Open Access
Research
Predicting RNA-binding sites of proteins using support vector
machines and evolutionary information
Cheng-Wei Cheng
†1,4, Emily Chia-Yu Su
†2,3,4, Jenn-Kang Hwang
2,
Ting-Yi Sung*
4and Wen-Lian Hsu*
1,4Address: 1Institute of Information Systems and Applications, National Tsing Hua University, Hsinchu, Taiwan, 2Institute of Bioinformatics, National Chiao Tung University, Hsinchu, Taiwan, 3Bioinformatics Program, Taiwan International Graduate Program, Academia Sinica, Taipei, Taiwan and 4Bioinformatics Lab., Institute of Information Science, Academia Sinica, Taipei, Taiwan
Email: Cheng-Wei Cheng - [email protected]; Emily Chia-Yu Su - [email protected];
Jenn-Kang Hwang - [email protected]; Ting-Yi Sung* - [email protected]; Wen-Lian Hsu* - [email protected] * Corresponding authors †Equal contributors
Abstract
Background: RNA-protein interaction plays an essential role in several biological processes, such
as protein synthesis, gene expression, posttranscriptional regulation and viral infectivity. Identification of RNA-binding sites in proteins provides valuable insights for biologists. However, experimental determination of RNA-protein interaction remains time-consuming and labor-intensive. Thus, computational approaches for prediction of RNA-binding sites in proteins have become highly desirable. Extensive studies of RNA-binding site prediction have led to the development of several methods. However, they could yield low sensitivities in trade-off for high specificities.
Results: We propose a method, RNAProB, which incorporates a new smoothed position-specific
scoring matrix (PSSM) encoding scheme with a support vector machine model to predict RNA-binding sites in proteins. Besides the incorporation of evolutionary information from standard PSSM profiles, the proposed smoothed PSSM encoding scheme also considers the correlation and dependency from the neighboring residues for each amino acid in a protein. Experimental results show that smoothed PSSM encoding significantly enhances the prediction performance, especially for sensitivity. Using five-fold cross-validation, our method performs better than the state-of-the-art systems by 4.90%~6.83%, 0.88%~5.33%, and 0.10~0.23 in terms of overall accuracy, specificity, and Matthew's correlation coefficient, respectively. Most notably, compared to other approaches, RNAProB significantly improves sensitivity by 7.0%~26.9% over the benchmark data sets. To prevent data over fitting, a three-way data split procedure is incorporated to estimate the prediction performance. Moreover, physicochemical properties and amino acid preferences of RNA-binding proteins are examined and analyzed.
Conclusion: Our results demonstrate that smoothed PSSM encoding scheme significantly
enhances the performance of RNA-binding site prediction in proteins. This also supports our
from Asia Pacific Bioinformatics Network (APBioNet) Seventh International Conference on Bioinformatics (InCoB2008) Taipei, Taiwan. 20–23 October 2008
Published: 12 December 2008
BMC Bioinformatics 2008, 9(Suppl 12):S6 doi:10.1186/1471-2105-9-S12-S6
<supplement> <title> <p>Seventh International Conference on Bioinformatics (InCoB2008)</p> </title> <editor>Shoba Ranganathan, Wen-Lian Hsu, Ueng-Cheng Yang and Tin Wee Tan</editor> <note>Proceedings</note> </supplement>
This article is available from: http://www.biomedcentral.com/1471-2105/9/S12/S6 © 2008 Cheng et al; licensee BioMed Central Ltd.
This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
assumption that smoothed PSSM encoding can better resolve the ambiguity of discriminating between interacting and non-interacting residues by modelling the dependency from surrounding residues. The proposed method can be used in other research areas, such as DNA-binding site prediction, protein-protein interaction, and prediction of posttranslational modification sites.
Background
RNA-protein interaction plays an important role in vari-ous biological processes, such as protein synthesis, gene expression, posttranscriptional regulation, and viral infec-tivity. The prediction results of RNA-binding sites in pro-teins can provide biological insights for investigating RNA-protein interaction. For instance, the ribosome is a protein synthesis complex consisting of ribosomal RNAs (rRNAs) and proteins. Sunita et al. [1] applied predicted RNA-binding sites to study the relationship between RNA methyltransferases RsmC and 16S rRNA. In addition, Bechara et al. [2] incorporated predicted results from a RNA-binding site predictor to inspect the connection between fragile X mental retardation protein and G-quar-tet RNA structure. Moreover, some RNA viruses, such as human immunodeficiency virus (HIV) and hepatitis C virus, have a RNA genome and replicate themselves by interacting with host proteins [3]. Therefore, identifica-tion of the RNA interacting residues in proteins provides valuable information for understanding the mechanisms of protein synthesis, gene regulation, and pathogen-host interaction.
In recent years, rapid advances in genomic and proteomic studies have yielded a tremendous amount of DNA and protein sequences. We used the keyword "RNA-binding" to search against the National Center for Biotechnology Information (NCBI) protein sequence database on June 9, 2008, and obtained 196,686 protein sequences. However, when searching against Protein Data Bank (PDB) [4] for molecular/chain type containing protein and RNA, we only retrieved 684 structures. In addition, experimental determination of RNA-protein interaction remains time-consuming and labor-intensive. Therefore, computational approaches for predicting RNA-binding sites in proteins have become increasingly important to understand the mechanisms of RNA-protein interaction.
Previous work
Extensive studies of RNA-protein binding site prediction have lead to the development of several methods, which can be classified as follows.
1. Amino acid composition-based methods
Jeong et al. [5] used an artificial neural network (ANN) to predict RNA-protein interacting residues based on amino acid compositions and predicted secondary structure ele-ments. It achieved Matthew's correlation coefficient
(MCC) of 0.29 and overall accuracy of 77.50% along with specificity of 87.29% and sensitivity of 40.30%. Terribilini
et al. [6] presented RNABindR using a Naïve Bayes
classi-fier on amino acid sequences to predict RNA binding sites in proteins. RNABindR attained MCC, overall accuracy, specificity, and sensitivity of 0.35, 84.80%, 93% and 38%, respectively.
2. Evolutionary information-based methods
Jeong and Miyano [7] applied an ANN to predict the RNA interacting residues based on evolutionary information from the position-specific scoring matrix (PSSM), and achieved MCC, overall accuracy, specificity, and sensitiv-ity of 0.39, 80.20%, 91.04%, and 43.40%, respectively. The MCC is further improved to 0.41 by the incorporation of weighted profiles. Kumar et al. proposed a predictor, PPRint [8], using PSSM profiles in a support vector machine (SVM) model, and it achieved MCC, overall accuracy, specificity, and sensitivity of 0.45, 81.16%, 89.55%, and 53.05%, respectively.
3. Hybrid methods
Wang and Brown [9] developed an SVM-based classifier, BindN, using features including relative solvent accessible surface area, hydrophobicity index, side chain pKa value, molecular mass, and BLAST results. The overall accuracy, specificity, and sensitivity of BindN are 74.25%, 75.70%, and 65.78%, respectively.
Challenges
Although many methods have been proposed for RNA-binding site prediction, several challenges still remain. First, many of previous methods yield low sensitivities in tradeoff for high specificities since some biological appli-cations, such as identification of critical residues for site-specific mutagenesis, emphasize more on site-specificities rather than sensitivities [6,8]. These methods could suffer from low coverage of RNA-binding sites in high-through-put proteomic analyses. Second, the MCC values of exist-ing methods remain in the range of 0.27~0.45, which presents a great scope for improvement in the comple-mentary measure of prediction performance. Finally, in most methods parameters such as the size of the sliding window are selected from test results evaluated by n-fold cross-validation, which may lead to overestimation of the prediction performance. Thus, the performance would be worse if a more rigorous procedure is applied for parame-ter selection and performance evaluation.
Our method and future applications
In this study, we propose a method, RNAProB (RNA-Pro-tein Binding site prediction), for prediction of RNA-bind-ing residues in proteins usRNA-bind-ing SVM classifiers and a new smoothed PSSM encoding scheme. Besides incorporation of upstream and downstream residues in a standard PSSM generated by PSI-BLAST, smoothed PSSM encoding also considers, for each amino acid in a sequence, the depend-ency effect from its neighboring amino acids. Similar to the spatial domain method used in the research field of image processing [10], smoothed PSSM encoding calcu-lates the evolutionary information of a central position based on the sum of those from surrounding residues. Experimental results show that the prediction perform-ance of smoothed PSSM encoding performs better than the state-of-the-art approaches on the benchmark data sets. Evaluated by five-fold cross-validation, RNAProB outperforms the other approaches by 0.10~0.23 in MCC, 4.90%~6.83% in overall accuracy, and 0.88%~5.33% in specificity. Most notably, our method significantly improves sensitivity by 26.90%, 26.62%, and 7.05% for the RBP86, RBP109, and RBP107 data sets, respectively. To avoid data overfitting, we also incorporate a three-way data split procedure to evaluate the prediction perform-ance of RNAProB. Our results show that our method not only achieves significant improvement on the perform-ance, but also attains a high prediction accuracy evaluated by a three-way data split procedure. Moreover, our analy-sis indicates that smoothed PSSM could serve as a more discriminative feature for distinguishing between interact-ing and non-interactinteract-ing residues. We believe that the pro-posed encoding scheme could be applicable to other research fields, such as DNA-binding sites, protein-pro-tein interaction, and prediction of posttranslational mod-ification sites.
Methods
Data sets
In this study, we apply three data sets used in previous studies to compare the performance of our method and other systems. Table 1 shows a summary of these data sets, which are detailed as follows and available in the supple-mentary material [see Additional files 1, 2, and 3].
1. RBP86
The RBP86 data set consists of 86 protein chains extracted from RNA-protein complexes with X-ray crystallography resolution better than 3 Å in PDB. Sequence redundancy in the data set is removed so that no protein pair has a sequence identity greater than 70%. In the RNA-protein complexes, a residue is regarded as interacting with RNA if the distance between an RNA molecule and the residue in the protein is less than 6 Å. The resultant data set con-tains 4,568 RNA interacting residues and 15,503 non-interacting residues. The RBP86 data set has been used in Terribilini et al. [6] and Kumar et al. [8]. In Kumar et al., it is also referred to as the "main" data set.
2. RBP109
The RBP109 data set contains 109 protein sequences obtained from 56 RNA-protein complexes with X-ray crys-tallography resolution better than 3.5 Å in PDB. For any two protein chains, the sequence identity is no more than 30%. The numbers of interacting and non-interacting res-idues are 3,581 and 21,526, respectively. The RBP109 data set is downloaded from RNABindR web server [11]. In Terribilini et al. [6], this is named as the "RB109" data set.
3. RBP107
Derived from 61 RNA-protein complexes in PDB, the RBP107 data set is comprised of 107 protein chains with X-ray crystallography resolution better than 3.5 Å and sequence identity no more than 25%. Based on the cut-off distance of 3.5 Å, the RBP107 data set contains 2,555 interacting residues and 19,496 non-interacting ones. Wang and Brown [9] applied this data set to construct and evaluated their approach. In Kumar et al. [8], it is referred to as the "alternate" data set.
Support vector machines (SVM)
SVM is a machine learning approach proposed by Vapnik [12] based on structural risk minimization principle of statistics learning theory. It can be used to deal with clas-sification or regression. Distinguishing RNA binding resi-dues form non-binding resiresi-dues in a protein could be regarded as a binary classification problem. For a set of given input data vectors xi (xi ∈ ⺢d, i = 1, 2,..., n) with
labels yi (yi ∈ {+1, -1}, i = 1, 2,..., n; where "+1" represents
a positive instance and "-1" denotes a negative instance), the mission in the training procedure is to optimize the following equation that maps input vectors into a higher dimensional feature space (i.e., Hilbert space), and seeks a separation hyperplane with a maximum margin to divide positive instances from negative ones. The calcula-tion of SVM is defined in Equacalcula-tion (1).
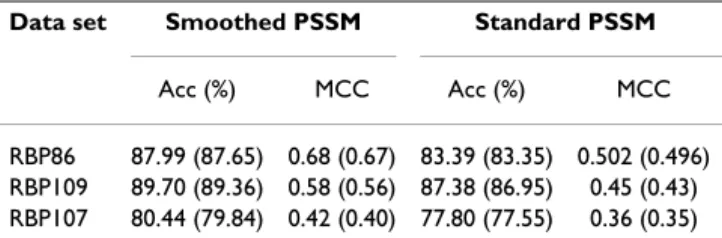
Table 1: Summary of three benchmark data sets
Data set RBP86 RBP109 RBP107
Number of protein chains 86 109 107
X-ray crystallography resolution >3 Å >3.5 Å >3.5 Å
Sequence identity ≤70% ≤30% ≤25%
Number of interacting residues 4,568 3,581 2,555 Number of non-interacting residues 15,503 21,526 19,496 Non-interacting/interacting residues 3.39 6.01 7.63
where w ∈ ⺢d is a weight vector, b is a bias (constant), and
Φ is a mapping function. For more flexible classification, SVM allows instance i positions at the wrong side of hyperplane with slack variable ξi and cost parameter C. In
SVM, a kernel function K(xi, xj), such as linear, polyno-mial, radial basis function (RBF), and sigmoid function, is used to present Φ(xi)·Φ(xj) where xi and xj are two data vectors. In this study, we use RBF as the kernel function in the SVM. The formulation of RBF is defined in Equation (2), where γ is a training parameter.
K(xi, xj) = exp(-γ||xi - xj||2) (2)
Developed by Lin et al. [13], LIBSVM is a powerful and well-known SVM package used by many researchers. We apply LIBSVM to implement our classifiers for prediction of RNA-binding sites in proteins.
Feature extraction and representation
Evolutionary information has been shown to be effective for RNA-binding site prediction [8]. For this reason, we use PSI-BLAST [14] to search against NCBI non-redundant (nr) database and generate a PSSM based on BLOSUM62 substitution matrix [15] for each protein with e-value as 0.001 and iteration number as 3. A PSSM is comprised of
L vectors (L denotes the length of the protein), in which
contain the log-likelihoods for different amino acids in a position. Next, we illustrate two different encoding schemes to represent the PSSM.
1. Standard PSSM encoding scheme
Standard PSSM has been used for RNA-binding site pre-diction by Kumar et al. [8]. For a PSSM profile, the feature representation of a residue αi at position i in a protein sequence is presented by an evolutionary information vec-tor Vi comprised of log-likelihoods for 20 different amino acids. Considering the surrounding residues of αi, we apply a sliding window of size w to incorporate the evolu-tionary information from upstream and downstream neighbors. The feature vector of a residue αi is represented by (Vi-(w-1)/2,..., Vi,..., Vi+(w-1)/2). For the N-terminal and
C-terminal of a protein, (w-1)/2 ZERO vectors, consisting of 20 zero elements, are appended to the hand or tail of a PSSM profile. The feature values in each vector are nor-malized to a range between -1 and 1. In our study, we apply different sliding window sizes ranging from 3 to 41 with a step as 2 (i.e., w = 3, 5,..., 41). Figure 1(A) shows an example of standard PSSM of a protein with e-value as 0.001 and iteration number as 3 in PSI-BLAST.
2. Smoothed PSSM encoding scheme
In addition to the consideration of neighbors of a residue αi, we propose a new encoding scheme to incorporate the dependency of surrounding residues. In a standard PSSM profile, the log-likelihood at each position is calculated based on an assumption that each position is independ-ent from the others. However, Terribilini et al. [6] observed that RNA binding residues tend to occur in
clus-Min w w C subject to y w x b w b T i i n i T i i i , , ( ( ) ) ξ ξ ξ 1 2 1 1 + ⎛ ⎝⎜ ⎞ ⎠⎟ + ≥ − =
∑
Φ ,,ξi ≥0,i=1 2, ,..., ,n (1)Examples of (A) standard PSSM and (B) smoothed PSSM generated by PSI-BLAST (e-value = 0.001, iteration number = 3)
Figure 1
ters. Their analysis revealed that 95% of interacting resi-dues in the RBP109 data set have at least one additional interacting residue among the four amino acids on either side, and 49% of those have at least four. Inspired by the consideration of adjacent pixels used in the spatial domain method from the research field of image process-ing [10], we present a new encodprocess-ing scheme to model the dependency or correlation among surrounding neighbors of a central residue. Similar to the feature representation in standard PSSM encoding, we use a sliding window of size w to incorporate the evolutionary information from upstream and downstream residues. In the construction of a smoothed PSSM, each row vector of a residue αi is rep-resented and smoothed by the summation of ws sur-rounding row vectors (Vsmoothed_i = Vi-(ws-1)/2 + ... + Vi + ... +
Vi+(ws-1)/2). For the N-terminal and C-terminal of a pro-tein, (w-1)/2 ZERO vectors, are appended to the hand or tail of a smoothed PSSM profile. Using the smoothed PSSM encoding scheme, the feature vector of a residue αi is represented by (Vsmoothed_i-(w-1)/2,..., Vsmoothed_i,...,
Vsmoothed_i+(w-1)/2). The feature values in each vector are normalized to a range between -1 and 1. Here, we apply different smoothing window sizes from 3 to 11 with a step as 2 (i.e., ws = 3, 5,..., 11). Figure 1(B) illustrates an exam-ple of a smoothed PSSM profile. At position 9, the corre-sponding value of amino acid 'A' represented by a smoothed PSSM encoding is the sum of [(-2)+(-2)+(-3)+5+(-2)+3+0].
Window size selection and parameter optimization In order to optimize the performance of RNAProB, we have to determine the best combination of several param-eters, including the sliding window size w, cost parameter
C and kernel parameter γ in the SVM classifier, the
smoothing window size ws, and the weight parameters w1 and w-1 in SVM. Table 2 shows the workflow of window size selection and parameter optimization. In our study, the best parameters are optimized with respect to overall accuracy. First, we test the performance of different sliding window sizes w from 3, 5, 7,..., 41 in standard PSSM
encoding scheme using default C and γ parameters in
SVM, and initial weight parameter w1 as 1 and w-1 as the ratio of the number of non-interacting residues to that of interacting residues in a data set. As shown in Table 1, the ratios of the numbers of non-interacting residues to those of interacting residues in the RBP86, RBP109, and RBP107 data sets are 1:3.39, 1:6.01, and 1:7.63, respec-tively. Second, based on the optimized sliding window size w selected from the first step, the best combination of cost parameter C and kernel parameter γ is determined
with initial weight parameters. The log2C and log2γ ranged from -3 to 12 and -3 to -15, respectively. Third, the prediction performance of different smoothing window sizes ws ranged from 3 to 11 with a step 2 is evaluated using initial weight parameters and previously selected parameters (i.e., w, C, and γ). Fourth, due to data set imbalance, the weight parameters w1 and w-1 are tuned with optimized w, C, γ, and ws. After these steps, the opti-mal parameters, including sliding window size w, cost parameter C, kernel parameter γ, smoothing window size
ws, and weight parameters w1 and w-1, are determined.
System architecture
The system architecture of RNAProB is shown in Figure 2. Given a protein sequence, RNAProB performs the follow-ing steps:
1. Apply PSI-BLAST to generate a standard PSSM of the protein.
2. Generate a smoothed PSSM of the protein using an optimized smoothing window size.
3. Construct a feature vector for each residue in the pro-tein sequence by an optimized sliding window size, and normalize all feature values in the vector into a range of -1 and -1.
Table 2: The workflow of window size selection and parameter optimization.
Sliding window size (w) C and γ Smoothing window size (ws) Weight parameter (w1 and w-1)
Step 1 3 ≤ w ≤ 41 (step = 2) Default - Default ratio
Step 2 Optimized w from step 1 -3 ≤ log2C ≤ 12 (step = 1) Default ratio
-3 ≤ log2γ ≤ -15 (step = -1)
Step 3 Optimized w from step 1 Optimized C and γ from step 2 3 ≤ ws ≤ 11 (step = 2) Default ratio Step 4 Optimized w from step 1 Optimized C and γ from step 2 Optimized ws from step 3 1 ≤ w1 ≤ 8# (step = 1), w-1 = 1
Final Optimized w from step 1 Optimized C and γ from step 2 Optimized ws from step 3 Optimized w1 and w-1 from step4
# In the RBP107 data set, we test w
System architecture of RNAProB
Figure 2
4. Use a trained SVM classifier with optimized parameters (C, γ, w1, w-1) to predict the interacting and non-interact-ing residues in the protein.
After the above steps, RNAProB outputs the correspond-ing interactcorrespond-ing or non-interactcorrespond-ing state of each residue in the protein.
Training and testing
The performance of RNAProB is assessed by n-fold cross-validation and three-way data split. To compare with other approaches, we use five-fold cross-validation to evaluate the performance of RNAProB. However, to pre-vent data-overfitting, a three-way data split procedure is applied to assess our predictor. The performance of RNAProB is evaluated as follows.
1. n-fold cross-validation
A data set is randomly divided into five distinct non-over-lapping sets of positive and negative instances (i.e., n = 5), four of which are used to train the predictor and the accu-racy of the predictor is evaluated on the remaining set. This procedure is repeated five times.
2. Three-way data split
To avoid over fitting, we use a more stringent three-way data split procedure [16,17] to evaluate the performance of RNAProB. A data set is randomly partitioned into three non-overlapping sets: a training set for classifier learning, a validation set for parameter selection, and a test set for performance evaluation. In this paper, we divide a data set into five distinct sets, three for training, one for validation, and one for testing. The procedure is also iterated 5 times. Performance evaluation measures
For comparison with other approaches, we follow the measures used in previous work [8,9,18], including specif-icity (Spec), sensitivity (Sens), MCC [19], and overall accuracy (Acc). Specificity and sensitivity measure how well the binary classifier recognizes negative and positive cases, respectively. A specificity of 100% and a sensitivity of 100% imply that the classifier identifies all non-inter-acting residues as non-internon-inter-acting and all internon-inter-acting resi-dues as interacting, correspondingly. When a predictor's specificity increases, its sensitivity often decreases. On the other hand, MCC, which considers both under- and over-predictions, gives a complementary measure of the predic-tion performance, where MCC = 1 denotes a perfect pre-diction, MCC = 0 indicates a completely random assignment, and MCC = -1 means a perfectly reverse cor-relation. Moreover, overall accuracy presents how well the classifier distinguishes true positives and true negatives, and 100% overall accuracy denotes a perfect prediction. The definitions of specificity, sensitivity, MCC, and over-all accuracy are defined in Equations (3), (4), (5), and (6),
respectively. In the equations, TP, TN, FP, and FN denote the numbers of true positives, true negatives, false posi-tives, and false negaposi-tives, correspondingly.
Specificity = TN/(TN + FP) × 100 (3) Sensitivity = TP/(TP + FN) × 100 (4)
Acc = (TP + TN)/(TP + TN + FP + FN) × 100 (6) In addition to the above measures, we also use the receiver operating characteristic (ROC) curve [20] and area under the ROC curve (AUC) [21] to evaluate the performance of standard and smoothed PSSM encoding schemes. In an ROC curve plot, the X-axis represents false positive rate (i.e., 1-specificity) and Y-axis denotes true positive rate (i.e., sensitivity). We incorporate different thresholds in the SVM classifier to plot the true positive rates against false positive rates in an ROC curve. Moreover, AUC cal-culates the area under an ROC curve and the maximum value of AUC is 1, which denotes a perfect prediction. A random guess results in an AUC value close to 0.5. To determine the thresholds in the SVM classifiers, we fol-low the criteria used in the previous work. We notice that the thresholds in other approaches are optimized with respect to different measures. For example, Kumar et al. [8] and Jeong and Miyano [7] both optimized their results in the RBP86 data set based on MCC. In addition, Terri-bilini et al. [6] also selected the thresholds with the best MCC for the RBP109 data set. On the other hand, Wang and Brown [18] determined the best thresholds in the RBP107 data set based on the average of specificity and sensitivity. Therefore, the thresholds in RNAProB are opti-mized with respect to MCC for the RBP86 and RBP109 data sets, while the threshold is determined by the average of sensitivity and specificity for the RBP107 data set.
Results
Effect of smoothed PSSM encoding scheme
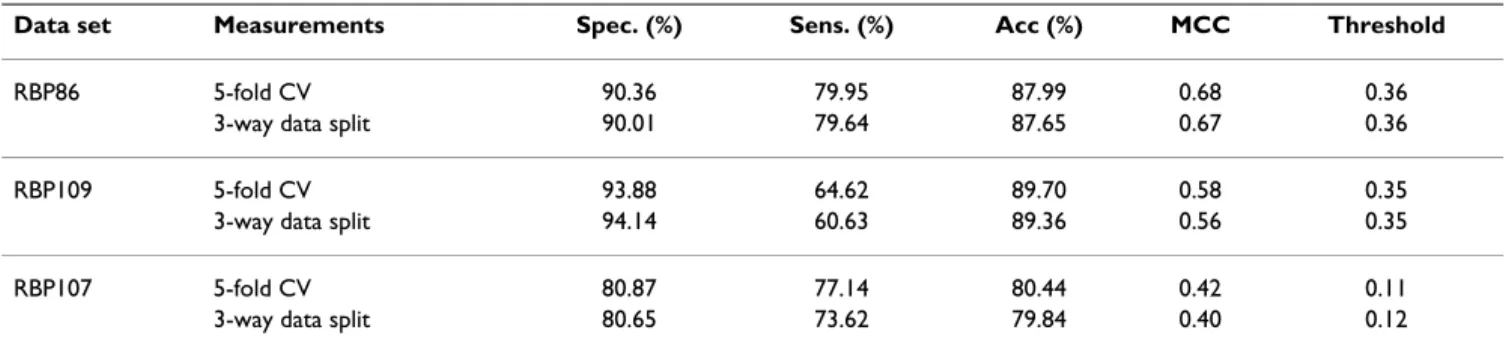
Here we compare the performance of smoothed PSSM and standard PSSM encoding scheme in terms of MCC, overall accuracy, ROC curve, and AUC for the benchmark data sets. Table 3 shows the performance comparison of standard PSSM and smoothed PSSM using five-fold cross-validation and three-way data split. Evaluated by five-fold cross-validation, smoothed PSSM encoding scheme attains overall accuracy of 87.99%, 89.70%, and 80.44% compared to 83.39%, 87.38%, and 77.80% by standard PSSM encoding for the RBP86, RBP109, and RBP107 data sets, respectively. Moreover, smoothed PSSM encoding
MCC=(TP×TN−FP×FN) / (TP+FP) (× TP+FN) (× TN+FP) (× TN+FN)
scheme achieves improvements of 0.06~0.178 in MCC compared to standard PSSM. Similarly, assessed by three-way data split, smoothed PSSM encoding also performs better than standard PSSM in terms of both overall accu-racy and MCC in the three data sets.
Figure 3(A), (B), and 3(C) illustrate the ROC curves and AUC of smoothed PSSM and standard PSSM encoding schemes for the three benchmark data sets. The solid blue line and dotted red line represent the ROC curves plotted according to the performance of smoothed PSSM and standard PSSM encoding schemes, respectively. When smoothed PSSM encoding scheme is used to represent the proteins, AUC achieve 0.929, 0.902, and 0.860 on the RBP86, RBP109, and RBP107 data sets, respectively; on the other hand, standard PSSM only attains AUC of 0.835, 0.824, and 0.817.
Experimental results demonstrate that our proposed smoothed PSSM encoding scheme not only achieves good prediction performance, but also yields a significant
improvement over standard PSSM encoding. Smoothed PSSM encoding scheme outperforms standard PSSM by 2.32%~4.60% in overall accuracy and 0.06~0.178 in MCC. The consideration of dependency among neighbor-ing residues works well in distneighbor-inguishneighbor-ing interactneighbor-ing resi-dues from non-interacting ones; accordingly, the prediction performance of smoothed PSSM encoding scheme is substantially improved. This supports our assumption that the incorporation of the correlation between surrounding residues in PSSM profiles can signif-icantly enhance the performance of RNA-binding site pre-diction.
RNAProB prediction performance on the benchmark data sets
For each data set, we used five-fold cross-validation and three-way data split to evaluate the prediction perform-ance, which is detailed below and summarized in Table 4.
1. Performance comparison with other approaches on the RBP86 data set
The window sizes, including the sliding window size w and smoothing window size ws, and other parameters in RNAProB are selected with respect to overall accuracy. First, Figure 4(A) shows the overall accuracy of applying different sliding window sizes on the RBP86 data set. The overall accuracy evaluated by both five-fold cross-valida-tion and three-way data split grows rapidly before it reaches 77%. However, a slow growth in the overall accu-racy is observed as the size of sliding window is greater than 25. Thus, the sliding window size w is set as 25 for the RBP86 data set. Next the prediction performance of different smoothing window sizes based on previously
Table 3: Performance comparison of standard PSSM and smoothed PSSM.
Data set Smoothed PSSM Standard PSSM
Acc (%) MCC Acc (%) MCC
RBP86 87.99 (87.65) 0.68 (0.67) 83.39 (83.35) 0.502 (0.496) RBP109 89.70 (89.36) 0.58 (0.56) 87.38 (86.95) 0.45 (0.43) RBP107 80.44 (79.84) 0.42 (0.40) 77.80 (77.55) 0.36 (0.35)
§ The performance of incorporating a three-way data split procedure
is shown in the parentheses.
ROC curves and AUC of the (A) RBP86, (B) RBP109, and (C) RBP107 data sets
Figure 3
ROC curves and AUC of the (A) RBP86, (B) RBP109, and (C) RBP107 data sets.
determined sliding window size (i.e. w = 25) is illustrated in Figure 4(B) and 4(C). In Figure 4(B), although there is a very slow growth in the overall accuracy, we observe that MCC is improved from 0.50 to 0.67 when the size of smoothing window is increased from 1 to 7. Nevertheless, the performance improvement in MCC (i.e. improvement < 0.01) is not significant as the size of smoothing window is greater than 7. Similar trends in MCC and overall accu-racy are also observed in Figure 4(C). Therefore, we use 7 as the smoothing window size ws in our method. As shown in Table 4, the performance of RNAProB evaluated by five-fold cross-validation achieves MCC, overall accu-racy, specificity, and sensitivity of 0.68, 87.99%, 90.36%, and 79.95%, (with sliding window size w = 25, smoothing window size ws = 7, cost parameter C = 4, kernel function parameter γ = 0.015625, weight parameter w1 = 4, w-1 = 1, and threshold value = 0.36), respectively. Besides, using a more rigorous three-way data split procedure, our method also attains MCC, overall accuracy, specificity, and sensi-tivity of 0.67, 87.65%, 90.01%, and 79.64%, (with w = 25,
ws = 7, C = 1, γ = 0.03125, w1 = 4, w-1 = 1, and threshold value = 0.36), correspondingly. The experimental results of window size selection and parameter optimization on the RBP86 data set are shown in the supplementary mate-rial [see Additional file 4].
The performance comparison with two other approaches developed on the same data set is shown in Table 5. Jeong and Miyano [7] used an ANN to incorporate evolutionary information and obtained MCC, overall accuracy, specifi-city, and sensitivity of 0.39, 80.20%, 91.04%, and 43.40%, respectively. The MCC of their proposed method was further improved to 0.41 based on a weighted profile approach. In addition, Kumar et al. developed PPRint [8], which incorporated PSSM profiles in an SVM model, and attained MCC, overall accuracy, specificity, and sensitivity of 0.45, 81.16%, 89.55%, and 53.05%, respectively. Com-pared to these approaches, our method not only achieves high overall accuracy but also significantly improves the sensitivity by 26.90%~36.55% using five-fold cross-vali-dation. Moreover, RNAProB achieves 0.68 in MCC, com-pared to 0.45 by PPRint and 0.41 by Jeong and Miyano.
2. Performance comparison with RNABindR on the RBP109 data set
Figure 5 illustrates the experimental results of different sliding and smoothing window sizes on the RBP109 data set. Similar to the RBP86 data set, the RBP109 data set exhibits a slow growth in the prediction performance when sliding window size w is greater than 25 or smooth-ing window size ws is larger than 7. Thus, we also select w as 25 and ws as 7 for this data set. Table 4 shows that RNAProB attains 0.58, 89.70%, 93.88%, and 64.62% in MCC, overall accuracy, specificity, and sensitivity using five-fold cross-validation (with w = 25, ws = 7, C = 4, γ =
0.015625, w1 = 4, w-1 = 1, and threshold value = 0.35), respectively. Besides, evaluated by three-way data split, our method obtains MCC, overall accuracy, specificity, and sensitivity of 0.56, 89.36%, 94.14%, and 60.63% (with w = 25, ws = 7, C = 8, γ = 0.015625, w1 = 4, w-1 = 1, and threshold value = 0.35), respectively. The prediction performance of different window sizes and parameters on the RBP109 data set is detailed in the supplementary material [see Additional file 5].
Table 5 illustrates the performance comparison with RNABindR [6,11], a Naïve Bayes based method developed on the same data set. Using five-fold cross-validation, RNAProB achieves 0.58, 89.70%, 93.88%, and 64.62% in MCC, overall accuracy, specificity, and sensitivity, respec-tively, compared favourably to 0.35, 84.80%, 93.00%, and 38.00% by RNABindR. Particularly, our method sig-nificantly outperforms RNABindR by 26.62% in terms of sensitivity.
3. Performance comparison with other approaches on the RBP107 data set
The prediction performance of different sliding and smoothing window sizes on the RBP107 data set is dem-onstrated in Figure 6. Similar to the RBP86 data set, we observe that the overall accuracy converges as sliding win-dow size is greater than 25 on the RBP107 data set in Fig-ure 6(B). Moreover, the MCC shows a slight peak when the smoothing window size reaches 7 in Figure 6(C). Thus RNAProB also selects w as 25 and ws as 7 for this data set. As illustrated in Table 4, our method reaches 0.42,
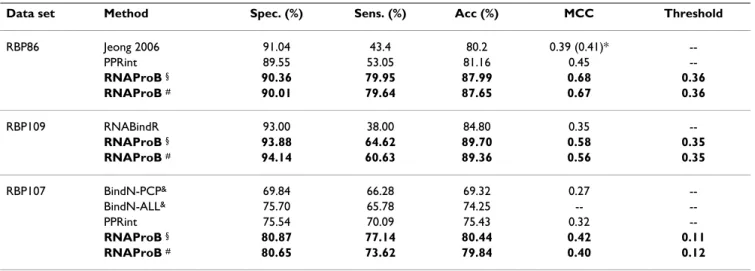
Table 4: Performance of five-fold cross-validation and three-way data split for the benchmark data sets.
Data set Measurements Spec. (%) Sens. (%) Acc (%) MCC Threshold
RBP86 5-fold CV 90.36 79.95 87.99 0.68 0.36
3-way data split 90.01 79.64 87.65 0.67 0.36
RBP109 5-fold CV 93.88 64.62 89.70 0.58 0.35
3-way data split 94.14 60.63 89.36 0.56 0.35
RBP107 5-fold CV 80.87 77.14 80.44 0.42 0.11
(A) Accuracy with respect to different sliding window sizes using five-fold cross-validation and three-way data split for the RBP86 data set, respectively
Figure 4
(A) Accuracy with respect to different sliding window sizes using five-fold cross-validation and three-way data split for the RBP86 data set, respectively. (B) The performance of the RBP86 data set with different smoothing window sizes by five-fold cross-validation. (C) The performance of the RBP86 data set with different smoothing window sizes by three-way data split.
(A)
(B)
80.44%, 80.87%, and 77.14% in MCC, overall accuracy, specificity, and sensitivity by five-fold cross-validation (with w = 25, ws = 7, C = 4, γ = 0.015625, w1 = 4, w-1 = 1, and threshold value = 0.11), respectively. In addition, RNAProB also attains MCC, overall accuracy, specificity, and sensitivity of 0.40, 79.84%, 80.65%, and 73.62% by three-way data split (with w = 25, ws = 7, C = 8, γ =
0.015625, w1 = 4, w-1 = 1, and threshold value = 0.12), cor-respondingly. The detailed experimental results on the RBP109 data set are summarized in the supplementary material [see Additional file 6].
Table 5 compares the performance of RNAProB with other approaches on the RBP107 data set. Based on physico-chemical properties, BindN (i.e. referred to as BindN-PCP in Table 5) attains MCC, overall accuracy, specificity, and sensitivity of 0.27, 69.32%, 69.84%, and 66.28%, respec-tively [9]. Incorporated with more biological features, BindN (i.e. denoted as BindN-ALL in Table 5) further improves specificity and accuracy by 5.86% and 4.93% with a slight decrease in sensitivity [18]. PPRint improves sensitivity to 70.09% with the other measures performed comparable to those of BindN-ALL. Our method signifi-cantly outperforms the-state-of-the-art approaches by 0.10, 5.10%, 5.33%, and 7.05% in MCC, overall accuracy, specificity, and sensitivity, respectively. This demonstrates that RNAProB not only achieves accurate performance, but also substantially improves sensitivity in the predic-tion of RNA-binding sites.
Discussion
Physicochemical preferences of interacting and non-interacting residues
In this section, we examine the physicochemical proper-ties of RNA interacting and non-interacting residues. Fig-ure 7(A), (B), and 7(C) show the amino acid compositions of interacting and non-interacting residues in the RBP86, RBP109, and RBP107 data sets, respectively. It is observed that interacting and non-interacting residues show preferences for different amino acids. RNA interact-ing residues tend to have high compositions for Arginine (R), Asparagine (N), Glutamine (Q), Glycine (G), Histi-dine (H), and Lysine (K). For example, there are relatively high proportions for Arginine (R) and Lysine (K), which may interact with negatively charged RNA with their pos-itive side chains. In addition, the smallest amino acid, Glycine (G), also has a high composition in interacting residues because it rotates easily and provides flexibility to interact with RNA molecules. Moreover, positively charged Histidine (H) can have an aromatic interaction with RNA molecules due to its specific pKa value and imi-dazole ring. On the other hand, non-interacting residues show slight preferences for Alanine (A), Aspartic acid (D), Glutamic acid (E), Isoleucine (I), Leucine (L), Phenyla-lanine (F), and Valine (V). Cysteine (C), Aspartic acid (D), and Glutamic acid (E) are favoured by non-interacting res-idues because of their negatively charged side chains. In addition, although Kumar et al. [8] reported that Aspartic acid (D) showed no preference for interacting or non-interacting residues in their main data set (i.e., the RBP86 data set in our study), we observed that the Aspartic acid (D) composition of non-interacting residues is signifi-cantly higher than that of interacting residues in both of
Table 5: Performance comparison of different approaches using five-fold cross-validation for the benchmark data sets.
Data set Method Spec. (%) Sens. (%) Acc (%) MCC Threshold
RBP86 Jeong 2006 91.04 43.4 80.2 0.39 (0.41)* --PPRint 89.55 53.05 81.16 0.45 --RNAProB § 90.36 79.95 87.99 0.68 0.36 RNAProB # 90.01 79.64 87.65 0.67 0.36 RBP109 RNABindR 93.00 38.00 84.80 0.35 --RNAProB § 93.88 64.62 89.70 0.58 0.35 RNAProB # 94.14 60.63 89.36 0.56 0.35 RBP107 BindN-PCP& 69.84 66.28 69.32 0.27 --BindN-ALL& 75.70 65.78 74.25 -- --PPRint 75.54 70.09 75.43 0.32 --RNAProB § 80.87 77.14 80.44 0.42 0.11 RNAProB # 80.65 73.62 79.84 0.40 0.12
§ presents the performance by five-fold cross-validation. # denotes the performance by a three-way data split procedure.
* indicates the performance of weighted profiles by Jeong and Miyano [7].
&BindN-PCP represents the results based only on physicochemical properties, while BindN-ALL shows the performance using physicochemical
(A) Accuracy with respect to different sliding window sizes using five-fold cross-validation and three-way data split for the RBP109 data set, respectively
Figure 5
(A) Accuracy with respect to different sliding window sizes using five-fold cross-validation and three-way data split for the RBP109 data set, respectively. (B) The performance of the RBP109 data set with different smoothing window sizes by five-fold cross-validation. (C) The performance of the RBP109 data set with different smoothing window sizes by three-way data split.
(A)
(B)
(A) Accuracy with respect to different sliding window sizes using five-fold cross-validation and three-way data split for the RBP107 data set, respectively
Figure 6
(A) Accuracy with respect to different sliding window sizes using five-fold cross-validation and three-way data split for the RBP107 data set, respectively. (B) The performance of the RBP107 data set with different smoothing window sizes by five-fold cross-validation. (C) The performance of the RBP107 data set with different smoothing window sizes by three-way data split.
(A)
(B)
Amino acid compositions of interacting and non-interacting residues in the benchmark data sets
Figure 7
Amino acid compositions of interacting and non-interacting residues in the benchmark data sets.
(A) The RBP86 data set
(B) The RBP86 data set
Grouped amino acid compositions of interacting and non-interacting residues in the benchmark data sets
Figure 8
Grouped amino acid compositions of interacting and non-interacting residues in the benchmark data sets.
(A) The RBP86 data set
(B) The RBP109 data set
the RBP109 and RBP107 data sets. Our analysis indicates that the finding from Kumar et al. could be a bias from the data set.
To further analyze the physicochemical properties of the RNA interacting and non-interacting residues, each amino acid is classified into one of the four groups: acidic (DE), basic (HKR), polar (CGNQSTY), and non-polar (AFILM-PVW) [22]. Figure 8 shows the grouped amino acid com-positions of interacting and non-interacting residues for the benchmark data sets. It is observed among the three data sets that basic and polar amino acids tend to interact with RNA, and acidic and non-polar amino acids are not favoured by RNA molecules. Particularly, our analysis shows that the compositions of basic amino acids exhibit significantly over-represented patterns for interacting resi-dues.
Furthermore, we inspect the amino acid compositions of proteins that interact with different RNA molecules. The proteins in the RBP109 data set are divided into four cat-egories according to the definition in Terribilini et al [6]. Figure 9(A), (B), (C), and 9(D) show the amino acid com-positions of (A) rRNA, (B) mRNA, snRNA, dsRNA, and siRNA, (C) tRNA, and (D) viralRNA, respectively. It is observed that viralRNA group shows a different amino acid composition compared to the other groups. Proteins that interact with viralRNA evolve fast and induce confor-mational changes in the active sites. Thus, these proteins exhibit a specific mechanism to interact with viralRNA. Comparison of smoothed PSSM and standard PSSM Here we examine the correlation between interacting and non-interacting residues for both smoothed PSSM and standard PSSM encoding schemes. We incorporate Pear-son correlation coefficient (PCC) [23] to measure the cor-relation between the evolutionary information of
Amino acid compositions of interacting and non-interacting residues in four different RNA groups of the RBP109 data set
Figure 9
Amino acid compositions of interacting and non-interacting residues in four different RNA groups of the RBP109 data set.
(A) The rRNA group (55 protein chains with 2,392
interacting and 5,302 non-interacting residues).
(B) The mRNA, snRNA, dsRNA, and siRNA group
(23 protein chains with 394 interacting and
3,320 non-interacting residues).
(C) The tRNA group (19 protein chains with 646
interacting and 9,095 non-interacting residues).
(D) The viralRNA group (12 protein chains with 149
interacting and 3,809 non-interacting residues).
interacting and non-interacting for an amino acid. For each amino acid a, we use two vectors, X and Y, to present the sum of PSSM evolutionary information vectors for
interacting and non-interacting amino acid a, respectively. The Pearson correlation coefficient for a series of n meas-urements for variables X and Y is defined in Equation (7).
Pearson correlation coefficient between interacting and non-interacting evolutionary vectors generated by different PSSM encoding schemes in the benchmark data sets
Figure 10
Pearson correlation coefficient between interacting and non-interacting evolutionary vectors generated by different PSSM encoding schemes in the benchmark data sets.
(A) The RBP86 data set
(B) The RBP109 data set
Figure 10 shows the Pearson correlation coefficient between interacting and non-interacting evolutionary information vectors based on different PSSM encoding schemes in the benchmark data sets. It is observed that the correlation coefficients calculated from smoothed PSSM encoding scheme are lower than those from standard PSSM, especially for Cysteine (C) and Tryptophan (W). In Figure 10(A), smoothed PSSM encoding attains lower cor-relation coefficients not only in interacting residues, such as arginine (R), asparagine (N), glutamine (Q), glycine (G), histidine (H), and lysine (K), but also in non-inter-acting residues, including alanine (A), aspartic acid (D), glutamic acid (E), isoleucine (I), leucine (L), phenyla-lanine (F), and valine (V). Similarly, Figure 10(B) and 10(C) also show lower correlation coefficients between interacting and non-interacting residues based on smoothed PSSM encoding. Furthermore, it is observed that the correlation coefficients calculated with smooth-ing window size ws = 7 are usually lower than those gen-erated by other smoothing window sizes. If an encoding scheme leads to a lower Pearson correlation coefficient, it indicates that the encoding scheme can better resolve ambiguity in discriminating interacting residues from non-interacting ones. Our analysis lends support to our assumption that smoothed PSSM encoding scheme can improve the recognition RNA interacting and non-inter-acting sites by modelling the dependency from surround-ing residues.
Conclusion
We present RNAProB, which combines a new smoothed PSSM encoding scheme with a SVM model for prediction of RNA-binding sites in proteins. In a standard PSSM pro-file, evolutionary information is calculated based on an assumption that each position is independent of others. However, the correlation or dependency from surround-ing residues is incorporated in the proposed smoothed PSSM encoding. Experimental results show that the pre-diction performance of smoothed PSSM encoding per-forms better than the state-of-the-art approaches on the benchmark data sets. Evaluated by five-fold cross-valida-tion, RNAProB outperforms the other approaches by 0.10~0.23 in MCC, 4.90%~6.83% in overall accuracy, and 0.88%~5.33% in specificity. Most notably, our method significantly improves sensitivity by 26.90%, 26.62%, and 7.05% for the RBP86, RBP109, and RBP107 data sets, respectively. Performance improvement in RNAProB not only demonstrates that smoothed PSSM can better resolve the ambiguity in discriminating RNA inter-acting and non-interinter-acting residues, but also supports our assumption that consideration of correlation between
neighboring residues can significantly enhance prediction accuracy. To prevent data over fitting, a rigorous three-way data split procedure is incorporated to evaluate our pre-diction performance. The proposed method can be used in other research topics, such as DNA-binding site predic-tion, protein-protein interacpredic-tion, and prediction of post-translational modification sites.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
CWC developed the method, implemented the system, and drafted the manuscript. ECYS provided biological knowledge, participated in the experimental design, and refined the manuscript. TYS and WLH coordinated this study. All of authors read and approved the final manu-script.
Additional material
PCC=rxy=(n
∑
x yi i−∑ ∑
xi yi) / n∑ ∑
xi2−( xi)2 n∑
yi2−(∑
yi)2(7)
Additional file 1
The RBP86 data set. Click here for file
[http://www.biomedcentral.com/content/supplementary/1471-2105-9-S12-S6-S1.doc]
Additional file 2
The RBP109 data set. Click here for file
[http://www.biomedcentral.com/content/supplementary/1471-2105-9-S12-S6-S2.doc]
Additional file 3
The RBP107 data set. Click here for file
[http://www.biomedcentral.com/content/supplementary/1471-2105-9-S12-S6-S3.doc]
Additional file 4
Detailed experimental results on the RBP86 data set. Click here for file
[http://www.biomedcentral.com/content/supplementary/1471-2105-9-S12-S6-S4.doc]
Additional file 5
Detailed experimental results on the RBP109 data set. Click here for file
[http://www.biomedcentral.com/content/supplementary/1471-2105-9-S12-S6-S5.doc]
Additional file 6
Detailed experimental results on the RBP107 data set. Click here for file
[http://www.biomedcentral.com/content/supplementary/1471-2105-9-S12-S6-S6.doc]
Publish with BioMed Central and every scientist can read your work free of charge "BioMed Central will be the most significant development for disseminating the results of biomedical researc h in our lifetime."
Sir Paul Nurse, Cancer Research UK Your research papers will be:
available free of charge to the entire biomedical community peer reviewed and published immediately upon acceptance cited in PubMed and archived on PubMed Central yours — you keep the copyright
Submit your manuscript here:
http://www.biomedcentral.com/info/publishing_adv.asp
BioMedcentral
Acknowledgements
We would like to express our sincere thanks to Dr. Kumar, Dr. Terribilini, and Dr. Jeong for kindly providing their data sets. We also thank Jia-Ming Chang, Allan Lo, Hua-Sheng Chiu, Ei-Wen Yang, Yi-Yuan Chiu, and Dr. Ming-Tat Ko for helpful discussion and useful suggestions.
This article has been published as part of BMC Bioinformatics Volume 9 Sup-plement 12, 2008: Asia Pacific Bioinformatics Network (APBioNet) Seventh International Conference on Bioinformatics (InCoB2008). The full contents of the supplement are available online at http://www.biomedcentral.com/ 1471-2105/9?issue=S12.
References
1. Sunita S, Purta E, Durawa M, Tkaczuk KL, Swaathi J, Bujnicki JM, Sivar-aman J: Functional specialization of domains tandemly dupli-cated within 16S rRNA methyltransferase RsmC. Nucleic Acids Res 2007, 35(13):4264-4274.
2. Bechara E, Davidovic L, Melko M, Bensaid M, Tremblay S, Grosgeorge J, Khandjian EW, Lalli E, Bardoni B: Fragile X related protein 1 isoforms differentially modulate the affinity of fragile X men-tal retardation protein for G-quartet RNA structure. Nucleic Acids Res 2007, 35(1):299-306.
3. McKnight KL, Heinz BA: RNA as a target for developing antivi-rals. Antivir Chem Chemother 2003, 14(2):61-73.
4. Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S, et al.: The Protein Data Bank. Acta Crystallogr D Biol Crystallogr 2002, 58(Pt 6 No 1):899-907. 5. Jeong E, Chung IF, Miyano S: A neural network method for
iden-tification of RNA-interacting residues in protein. Genome Inform 2004, 15(1):105-116.
6. Terribilini M, Lee JH, Yan C, Jernigan RL, Honavar V, Dobbs D: Pre-diction of RNA binding sites in proteins from amino acid sequence. RNA 2006, 12(8):1450-1462.
7. Jeong E, Miyano S: A Weighted Profile Based Method for Pro-tein-RNA Interacting Residue Prediction. Transactions on Com-putational Systems Biology 2006:123-139.
8. Kumar M, Gromiha MM, Raghava GP: Prediction of RNA binding sites in a protein using SVM and PSSM profile. Proteins 2008, 71(1):189-194.
9. Wang L, Brown SJ: BindN: a web-based tool for efficient predic-tion of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res 2006:W243-248.
10. Gonzalez RC, Woods RE: Digital Image Processing. Prentice Hall; 2002.
11. Terribilini M, Sander JD, Lee JH, Zaback P, Jernigan RL, Honavar V, Dobbs D: RNABindR: a server for analyzing and predicting RNA-binding sites in proteins. Nucleic Acids Res 2007:W578-584. 12. Vapnik VN: The Nature of Statistical Learning Theory.
Springer; 1995.
13. LIBSVM: a library for support vector machines [http:// www.csie.ntu.edu.tw/~cjlin/libsvm/]
14. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lip-man DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25(17):3389-3402.
15. Henikoff S, Henikoff JG: Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA 1992, 89(22):10915-10919. 16. Ritchie MD, White BC, Parker JS, Hahn LW, Moore JH: Optimiza-tion of neural network architecture using genetic program-ming improves detection and modeling of gene-gene interactions in studies of human diseases. BMC Bioinformatics 2003, 4:28.
17. Su EC, Chiu HS, Lo A, Hwang JK, Sung TY, Hsu WL: Protein sub-cellular localization prediction based on compartment-spe-cific features and structure conservation. BMC Bioinformatics 2007, 8:330.
18. Wang L, Brown SJ: Prediction of RNA-binding residues in pro-tein sequences using support vector machines. Conf Proc IEEE Eng Med Biol Soc 2006, 1:5830-5833.
19. Matthews BW: Comparison of the predicted and observed sec-ondary structure of T4 phage lysozyme. Biochim Biophys Acta 1975, 405(2):442-451.
20. Swets JA: Measuring the accuracy of diagnostic systems. Sci-ence 1988, 240(4857):1285-1293.
21. Bradley AP: The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognition 1997, 30(7):1145-1159.
22. Yu CS, Chen YC, Lu CH, Hwang JK: Prediction of protein subcel-lular localization. Proteins 2006, 64(3):643-651.
23. Chang JM, Su EC, Lo A, Chiu HS, Sung TY, Hsu WL: PSLDoc: Pro-tein subcellular localization prediction based on gapped-dipeptides and probabilistic latent semantic analysis. Proteins 2008, 72(2):693-710.