum overlap with the previous one, which prevents switching of orbital occupation during the SCF iterative procedure. All calculations reported in this work were performed using the Gaussian 94 package.30
III. Results and Discussions
A. Energies of the CTTS Precursor States. For the discussion of the energetic properties of the CTTS precursor states I-(H2O)n*, two physical quantities are calculated for
various cluster sizes and structures. The results are presented in Table 1. One is the excitation energy (hνmax) which is defined
as the difference between the total electronic energy of the lowest CTTS precursor state and the ground anionic state at the geometry of the ground anionic state I-(H2O)n. This quantity
can be directly compared with the absorption maximum in experiment.22The other quantity is the vertical binding energy
(VBE) which is the total electronic energy difference of the neutral I(H2O)nand CTTS states I-(H2O)n* at the geometry of
I-(H2O)n. This quantity can be directly measured in
pump-probe experiments or be indirectly evaluated by subtracting the excitation energy from the ionization potential (IP) of the ground anionic state I-(H2O)n. The dipole moments of the neutral
molecular cluster frames I(H2O)n, including the contribution
from the iodine atom, calculated at the HF level are also listed in Table 1 for later discussions. Although higher levels of calculation can provide more accurate dipole moments, they are not done here because it is not our purpose to obtain highly accurate values.
For surface structures denoted as (S) (cf. Figure 1), only one excited state is located in clusters n ) 2 and 3, while two excited states are found at n g 4 (Table 1). Except for the n ) 6 cluster, the VBE of (S)-I-(H2O)n* approximately increases with the
dipole moment of the corresponding neutral molecular frame (S)-I(H2O)n. Although the dipole moment of (S)-I(H2O)6is larger
than that of (S)-I(H2O)5(7.312 vs 6.636D), the VBE of (S)-I-
-(H2O)6* is smaller than that of (S)-I-(H2O)5* (0.069 vs 0.102
eV). This exception was also previously observed for the IP of the ground state (S)-I-(H2O)n.24 In a recent study of water
hexamer anions,31 Kim et al. pointed out that the VBE was
highly correlated with the e-‚‚‚Hdinteraction (where Hdstands
for a dangling hydrogen), while the e-‚‚‚dipole interaction only
(27) Szabo, A.; Ostlund, N. S. Modern Quantum Chemistry; McGraw-Hill: New York, 1989; p 123.
(28) Bacskay, G. B. Chem. Phys. 1981, 61, 385.
(29) Seeger, R.; Pople, J. A. J. Chem. Phys. 1976, 65, 265.
(30) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.; Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T. A.; Petersson, G. A.; Montgomery, J. A.; Raghavachari, K.; Al-Laham, M. A.; Zakrzewski, V. G.; Ortiz, J. V.; Foresman, J. B.; Cioslowski, J.; Stefanov, B. B.; Nanayakkara, A.; Challacombe, M.; Peng, C. Y.; Ayala, P. Y.; Chen, W.; Wong, M. W.; Andres, J. L.; Replogle, E. S.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Binkley, J. S.; Defrees, D. J.; Baker, J.; Stewart, J. P.; Head-Gordon, M.; Gonzalez, C.; Pople, J. A. Gaussian 94, revision C.3; Gaussian Inc.: Pittsburgh, PA, 1995.
(31) (a) Lee, S.; Lee, S. J.; Lee, J. Y.; Kim, J.; Kim, K. S.; Park, I.; Cho, K.; Joannopoulos, J. D. Chem. Phys. Lett. 1996, 254, 128. (b) Kim, K. S.; Park, I.; Lee, S.; Cho, K.; Lee, J. Y.; Kim, J.; Joannopoulos, J. D. Phys. ReV. Lett. 1996, 76, 856. (c) Lee, S.; Kim, J.; Lee, S. J.; Kim, K. S. ibid. 1997, 79, 2038.
Table 1. Vertical Binding Energies (VBE) and Excitation Energies (hνmax) of the CTTS Excited Precursor Statesa
(S)-I-(H2O)2 (S)-I-(H2O)3 (S)-I-(H2O)4 (I)-I-(H2O)4
HF MP2 HF MP2 HF MP2 HF MP2
VBE
1st CTTS 0.004 0.009 0.009 0.020 0.026 0.055 0.001 0.008 2nd CTTS not found not found 0.0002 0.0004 not found hνmaxb 3.209 3.849 3.522 4.176 3.749 4.396 3.894 4.557 µc 4.429 4.900 5.926 2.449 (S)-I-(H2O)5 (I)-I-(H2O)5 HF MP2 HF MP2 VBE 1st CTTS 0.051 0.102 0.017 0.086 2nd CTTS 0.0005 0.001 not found hνmaxb 3.906 4.537 4.115 4.726 µc 6.636 3.531
(S)-I-(H2O)6 (I)-I-(H2O)6 (V)-I-(H2O)6
HF MP2 HF MP2 HF MP2 VBE 1st CTTS 0.038 0.069 0.011 0.132 0.128 0.214 2nd CTTS 0.0005 0.001 not found 0.004 0.006 hνmaxb 3.793 4.442 4.450 5.020 4.003 4.608 µc 7.312 0.086 9.917 aVBE and hν
maxare in eV. Dipole moments are in D. (S), (I), and
(V) stand for the surface, interior, and V-shaped structure, respectively bExcitation energy of the first (lowest) CTTS precursor state.cDipole moment of the corresponding neutral frame I(H2O)n.
slightly enhanced the VBE when the number of clustered Hd’s
is low. Based on this point of view, the unexpected low VBE of (S)-I-(H2O)6* can be rationalized by noting the fact that in
this structure only three clustered Hd's point directly toward the
excited electron while the other three Hd’s do not point toward
the excited electron and are at a greater distance from the electron (cf. Figures 1e and 4 in ref 24). In comparison, there are five clustered Hd’s to stabilize the excited electron in
(S)-I-(H2O)5* (cf. Figures 1d and 3 in ref 24). In addition Table 1
also reveals that the electron correlation effect contributes significantly to the VBE of (S)-I-(H2O)n* (∼50% for MP2
correction). This observation is consistent with the recent finding in the study of dipole-bound anions32that the dispersion force
between the loosely bound electron (lbe) and the electrons of the corresponding neutral frame is important for stabilizing the dipole-bound anion. This is due to the large polarizability of the lbe.
For interior structures denoted as (I) (cf. Figure 2), only one excited state is obtained for n ) 4-6. In contrast to the surface structures, there is little correlation between the VBE and the dipole moment of the corresponding neutral frame. Instead, the VBE of (I)-I-(H2O)n* seems to correlate only with the number
of clustered Hd’s. The contribution of electron correlation effect
to the VBE of (I)-I-(H2O)n* is found to be larger (∼80%) than
that for (S)-I-(H2O)n*. This may be due to the fact that the
distribution of the excited electron in (I)-I-(H2O)n* is closer to
the water molecules (cf. Figures 1 and 2) and hence the dispersion interactions between the excited electron and the electrons of water molecules become larger.
It is appropriate to compare the present results with the experimental results for I-(H2O)n)2-4reported by Johnson and
co-workers.22 The experimental VBE of I-(H
2O)n*, (i.e., the
energy gap between the absorption maximum and IP of I-(H2O)n
indicated by upward arrow in ref 22) are estimated to be 0.03, 0.08, and 0.16 eV for clusters n ) 2, 3 and 4, respectively. While our best results calculated at the MP2 level are qualita-tively in accord with the experimental results (cf. Table 1 and Figure 3), the magnitudes are somewhat smaller. However, this
(32) (a) Gutowski, M.; Skurski, P.; Boldyrev, A. I.; Simons, J.; Jordan, K. D. Phys. ReV. A 1996, 54, 1906. (b) Gutowski, M.; Skurski, P. J. Chem. Phys. 1997, 107, 2968. (c) Gutowski, M.; Skurski, P. J. Phys. Chem. B
1997, 101, 9143. (d) Gutowski, M.; Jordan, K. D.; Skurski, P. M. J. Phys.
Chem. A 1998, 102, 2624.
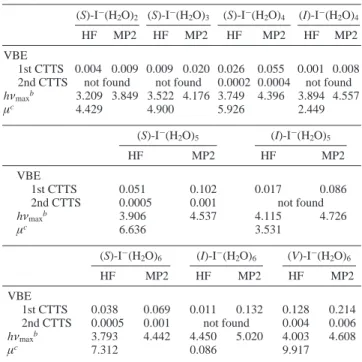
Figure 1. Isosurfaces of the HOMO of the lowest CTTS precursor states,ΨCTTS, for I-(H2O)nat surface and V-shaped structures. (a) The 0.003 isosurface for (S)-I-(H2O)2. (b) The 0.004 isosurface for (S)-I-(H2O)3. (c) The 0.0065 isosurface for (S)-I-(H2O)4. (d) The 0.0085 isosurface for
(S)-I-(H2O)5. (e) The 0.0065 isosurface for (S)-I-(H2O)6. (f) The 0.009 isosurface for (V)-I-(H2O)6. Note that different isosurfaces are plotted for
a better visual display. The stick-and-ball molecular frames are also plotted as a visual aid. See the text for explanation.
is not unexpected due to the very weak binding energies of the CTTS precursor states and the level of theory used. Based on experience studying the water dimer anion,33the calculations
at higher levels of theories, such as MP4 or CCSD(T), are expected to improve the calculated VBE of I-(H2O)n*.
More-over, these experimental values are not very accurate due to broadness of the absorption peak.22Another experimental VBE
can be obtained from the electron kinetic energy (eKE) of the photodetached electron created by a probe laser in the time-resolved dynamics study of the CTTS precursor states in I-(H2O)nand I-(D2O)nclusters done by Lehr et al.14Extracted
from the peak of the eKE distributions at the earliest delay times around 100-200 fs, the VBE are estimated to be about 0.12 and 0.23 eV for the n ) 5 and n ) 6 clusters, respectively, which are close to our calculated VBE of 0.102 eV for (S)-I- -(H2O)5* and 0.214 for (V)-I-(H2O)6*, respectively (cf. Table
1). These results suggest that the initial cluster isomers are closer to (S)-I-(H2O)5and (V)-I-(H2O)6, respectively. The implications
for the microsolvation environment of I-(H2O)6 will be
dis-cussed later. As for the n ) 4 cluster, the experimental VBE is about 0.2 eV. Instead of I-(H2O)4*, this value is close to the
ground-state VBE of the (S)-e-(H2O)4cluster (0.21 eV in Table
2, to be discussed later). This may be attributed to the creation of the water cluster anions in the dynamics, which will be further investigated in a future publication.
On the other hand, the experimental absorption maxima,22
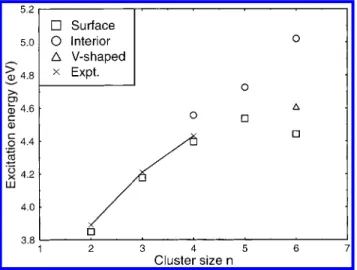
which are approximately located at 3.89, 4.21 and 4.43 eV for n ) 2, 3, and 4, respectively, are in excellent agreement with the present results of the excitation energies hνmaxof the surface
structure at 3.89, 4.18 and 4.40 eV for n ) 2, 3 and 4, respectively (cf. Table 1 and Figure 4). It is interesting to note that although only a limited number of water molecules are in the clusters considered, the largest hνmaxvalue of 5.02 eV, found
in the interior I-(H2O)6, is very close to the experimental value
of 5.48 eV, found in the bulk.34 This indicates that a large
portion of the solvation energy for the lowest CTTS state of the aqueous iodide can be attributed to the first hydration shell. Therefore, the configuration of the first-shell solvent molecules plays an important role in forming the CTTS states. This indicates that the early mean field theories3-8are inadequate in
describing the CTTS states in the bulk, as previously discussed by Sheu and Rossky.9
(33) Chen, H. Y.; Sheu, W. S. J. Chem. Phys. 1999, 110, 9032. (34) Fox, M. F.; Hayon, E. J. Chem. Soc., Faraday Trans. 1 1977, 73, 1003.
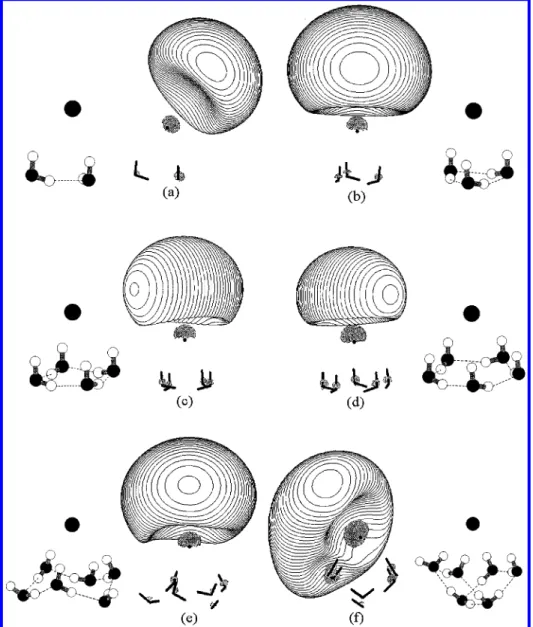
Figure 2. Isosurfaces ofΨCTTSfor I-(H2O)nat interior structures. (a) The 0.003 isosurface for n ) 4. (b) The 0.0085 isosurface for n ) 5. (c) The 0.0095 isosurface for n ) 6. Again, different isosurfaces are plotted. See the text for explanation.
Figure 3. Cluster size dependence and isomer specificity of the calculated vertical binding energies (VBE) of the lowest CTTS precursor states for I-(H2O)n. The experimental data are from ref 22.
Table 2. Vertical Binding Energies (VBE) of the Electronic States for the Corresponding Water Cluster Anions of I-(H2O)na
(S)-e-(H2O)2 (S)-e-(H2O)3 (S)-e-(H2O)4 (I)-e-(H2O)4
HF MP2 HF MP2 HF MP2 HF MP2
VBE
G.S. 0.016 0.032 0.046 0.089 0.128 0.210 0.178 0.296 1st E.S. not found not found 0.0005 0.001 not found
µb 3.971 4.300 5.361 2.200
(S)-e-(H2O)5 (I)-e-(H2O)5
HF MP2 HF MP2
VBE
G.S. 0.229 0.345 0.304 0.448
1st E.S. 0.002 0.003 not found
µb 6.064 3.322
(S)-e-(H2O)6 (I)-e-(H2O)6 (V)-e-(H2O)6
HF MP2 HF MP2 HF MP2
VBE
G.S. 0.125 0.213 0.475 0.657 0.359 0.508 1st E.S. 0.001 0.002 not found 0.011 0.017 2nd E. S. not found not found 0.0003 0.0005
µb 6.683 0.081 9.450
aVBE are in EV and dipole moments are in D.bDipole moment of the corresponding neutral frame (H2O)n.
Since more bound states are found when more water molecules are in the clusters, the CTTS precursor states may not be fully developed for I-(H2O)nwhen ne 6. In addition,
the bound excited states for monohydrated iodide I-(H2O) are
not found, probably due to the small dipole moment of its corresponding neutral frame to bind the excess electron.22
Therefore, it is concluded that the CTTS precursor states exist only in clusters n g 2, as a result of the cooperative effect of the water molecules in the clusters.
Recently, Majumdar et al.35also reported the CTTS energies
of small clusters, X-(H2O)n)1-4 (X ) F, Cl Br, and I), at
molecular configurations similar to, but slightly different from, those of Combariza.24 They used time-dependent density
functional theory (TDDFT) and adopted the basis sets of Combariza et al.24 without augmenting additional diffuse
functions, which are essential to describe the diffuse nature of the CTTS precursor states as discussed in section II. A. Although TDDFT is known to be cost-efficient, it appears to work best for low-lying excited states of clear valence type.36Its
applica-tion to treat the high-lying loosely bound states, as in the present case of the CTTS precursor states, is still to be documented. In addition, it is difficult to extract the excited-state wave functions from the TDDFT approach. With these understandings, Ma-jumdar et al. reported hνmax, but no VBE. For I-(H2O)n, their hνmaxare 3.74, 4.08, 4.29, and 4.44 eV for (S)- I-(H2O)nof n ) 1, 2, 3, and 4, respectively, which are close to, but consistently larger than, our values for n ) 2-4. It should be noted that some states of their calculation for small clusters may be unbound. For example, the n ) 1 cluster is not supposed to support bound excited states due to its small dipole moment (1.9D).22
B. Electronic Distributions of the CTTS Precursor States. To better understand the nature of the CTTS precursor states I-(H2O)n*, the isosurface plot of the highest occupied molecular
orbital (HOMO) of the lowest CTTS precursor state for I-(H2O)n* (hereafter denoted asΨCTTS) has been drawn and is
displayed in Figures 1 and 2. Note that the orbitals are very diffuse and located almost outside of the neutral frame. In addition, there is an obvious nodal plane between the ΨCTTS
and the iodine atom, which results from the exchange repulsion between the excited electron and the valence electrons of the iodine atom. The exclusion property of the iodine atom against
the excited electron distribution will have a pronounced effect on the CTTS precursor state energies and will be discussed in the next section.
The spatial distribution ofΨCTTSis also found to be highly
correlated with the symmetry of the potential provided by the water molecules. For asymmetrically hydrated structures, a two-center picture, where the two-center ofΨCTTSis separate from the
iodine atom center, is observed (cf. Figure 1 and Figure 2a-b). However, the center ofΨCTTScoincides with the iodine atom
when the hydration is symmetric (cf. Figure 2c). This observa-tion implies that the two-center model is more likely for the bulk CTTS states because the cavity occupied by the ion should not be ideally symmetric due to the thermal fluctuation. However, in the bulk phase, the deviation of the electronic distribution center from the iodine atom should not be too large, as shown in the recent simulations.15-17 Furthermore, the
capability of the free hydrogen (i.e., H that does not involve hydrogen bonding with water or iodide) to attract the electronic distribution ofΨCTTStoward itself is noteworthy (cf. Figure 2a
and b). This is similar to the case of water cluster anions.31
C. Effect of the Iodine Atom on the CTTS Precursor States. As discussed in the Introduction, the early models for the CTTS states in the bulk neglected the effect of the neutral halogen core on these states.3-8However, recent MD
simula-tions of Sheu and Rossky,9using a simplified one-electron model
pseudopotential, showed that the neutral halogen core played an important role in determining the CTTS state energies. Therefore, it is interesting to assess the effect of the halogen core on the CTTS precursor states using the more accurate ab initio approach. One way to examine the effect is to compare the CTTS precursor states with the states of the water cluster anions at the same configuration, since the effect of the iodine core is extracted in the latter case. The electronic states of the corresponding water cluster anions e-(H2O)n were calculated
and are shown in Table 2. These calculations were performed at the geometry of I-(H2O)n. Although the iodine atom is absent
in these calculations, the basis sets used for describing the iodine atom were still retained to avoid the artificial basis set superposition error (BSSE). It should be noted that if the iodine core had no effect on the CTTS precursor states, the lowest CTTS precursor state of I-(H2O)nshould have the same energy
as the ground state of the corresponding e-(H2O)nat the same
water configuration. However, this is not the case as to be discussed later.
Table 2 shows that the number of bound electronic states for the water cluster anions increases with the dipole moment of the corresponding neutral water cluster molecular frame. The dipole moment of the (H2O)n (cf. Table 2) is found to be
consistently smaller than that of I(H2O)n(cf. Table 1) since there
is no contribution from the induced dipole moment of the iodine atom in the (H2O)n cluster. The critical value of the dipole
moment for the existence of more than one dipole-bound state (DBS) was previously addressed by charge-dipole models.37
Recently, Bartlett et al. studied the DBS of polar diatomic anions by using the electron-attachment equation-of-motion coupled-cluster (EA-EOMCC) method.38They reported that the critical
dipole moments of about 5, 7, and 9.5 D are needed for sustaining the second, third, and fourth DBS, respectively. Our results in Table 2 agree well with these critical values. In addition, the VBE of e-(H2O)n correlates not only with the
dipole moment of the corresponding neutral frame (H2O)nbut
(35) Majumdar D.; Kim J.; Kim K. S. J. Chem. Phys. 2000, 112, 101. (36) Casida M. E.; Jamorski C.; Casida K. C.; Salahub D. R. J. Chem. Phys. 1998, 108, 4439.
(37) (a) Garrett, W. R. J. Chem. Phys. 1982, 77, 3666. (b) Clary D. C.; Henshaw J. P. Int. J. Mass Spectrom. Ion Processes 1987, 80, 31.
(38) Gutsev, G. L.; Nooijen, M.; Bartlett, R. J. Chem. Phys. Lett. 1997, 276, 13.
Figure 4. Cluster size dependence and isomer specificity of the calculated excitation energies (hνmax) of the lowest CTTS precursor
states for I-(H2O)n. The experimental data are from ref 22.
also with the number of clustered Hd’s, as seen in the case of
I-(H2O)n*. It should be also noted that the bound excited state
of e-(H2O)n for the water molecules arranged at the stable
equilibrium geometry of I-(H2O)nfirst emerges at n ) 4. Since
the water molecules at this geometry are not fully relaxed for e-(H2O)n, the electronic states for the water configuration of
the equilibrated e-(H2O)nshould be lower in energy than for
the water configuration arranged as in equilibrated I-(H2O)n.
Hence, the bound excited state of the equilibrated e-(H2O)n
should also emerge at n ) 4; i.e., the cluster size of n ) 4 is the upper bound for the bound excited states of the equilibrated e-(H2O)nto first appear.
Comparison of the VBE of the e-(H2O)nand I-(H2O)n* in
Table 1 shows that the CTTS precursor states are higher in energy than those of the corresponding water cluster anions. The reason for this phenomenon can be understood by inspecting the HOMO of I-(H2O)n* and e-(H2O)n. The representative
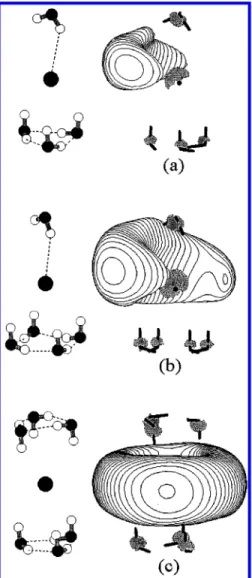
contour plots of the HOMO of the lowest CTTS precursor state of I-(H2O)n* (i.e., ΨCTTS) and the ground state of e-(H2O)n
(denoted asΨWCA) are displayed in Figure 5. It is clearly seen
that ΨCTTSis more diffuse than ΨWCA, as manifested by the
larger spacing between contour lines ofΨCTTS. This is further
seen by examining the electronic spatial extent〈r2〉1/2. The value
of〈r2〉1/2 is always found to be larger for I
-(H2O)n* than for
the corresponding e-(H2O)n, for example, 30 vs 18 Å for n )
3 and 70 vs 24 Å for the interior structure of n ) 4. Moreover,
the distributions of ΨCTTS and ΨWCA have very different
features. Figure 5 shows that the maximum ofΨWCAcenters
around the position of the imaginative iodine. This is because the geometry used in the calculation of e-(H2O)nis optimized
for the ground state of I-(H2O)n, at which I-is situated at the
most electrostaticly stable region for the negative charge, and hence the excess electron of e-(H2O)n prefers to distribute
around this region. Nevertheless, the presence of the iodine core in I-(H2O)n* excludes the electronic distribution ofΨCTTSfrom
the core region due to the wave function orthogonality and the exchange repulsion between the excess electron and the electrons of the iodine atom. This repulsion makes the potential experi-enced by the excess electron less attractive for I-(H2O)n* than
e-(H2O)n. This effect explains whyΨCTTSis more diffuse than ΨWCAand the VBE of I-(H2O)n* is higher in energy than that
of e-(H2O)n.
The above conclusion is at odds with the simulation results of the CTTS states in the bulk done by Sheu and Rossky.9Using
only a prescribed attractive pseudopotential between the excess electron and the iodine core,9(originated from the incomplete
shielding of the nuclear charge by the electrons of the atom), they pointed out that the iodine atom provides an additional stabilization potential for the CTTS states since considerable charge (∼50-60%) remains within the attractive potential of the iodine core. However, from the previous discussions for the I-(H2O)nclusters, the attractive force from the incompletely Figure 5. Contour plots of the HOMO ofΨCTTSandΨWCA, the ground states of the corresponding water cluster anions. (a) (S)-I-(H2O)3* (b)
(S)-e-(H2O)3(c) (I)-I-(H2O)4* (d) (I)-e-(H2O)4. The increment of the plotted contour lines is 0.0005/Å3/2. The heavy dot near the center of each
plot is the iodine atom in I-(H2O)n* or the imaginative iodine in e-(H2O)n. Note that the electronic distribution is largely excluded from the core region and is more diffuse forΨCTTS.
shielded charge of the iodine atom seems to be outweighed by the exclusion repulsion force so that the VBE of I-(H2O)n* is
higher than that of the corresponding e-(H2O)n.
Based on previous discussions, it is interesting to assess how the effect of the iodine core onΨCTTSaffects its VBE value.
For this purpose, VBECTTS/VBEWCA, the ratio of the VBE of
the lowest CTTS precursor state to that of the ground state of the corresponding water cluster anion, is proposed to be as a measure of the effect of the iodine core on the VBE and is tabulated in Table 3. This is because the difference between these two VBE is solely due to the existence of the iodine core: the lower the ratio, the larger effect of the iodine core on the VBE. By this criterion, the iodine core seems to have a larger effect on the interior cluster structure for a reason to be discussed below. To assess the effect of the iodine core on ΨCTTS, the integrated electronic probability of the wave function
within the iodine core is first examined. This can be done using a simplified method similar to the Mulliken population analysis. In this method, a one-electron charge-density bond-order matrix P is constructed from the coefficients ofΨCTTS. The diagonal
elements of the product of the charge-density bond-order matrix P and the overlap matrix of the basis sets S are partitioned into two parts (PS)µµand (PS)νν. Here the indexµ represents the basis functions used to represent the iodide and the index ν represents the supplemental diffuse sets 6sp and the 6-31++G* for the water molecules. The sum of (PS)µµis attributed to the charge remaining within the effective iodine core, which is counted as the sum of the integrated electronic probability within approximately 2.4 Å of the iodine core based on the maximum location of the outmost basis function used to represent the iodide. The results show that forΨCTTSonly a very small charge
remains within the iodine core, denoted as QCTTS, (cf. Table
3). However, QCTTSincreases slowly with the number of water
molecules, which may be due to the larger dipolar attractive force created by more water molecules acting against the previously discussed exclusion force. It is noteworthy that the charge within the iodine core for the interior structure is significantly larger than for the surface structure. The experi-mental implication of this point will be addressed in the next section. The same analysis is also applied toΨWCAof e-(H2O)n.
Compared toΨCTTS, forΨWCAa much larger charge is found
to reside within the imaginative iodine core, denoted as QWCA,
as expected due to the lack of the previously discussed exclusion repulsion for QWCA. It is seen that little correlation can be found
between QCTTSand VBECTTS/VBEWCAbecause, for example,
given that QCTTS) 0.2 for both the surface and interior structure
of I-(H2O)4, their VBECTTS/VBEWCA ratios differ by about
7-fold. In addition, QCTTS/QWCA shows no correlation with
VBECTTS/VBEWCA. This is not surprising since the exclusion
effect of the iodine core affects not only the wave function within the core but also the wave function outside the core, as shown in Figure 5. To include this factor, the overlap ofΨCTTS
withΨWCAis proposed as a new measure of the wave function
distortion due to the iodine core and the results are also listed
in Table 3. The larger the overlap, the smaller the distortion of ΨCTTS and hence a larger ratio VBECTTS/VBEWCA is to be
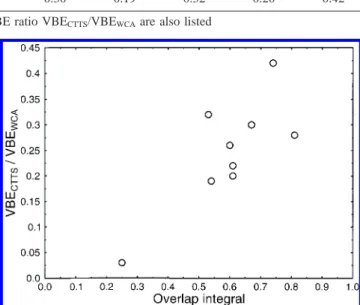
expected. This is indeed roughly the case shown in Table 3. To better demonstrate this point, Figure 6 is plotted. Although it does not form a straight line, Figure 6 shows that VBECTTS/
VBEWCAis well correlated with the overlap, indicating that the
localized exclusion effect from the iodine core has a global influence on the electronic distributions of the CTTS precursor states.
Before ending this section, we note that our small QCTTSis
in contrast with the very large charge on the iodine atom as reported by Majumdar et al.35One of the major reasons for this
discrepancy may be their use of small basis sets, as discussed in section III. A.
D. Determining Microscopic Hydration Environment. Microscopic hydration phenomena have long been a fascinating subject for physical chemists. One of many interesting questions in this field is the number of water molecules required to construct the first hydration shell (i.e., the critical cluster size for the (S) f (I) transition). By using a photoelectron technique, Markovich et al. measured the stabilization energies Estabfor
I-(H2O)n)1-1623and found that Estableveled off at n ) 6 and
concluded the first hydration shell of I-consisting of six water molecules. On the other hand, the reactivity of I-(H2O)nwith
Cl2 is used by Viggiano et al. to show that I-resides on the
surface of water clusters up to n ) 15.39 From a theoretical
point of view, the ab initio results of vertical ionization potentials calculated by Combariza and co-workers24are in line with the
experimental conclusion of Markovich et al. They compared the cluster size dependence and isomer specificity of the calculated vertical IP of the ground state with experimental data. The conclusion was that the surface structures are prevalent for
(39) Viggiano A. A.; Arnold S. T.; Morris R. A. Int. ReV. Phys. Chem.
1998, 17, 147.
Table 3. Integrated Probability of FindingΨCTTSandΨWCAwithin the Effective Iodine Core for I-(H2O)n, Denoted as QCTTSand QWCA,
Respectivelya
n ) 2 n ) 3 n ) 4 n ) 5 n ) 6
(S) (S) (S) (I) (S) (I) (S) (I) (V)
QCTTS 0.0 0.0 0.2 0.2 0.7 2.7 0.3 3.8 0.6
QWCA 2.8 9.0 19.7 30.8 29.8 39.8 16.7 47.3 23.0
〈ΨWCA|ΨCTTS〉 0.81 0.61 0.60 0.25 0.67 0.54 0.53 0.61 0.74
VBECTTS/VBEWCA 0.28 0.22 0.26 0.03 0.30 0.19 0.32 0.20 0.42
aIn addition, the overlap integrals betweenΨ
CTTSandΨWCAand the VBE ratio VBECTTS/VBEWCAare also listed
Figure 6. The energy deviation ratio VBECTTS/VBEWCAplotted as a
function of the overlap integral ofΨCTTSwithΨWCA, indicating the
global effect of the iodine core onΨCTTS.
Cl-(H2O)n(n ) 2-6), Br-(H2O)n(n ) 2-6), and I-(H2O)n(n ) 2-5), while an (S)f(I) transition might occur for I
-(H2O)6.
On the other hand, molecular dynamic (MD) simulations for halide water clusters showed conflicting results with the prevalence of surface structures for the cluster size n ) 2-15.40
Since the CTTS precursor electron in I-(H2O)n* is
predomi-nantly distributed outside the iodine core, the CTTS precursor states should be more sensitive to their hydration environment than the ground state, and should be better candidates to be used as diagnostic tools to distinguish between the surface and interior structures. As discussed in section III. A., the VBE, measured after 100-200 fs delay times in the experiment of Lehr et al., indicates that the initial cluster isomers are closer to (S)-I-(H2O)5for the n ) 5 and (V)-I-(H2O)6, another form
of surface structure (cf. Figure 1), for the n ) 6 cluster. (Since there are two surface structures for n ) 6 cluster, the term “surface structure” can refer to the (S) or (V) isomer for I-(H2O)6
in the discussion of the surface to interior transition.) Hence, they both belong to surface structures and hence are consistent with the results of Viggiano et al.39However, the assignment
should be viewed as tentative due to the MP2 level of theory used in the present calculation and the uncertainty of the iodine atom and water molecular motion during the machine response time and the 100-200 fs delay times. To further identify the critical cluster size for the surface-to-interior transition, it is better to examine the trend of how CTTS-related quantities vary with the cluster size due to the cancellation of higher-order electron correlation neglected in the MP2 calculation.24Figure
4 displays the excitation energy from the ground state to the lowest CTTS precursor state hνmaxas a function of the cluster
size and structure. Although the calculated hνmaxare somewhat
smaller than the experimental values (Figure 4), the trend for the surface structures of the cluster size of n ) 2-4 is consistent with experiments.22,23,39 This observation indicates the
preva-lence of the surface structures for n ) 2-4, which is in harmony with previous studies.24,40It is further noted that the calculated hνmaxfor the surface structures, (S) or (V), increases
monotoni-cally up to n ) 5, but drops or levels off at n ) 6. However, if the (S)f(I) transition occurs at n ) 6, the hνmaxshould sharply
increase with the cluster size up to n ) 6. Therefore, hνmax
provides a very valuable diagnostic tool to distinguish between the surface and interior structures. If the experimental hνmaxof
I-(H2O)ndisplays a drop or level-off at n ) 6, the surface cluster
structure is predominant up to n ) 6; otherwise, if there is a sharp increase up to n ) 6, the upper limit for the (S) f (I) transition should be at n ) 6. In addition, the transition intensity from the ground state to the lowest CTTS precursor state can be used as a further diagnostic tool. It has been previously noted that the distribution ofΨCTTShas a larger probability within
the effective iodine core in interior structures than in surface structures (cf. Table 3). Hence, the overlap betweenΨCTTSand
iodide 5p orbitalΨ5pis better for interior structures than surface
structures, which, in turn, enhances the electronic transition moment from theΨ5ptoΨCTTS(i.e.,〈Ψ5p|rel|ΨCTTS〉). Therefore,
a dramatic enhancement in absorption intensity will be seen at the (S) f (I) transition. On the basis of these observations, experimental hνmaxor absorption intensities for n ) 5-6 are
needed to determine at what size the surface-to-interior structure transition will occur. If the iodide is determined to reside on the surface of the n ) 6 clusters, the VBE of the lowest CTTS precursor state can be used to further determine whether it is
the (S) or (V) isomer, due to the marked difference of these two VBE (cf. Table 1). However, owing to the similarity of the structures for (S)-I-(H2O)6and (V)-I-(H2O)6(cf. Figure 1),
the VBE should be measured before any significant conforma-tion distorconforma-tion takes place.
IV. Conclusions
We have studied the CTTS precursor states in I-(H2O)n
clusters via the ab initio molecular orbital method. The results show that while there is no bound excited state in monohydrated iodide I-(H2O), the onset of the CTTS precursor states
I-(H2O)n* is at n ) 2, consistent with the experimental
observation.22In addition, two or more bound excited states
are found for larger clusters. For the same structure, the absorption maximum hνmaxfrom the ground state of I-(H2O)n
to the lowest CTTS precursor state increases with the size of the cluster, with the exception of the surface state at n ) 6. In addition, the hνmaxof the interior structure at n ) 6 is 5.02 eV,
comparable to the experimental value of 5.48 eV found in the bulk.34This indicates that a large portion of the solvation energy
for the lowest CTTS state of the aqueous iodide can be attributed to the first hydration shell and the molecular details of solvent molecules play an important role in forming the CTTS states. Nevertheless, the CTTS precursor states are not fully developed for I-(H2O)nwhen n e 6.
The effect of the iodine atom on I-(H2O)n* was investigated
by comparing I-(H2O)n* with the corresponding water cluster
anions e-(H2O)n. Because of the exchange repulsion between
the excited electron and the valence electrons of the iodine atom, the remaining iodine atom excludes the CTTS precursor wave function from the core region, which, in turn, results in higher energy for I-(H2O)n* compared with e-(H2O)n. It is also shown
that the localized exclusion force shows a global effect on the electronic distributions of the CTTS precursor states in that it disturbs not only the electronic distributions within the core but also the distributions outside the core.
The cluster size dependence and isomer specificity of the excitation energies (hνmax) and absorption intensities of the
CTTS precursor states I-(H2O)n* are also proposed to be
diagnostic tools to distinguish between surface and interior structures. For hνmax, if (S) f (I) transition occurs at n ) 6, the
trend of a sharp increase with the cluster size should be observed. Otherwise, if the surface structures are prevalent throughout n ) 2-6, a trend of a monotonic increase of hνmax
only up to n ) 5, followed by a descent or level-off at n ) 6 is expected. In addition, a dramatic increment of absorption intensity will mark the occurrence of the (S) f (I) transition. Therefore, experiments should be designed to measure the hνmax
or absorption intensities of the I-(H2O)nclusters to clarify at
what point the surface-to-interior structure transition will occur. The present study of the I-(H2O)n clusters serves as a
prototype to understand how the CTTS precursor states evolve toward the bulk CTTS states. Many of present findings about the CTTS precursor states of I-(H2O)n are expected to be
applicable to other ion clusters formed with different solvents. This is because common features exist in all CTTS spectra.1
Acknowledgment. This research has been funded by the National Science Council, ROC under Contract No. NSC 89-2113-M-030-013 and this support is gratefully acknowledged.
JA000207S (40) Perera, L.; Berkowitz, M. L. J. Chem. Phys. 1993, 99, 4222.