Adsorption and Dissociation of the HCl and Cl
2Molecules on W(111) Surface: A
Computational Study
Hui-Lung Chen,†Shin-Pon Ju,*,‡Hsin-Tsung Chen,§Djamaladdin G. Musaev,*,†and M. C. Lin*,†,§
Cherry L. Emerson Center for Scientific Computation and Department of Chemistry, Emory UniVersity, Atlanta, Georgia 30322, Department of Mechanical and Electro-Mechanical Engineering, Center for Nanoscience and Nanotechnology, National Sun-Yat-Sen UniVersity Kaohsiung, Taiwan 804, and Center for Interdisciplinary Molecular Science, Institute of Molecular Science, National Chiao Tung UniVersity, Hsinchu, Taiwan 300 ReceiVed: January 12, 2008; ReVised Manuscript ReceiVed: April 21, 2008
The adsorption and dissociation of Cl2and HCl molecules on W(111) surface have been studied at the density functional theory (DFT) level in conjunction with the projector augmented wave (PAW) method. The molecular structures and surface-adsorbent interaction energies of W(111)/Cl, W(111)/H, W(111)/Cl2, and W(111)/HCl systems are predicted. In these studies, four adsorption sites, such as top (T), bridge (B), shallow (S), and deep (D) sites, of the W(111) surface are considered. It is shown that the Cl2and HCl molecules adsorb to the W(111) surface by the end-on manner (by their Cl-Cl or H-Cl bonds perpendicular to the W surface), and their dissociative adsorptions occur without intrinsic energy barriers and are exothermic by 80.46 and 53.72 kcal/mol, for Cl2and HCl, respectively. Molecular dynamics studies show that the dissociation of Cl2 and HCl molecules on the W(111) surface occur in asymmetric fashion: at the beginning adsorbate forms a strong bond between one of their atoms and W centers, followed by the dissociation of the Cl-Cl (and/or H-Cl) bond and formation of the second bond between the atoms of adsorbate and the W center. For the Cl2 molecule, both Cl atoms are preferred to adsorb at the top W centers. For the HCl molecule, after the dissociation of the H-Cl bond the Cl atom still occupies the top adsorption site, but the H atom prefers to move to the position between the top and shallow W centers. The rate constants for the dissociative adsorption of Cl2and HCl have been predicted with variational RRKM theory.
Introduction
Understanding mechanism and controlling factors of the tungsten (W) and tungsten-based alloys with HCl and Cl2 molecules is vital for corrosion prevention at high temperature in waste incinerator plants or power plants1and in the field of integrated circuitry (for miniaturization of devices).2–4However, the limitations of the available experimental equipments and rapid dynamical motion of the molecules make an experimental study of the molecular and electronic properties of W(111) + HCl/Cl2 reactions rather difficult. In this case, the use of computer simulation based on reliable computational techniques has proven to be very useful.5–7
Previously, Michaelides et al.5have applied density functional theory (DFT) to investigate the variation of the workfunction of the W(100) surface with N adatoms. They have elucidated the reasons why N adatoms on the W(100) surface cause a significant decrease in the work function. In a recent work reported by Chen et al.,6 the adsorption, dissociation, and diffusion mechanisms of CO on the W(111) surface have been investigated by the DFT method. They have calculated the potential energy surface (PES) of the reaction of the CO molecule with the W(111) surface and have elucidated the reason of why the preadsorbed C atoms on the W(111) surface prevent the dissociation of CO on W(111). Also, density
functional theory was previously applied to study HCl adsorption on diamond(100),8ice(0001),9,10R-Al
2O3(0001),11and Ge(001)12 surfaces.
To the best of our knowledge, however, no theoretical study on the reaction of Cl2 and HCl molecules with the W(111) surface has been performed. In the present paper, we apply DFT to study the adsorption and dissociation of Cl2 and HCl molecules on the W(111) surface.
Computational Detalis
All present calculations are performed with the DFT plane-wave method utilizing the Vienna ab initio simulation package (VASP) with and without spin polarization.13–16 In these calculations, we use the projector-augmented wave method (PAW)17,18in conjunction with the Perdew-Wang (PW91)19,20 and the revised Perdew-Burke-Ernzerhof (rPBE)21,22densitiy functionals. The Brillouin zone is sampled with the Monkhorst-Pack grid.23The calculations are carried out using the (4× 4 × 4) and (4 × 4 × 1) Monkhorst-Pack mesh k-points for bulk and surface calculations, respectively. A 400 eV cutoff energy, which allows convergence to 1× 10-4eV in total energy, is used.
The side and top views of the model of the W(111) surface used in this paper are shown in Figure 1, panels a and b, respectively. Previously,24a we have carefully evaluated the model used in the present study, and have demonstrated that the adsorption energies of H2O molecule on p(2× 2) and p(3 × 3) lateral cells of the W(111) surface are quite consistent: the difference in the calculated adsorption energies is negligible
* Corresponding authors. E-mail: [email protected], jushin-pon@ mail.nsysu.edu.tw, and [email protected].
†Emory University.
‡National Sun-Yat-Sen University Kaohsiung. §National Chiao Tung University.
10.1021/jp8002992 CCC: $40.75 2008 American Chemical Society Published on Web 07/17/2008
(0.2 kcal/mol). Therefore, in the present study, we only use the computationally less expensive p(2× 2) model of the W(111) surface. The p(2 × 2) lateral cell of the W(111) surface is modeled as periodically repeated slabs with 6 layers, as shown in Figure 1a. The bottom three atomic layers are kept frozen and set to the experimentally estimated bulk parameters, whereas the remaining layers are fully relaxed during the calculations. The lateral cell has dimensions of a ) b ) 9.00 Å and c ) 17.47 Å, which includes a vacuum region of thickness ca. 15 Å and guarantees no interactions between the slabs. In this study, we calculate adsorption energies according to the following equation:
∆Eads) E[slab + adsorbate] - (E[slab] + E[adsorbate])
(1) where E[slab + adsorbate], E[slab], and E[adsorbate] are the calculated electronic energies of adsorbed species on a W(111) surface, a clean W(111) surface, and a gas-phase molecule, respectively.
The nudged elastic band (NEB) method25–27 is applied to locate transition states, and minimum energy pathways (MEP) are constructed accordingly. The NEB method is an efficient method for finding the minimum energy path (MEP) between the given initial and final state of a transition state. At least eight images are used to locate each calculated TS.
The rate constants for the dissociative adsorption of HCl and Cl2on the W(111) surface are calculated using the variational RRKM theory28a as implemented in the Variflex program.28b
Results and Discussion
A. Calculations of Bulk W and Gas-Phase Cl2and HCl Molecules. In order to ascertain the computational approaches
used in this paper, we calculate properties of bulk W and gas-phase Cl2and HCl molecules. Previously,24awe have computed the lattice constants of bulk tungsten by using PW91 and rPBE, and found it to be 3.183 and 3.181 Å, respectively. These values are in good agreement with the experimental value of 3.165 Å.29In addition, the W-W bond distance is calculated to be within the 2.750∼2.754 Å range, which also closely match as the experimental value of 2.741 Å.29 The structures and frequencies of the isolated gas-phase molecules, HCl and Cl2, are examined by embedding of isolated HCl and Cl2molecules in a large unit cell of 25× 25 × 25 Å3dimensions. In Table 1, we present the calculated and experimental properties of these molecules. The comparison of these data for HCl and Cl2 molecules clearly shows that the calculated properties of HCl and Cl2are in good agreement with the experimental values.30,31
B. Adsorption of Cl, H, Cl2, and HCl on the W(111) Surface. In order to locate possible intermediates, such as
W(111)/Cl, W(111)/H, W(111)/Cl2, and W(111)/HCl, we have placed Cl2, Cl, HCl, and H species on various sites of the W(111) surface as shown in Figure 1b. Four different adsorption sites of W(111) are considered: On-top (T), bridge (B), 3-fold-deep (D), and 3-fold-shallow (S) sites. For the On-top site (T), the molecule adsorbs on top of the first-layer W atom of W(111). At the bridge site (B), the molecule adsorbs above the center of the W-W bond of the two nearest W sites. At the 3-fold-deep site (D), the molecule adsorbs above the third-layer W atom. At the 3-fold-shallow site (S), the molecule coordinates above the second-layer W atom. All optimized local minima are listed in Tables 2–4.
In Table 2, we list the rPBE and PW91 optimized geometries and adsorption energies of Cl and H atoms at four different adsorption sites of W(111). As it could be expected, the radical adsorbates, Cl and H atoms, strongly interact with the W(111) surface. As seen in Table 2, the top site (T) is the most favorable binding site for the Cl atom, whereas the bridge site (B) is the most favorable binding site for the H atom, at the both PW91 and rPBE levels of theory. This could be attributed to the better overlap between the directional atomic d orbitals of the W atom with the p orbitals (which are directional too) of the Cl atom, and the spherical s orbital of the H atom with a diffuse W-W orbital.
The PW91-calculated W(111)-H and W(111)-Cl adsorption energies are about 3.7–11.0 kcal/mol greater than those com-puted at the rPBE level. This result is in a good agreement with the previous findings concluding that the PW91 method predicts larger adsorption energies of small molecules on metal surfaces compared with the rPBE and experimental data.32The PW91 and rPBE methods provide similar values for the W-Cl and W-H bond lengths.
For the Cl2 and HCl molecules, we have investigated both end-on (perpendicular to the W(111) surface with one atom of Cl2or HCl molecules) and side-on (Cl-Cl and H-Cl bonds are parallel to the W(111) surface) coordinations of the molecules to various adsorption sites of W(111). From our calculated results, it is found that both Cl2and HCl molecules prefer to adsorb on W(111) in the end-on configuration. The side-on coordination, however, leads to dissociation of these molecules to separate (Cl and Cl, and H and Cl) atoms. In Table 3, we have summarized the optimized geometries and adsorption energies of the W(111)/Cl2species.
As seen from this table, the calculated Cl-Cl bond lengths in four different adsorption configurations of W(111)/Cl2 are about 31-37% longer than that in the gas phase. Among all of the calculated adsorption configurations of W(111)/Cl2, the energetically most stable constructions (with ca. 48.0 kcal/mol adsorption energies) are those corresponding to the coordination of Cl2to the top and bridge sites of W(111).
Figure 1. Schematic presentation of W(111) surface used in the present
studies. The T, D, and S represent top, deep, and shallow sites, while the middle of two top sites is considered as a bridge site and labeled as B.
TABLE 1: Calculated (Both at the PW91 and rPBE Levels) and Experimental Values of the Geometry Parameters (Bond Lengths in Å and Angles in deg) and Vibrational
Frequencies (in cm-1) of HCl and Cl2Molecules
HCl (C∞V) Cl2(D∞V) parameters calc.a expt.b calc.a expt.b
r(H-Cl, Cl-Cl) 1.284(1.288)c 1.275 1.999(2.005)c 1.988
V 2908(2914)c 2991 536(545)c 560 aVibrational frequencies were not scaled.bFrom refs 30 and 31,
respectively.cThe data given in parentheses are at the PW91 level
of theory.
For the W(111)/HCl system, as shown in Table 4, the coordination of HCl with its Cl atom to W(111) is energetically more favorable among all of the calculated adsorption configu-rations. The calculated adsorption energies of W(111)-HCl are less than 3 kcal/mol, among which the most stable one is the on-top (T) configuration with a 2.23 kcal/mol bonding energy. Coordination of HCl with its Cl atom to W(111) is around 0.3-3.5 kcal/mol more favorable than that with the H atom. Since the W(111)/HCl interaction is very weak, the calculated H-Cl bond length of HCl molecule is almost the same with its free and adsorbed states. However, one should note that the calculated W(111)-HCl adsorption energy is within DFT preci-sion and because there are no experimental data available for comparison the result should be taken with caution.
As radical species are produced in the dissociative adsorption of Cl2 and HCl, spin polarization effects on the adsorption energies of various adsorbates such as Cl2(a), HCl(a), Cl(a), H(a), 2Cl(a), 2H(a), and Cl(a)+ H(a)on the W(111) surface have been examined; it is found that effects of spin polarization are negligible (smaller than 0.10 eV even in the case the atomic
adsorbates). In addition, we also use Bader’s method33 to examine the partial charge densities of the Cl and H atoms (with both the spin restricted and the spin polarized calculations) for the aforementioned adsorption configurations, giving rise to only a slight change in their relevant charge densities (less than 0.003 |e|).
C. Potential Energy Surfaces (PESs) of Dissociative Adsorption of Cl2 and HCl on W(111) Surface. The NEB
method is used to obtain the minimum energy pathways (MEP) of the reactions
Cl2+ W(111) f Cl/W(111)/Cl (2)
and
HCl + W(111) f H/W(111)/Cl (3)
for the H and Cl adsorption sites in the product structures Cl/ W(111)/Cl and H/W(111)/Cl, we use the energetically most stable sites (T, B, D, and S) of the W(111)/Cl and W(111)/H species discussed above. Since the products Cl/W(111)/Cl and H/W(111)/Cl have two adsorbed atoms located at two different
TABLE 2: Calculated (Both at the PW91 and rPBE Levels) Important Bond Distances (in Å) and Adsorption Energies (in kcal/mol) of Cl and H Atoms Adsorbed on the W(111) Surface
PW91 rPBE
adsorption site bond length adsorption energy bond length adsorption energy For W(111)/Cl on-top (T) 2.283 -77.21 2.291 -68.90 bridge (B) 2.800, 2.825 -70.37 2.775, 2.788 -59.37 3-fold-deep (D) 2.520 -50.69 2.516 -39.99 3-fold-shallow (S) 2.506 -66.46 2.513 -56.26 For W(111)/H on-top (T) 1.784 -73.23 1.787 -69.52 bridge (B) 2.324, 2.336 -80.14 2.330, 2.338 -74.80 3-fold-deep (D) 1.886 -61.59 1.883 -56.15 3-fold-shallow (S) 1.840 -75.24 1.848 -70.35
TABLE 3: Calculated (Both at the PW91 and rPBE Levels) Important Bond Distances (in Å) and Adsorption Energies (in kcal/mol) of Cl2Molecule Adsorbed on the W(111) Surface
PW91 rPBE
adsorption site bond lengtha adsorption energy bond lengtha adsorption energy
on-top (T) 2.231(2.799) -47.84 2.244(2.775) -47.35 bridge (B) 2.690, 2.694 -48.71 2.729, 2.737 -43.90
(2.711) (2.802)
3-fold-deep (D) 2.792(2.693) -32.25 2.887(2.713) -27.16 3-fold-shallow (S) 2.478(2.805) -41.71 2.480(2.889) -37.89
aThe values given in parentheses are for Cl-Cl bond length, whereas others are for W-Cl bond length.
TABLE 4: Calculated (Both at the PW91 and rPBE Levels) Important Bond Distances (in Å) and Adsorption Energies (in kcal/mol) of HCl Molecule Adsorbed on the W(111) Surface
PW91 rPBE
bond lengtha adsorption energy bond lengtha adsorption energy
Coordinated with H Atom of HCl
on-top (T) 2.592(1.309) -0.44 2.554(1.309) 1.31
bridge (B) 2.971, 2.889 -4.40 2.975, 2.951 (1.327) -1.91 (1.330)
3-fold-deep (D) 2.818(1.350) -3.69 2.901(1.324) -0.60 3-fold-shallow (S) 2.708(1.325) -4.20 2.662(1.330) -1.91 Coordinated with Cl Atom of HCl
on-top (T) 3.547(1.291) -2.85 3.593(1.289) -2.23
bridge (B) 3.563, 3.558 -0.76 4.034, 4.052 0.62
(1.296) (1.284)
3-fold-deep (D) 4.540(1.287) -0.39 4.437(1.290) 1.52 3-fold-shallow (S) 3.818(1.297) -0.83 3.877(1.291) 0.82
adsorption sites, below we denote these structures as XCl/ W(111)/ClY and XH/W(111)/ClY, where X and Y stand for adsorption sites and could be T, B, D, and S.
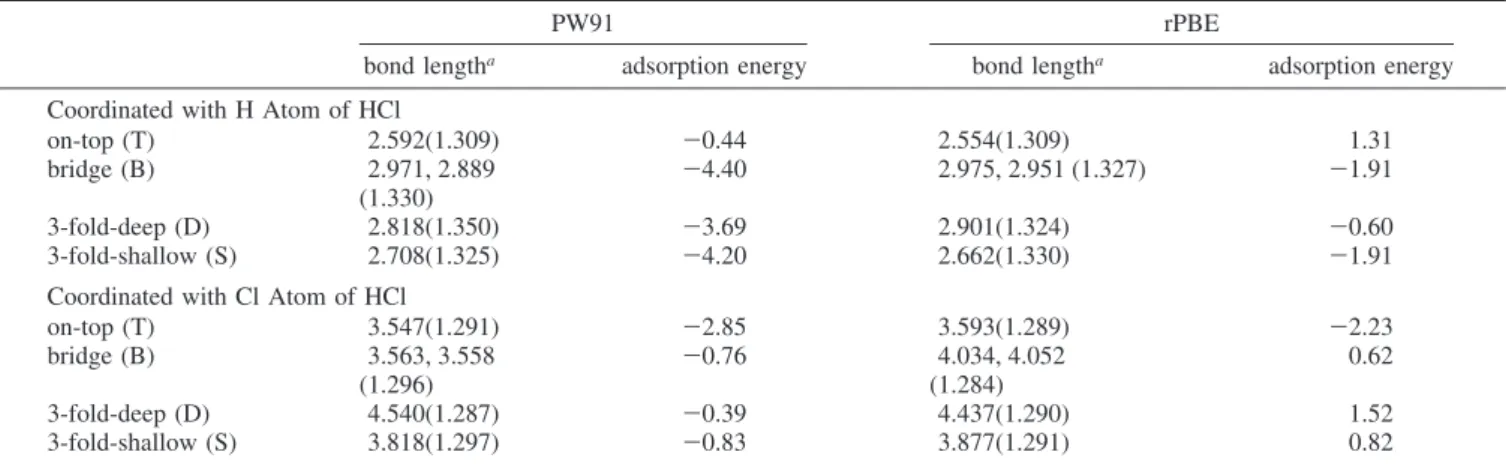
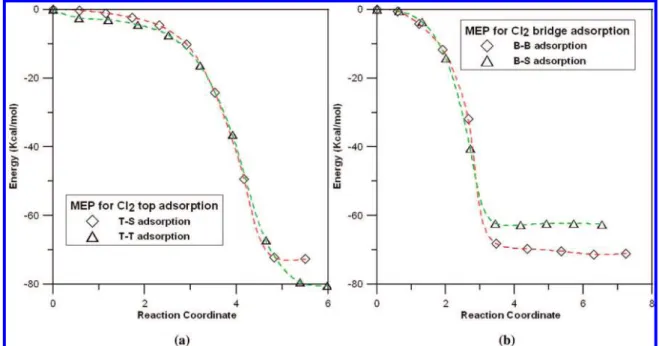
As shown in Figures 2 and 3, it is found clearly that no energy barriers are associated with the decomposition of the side-on approached Cl2and HCl molecules on the W(111) surface. As seen in Table 5, where we present the most favorable structures of XCl/W(111)/ClY andXH/W(111)/ClY, and dissociative ad-sorption energies of the reactions (2) and (3). Among which, the most stable product ofXCl/W(111)/ClYisTCl/W(111)/ClT structure with a 80.46 kcal/mol adsorption energy (relative to the Cl2+ W(111) reactants). We also evaluate the dissociation energies of Cl2 and HCl molecules (gas phase) by DFT calculation. The obtained results are summarized in the footnote of Table 5. As seen from this table the calculated Cl-Cl and H-Cl dissociation energies are 64.13 and 105.97 kcal/mol, which are in reasonable agreement with their experimental
values, 58.70 and 103.25 kcal/mol, respectively. In the case of reaction HCl + W(111), the most favorable product is SH/ W(111)/ClT with a 53.72 kcal/mol adsorption energy. The Figure 2. Minimum energy profile (MEP) of the end-on coordination of Cl2molecule to the (a) top and (b) bridge sites of W(111) surface.
Figure 3. Minimum energy profile (MEP) of the end-on coordination of HCl molecule to the (a) top and (b) bridge sites of W(111) surface.
TABLE 5: Calculated (at the rPBE Level of Theory) Adsorption Energies (in kcal/mol) of the Dissociative Adsorption of Cl2and HCl Molecules on W(111)a
configuration Cl2 HCl T-S -72.70 T-T -80.46 B-B -71.06 -48.36 B-S -62.70 -48.90 S-B -47.41 S-S -31.66 S-T -53.72
aThe Cl-Cl and H-Cl bond energies are calculated to be 64.13
and 105.97 kcal/mol vs 58.70 and 103.25 kcal/mol of their experimental values, respectively.
calculated 80.46 kcal/mol energy of reaction (2) is almost 6.79 kcal/mol larger than that (73.67 kcal/mol) estimated by using the following equation (see Tables 2 and 5 for details): ∆E[TCl/W(111)/ClT] ) 2{∆E[W(111)/ClT]} -∆E(Cl - Cl)
(4) These data demonstrates that coordination of the first Cl atoms to the W(111) surface changes surface properties and makes it more nucleophilic to the subsequent Cl atom.
Similarly, the calculated 53.72 kcal/mol adsorption energy for the reaction HCl + W(111) fSH/W(111)/ClTis 20.44 kcal/ mol larger than that (33.28 kcal/mol) estimated by using the following equation:
∆E[SH/W(111)/ClT] ) {∆E[W(111)/HS] +
∆E[W(111)/ClT]} -∆E(H - Cl) (5) These data demonstrate that the coordination of H (or Cl) atom on W(111) enhances the adsorption property of the surface. However, on the aforementioned observations, one should note that the adsorption energy of a single atom (Cl or H) is not exactly in the same technical conditions with that for dissociated molecules (Cl2or HCl).
D. Rate Constant Calculations. Based on the
aforemen-tioned PESs for the dissociative adsorption of Cl2and HCl on the W(111) surface, we have predicted the rate coefficients for the following reactions: Cl2+ W(111) f Cl/W(111)/Cl and HCl + W(111) f H/W(111)/Cl, respectively. For this purpose, the minimum energy paths (MEP) of these reactions are calculated along the reaction coordinate of M-Cl and M-H, which are stretched from its equilibrium value to 5.0 Å with the step size of 0.1 Å. At both fixed M-Cl and M-H distances, the bottom three atomic layers of the W(111) surfaces are fixed, while atoms in the remaining layers and Cl2as well as HCl are fully optimized at the rPBE level. The obtained MEP can be approximated with a Morse potential, V(r) ) De{1 - exp[-β(R - R0)]}2, where R is the reaction coordinate, R0 is the equilibrium W-Cl (or W-H) bond distance, and Deis the bond energy without zero-point energy corrections. The parameters obtained by fitting the Morse potential to the MEP are R0) 2.244,β ) 2.995 Å-1, and De) 46.54 kcal/mol for dissociative adsorption of Cl2, and R0) 2.951, β ) 3.527 Å-1, and De) 1.87 kcal/mol for dissociative adsorption of HCl, respectively. Our calculations have been carried out for the temperature of 200∼3000 K. The predicted rate coefficients (in units of cm3 molecule-1 s-1) in the broad temperature range can be repre-sented, respectively, by:
kCl2) 3.71 × 10
-8T-1.097exp(-0.12 kcal mol-1/RT) for the dissociative adsorption of Cl2
kHCl ) 7.87 × 10-8 T-0.994exp(-0.18 kcal mol-1/RT) for the dissociative adsorption of HCl
The rate coefficients for the dissociative adsorption processes of Cl2and HCl on W(111) are both defined by34
d[X ]surf/dt ) k(θ/As)[X ]g (6) which have the unit of a flux, molecule cm-2 s-1. In the equation, θ, As, and [X]g represent the fraction of available surface sites, the surface area, and the gas-phase concentration of Cl2and HCl in molecules/cm3, respectively. At 298 K, the values of the rate coefficients can be represented by kCl2) 5.79
× 10-11 and k
HCl ) 2.02 × 10-10 cm3 molecule-1 s-1, respectively.
E. Molecular Dynamics Simulation for Cl2 and HCl Molecules on W(111). In order to study in detail of dissociation
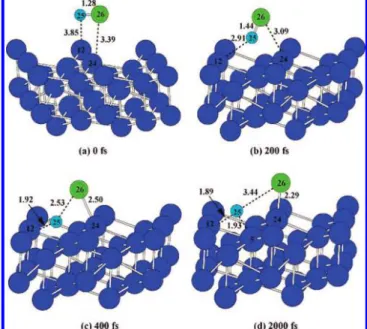
of Cl2and HCl on the W(111) surface, the molecular dynamics (DFT-MD) simulation at 10 K is carried for reactions (2) and (3). In these studies, we use the canonical ensemble (NVT) in which the Verlet algorithm is employed to calculate the trajectories of the atoms with a time step∆t ) 2 fs for a total time period of 2000 fs. As noted above, we have studied the MEP of reactions (2) and (3) when Cl-Cl and H-Cl coordi-nates to the W(111) surface in the side-on (in parallel) manner. However, when these adsorbates coordinate in the end-on (with one atom) fashion to the surface, it requires only very small amount of energy to rotate a specific angle toward the surface and then follow the dissociation pathway. Since W(111)-Cl2/ HCl interaction energies are very small, molecules can easily rearrange from their end-on coordination modes to their side-on coordinatiside-on modes side-on the W(111) surface. The barrier separating these two coordination modes is expected to be very small and, therefore, is not studied. In the DFT-MD simulation, which is performed at 10 K, molecules in the end-on coordina-tion mode should possess enough kinetic energy to overcome this small energy barrier. For the initial configurations of MD simulation, Cl2and HCl molecules are placed parallel to the W(111) surface at about 3.5 Å above the surface. It should be mentioned that in our MD simulations, we have chosen the different initial orientations of Cl2and HCl molecules relative to the W(111) surface. The obtained results indicate that these dissociation processes are orientation independent. Figure 4 shows several snapshots from the initial configuration at 0 fs to the final configuration at 2000 fs for the dissociation process of the Cl2molecule.
In these snapshots, atoms 25 and 26 are two Cl atoms of the Cl2molecule and the others are the W atoms of the tungsten slab. As seen from this Figure, as well as from Figure 5 (which shows the variations of bond lengths and energy with the simulation time step during the course of Cl2dissociation, where the energy and bond lengths at 0 fs are used as referenced values), the bond length of the Cl2molecule elongates with the simulation time step.
At the final stage (Figure 4d), atoms 25 and 26 form bonds with the W atoms of 24 and 18, respectively. As seen in Figure Figure 4. Snapshots of the Cl2dissociation on the W(111) surface at
(a) 0, (b) 200, (c) 600, and (d) 2000 fs. The blue and green spheres represent W and Cl, respectively. The bond lengths are given in Å.
5 (where the positive and negative values represent the increase or decrease of energy or bond length, compared with those at 0 fs), the energy of the system significantly decreases when the Cl2molecule starts to dissociate on the W(111) surface for the first 500 fs. The bond length between the atoms 25 (Cl) and 26 (Cl) increases with the approach to atoms 24 (W) and 18 (W), respectively. After 500 fs, the bond length between the atoms 24 and 25 remains almost constant, indicating the completion of the bond formation between the atoms 24 and 25. The bond length between the atoms 25 and 26 then increases more significantly. This indicates that the bond formation between the Cl atom and the top W atom weakens the Cl-Cl bond and facilitates the dissociation of Cl2. As a result, atom 26 moves from its original position and then forms bond with another top W atom. After 1600 fs, the bond length between atoms 18 and 26 and the energy variation remain almost constant, indicating the formation of a stable bond. Finally, after the dissociation, the two Cl atoms adsorb on two top W atoms, which is the most favorable adsorption site, as shown in Table 2.
Several snapshots during the course of the MD simulation of the HCl + W(111) reaction, are given in Figure 6.
In this figure, atoms 25 and 26 are H and Cl atoms of an HCl molecule. After the completion of the dissociation process, the H atom moves to the position between the atoms 12 (top W) and 5 (shallow W), and the Cl atom is adsorbed at the top site (see Figure 6d). Since the distance between the top site and the closest bridge site is too short, the H atom can not adsorb at its favorable adsorption site. Therefore, after the dissociation of H-Cl bond the H atom moves to the position between the top and shallow W atoms, instead of the bridge site (B). In addition, the Figure 7 records the variations of bond lengths and energy during the course of the simulation and it is found that the dissociation process of the HCl molecule on the W(111) surface occurs via a similar pathway to that of the Cl2molecule as discussed above.
Conclusions
We use the DFT method to investigate the adsorption and dissociation of Cl2and HCl molecules on the W(111) surface. The calculated results show that the Cl2and HCl molecules on the W(111) surface can only exist in the end-on adsorption mode
and the MEP profiles from NEB calculations indicate that there are no intrinsic barriers for the dissociations of Cl2and HCl molecules. We also have predicted the rate constants for Cl2 and HCl dissociative adsorption on W(111). In addition, the DFT-MD modeling has been shown to be useful for elucidating the mechanism of Cl2 and HCl on the W(111) surface. By observing the variations of energy and bond lengths, one can see that the interaction between Cl2(and/or HCl) and the W centers will result in weakening the Cl-Cl (and/or H-Cl) bond, giving rise to the dissociation process.
Acknowledgment. We gratefully acknowledge: (1) financial
support from the Office of Naval Research under a MURI grant (Prime Award No. N00014-04-1-0683 and Subaward No. 2794-EU-ONR-0683), (2) the Emerson Center for the use of its resources, (3) the use of CPUs from National Center for High-Figure 5. Variations of energy of the system and specific bond lengths
during the dissociation (MD simulation) of Cl2molecule on the W(111)
surface. Figure 6. Snapshots of the HCl dissociation on the W(111) surface at(a) 0, (b) 200, (c) 400, and (d) 2000 fs. The blue, green, and sky blue spheres represent W, Cl and H, respectively. The bond lengths are given in Å.
Figure 7. Variations of energy of the system and specific bond lengths
during the dissociation (MD simulation) of HCl molecule on the W(111) surface.
performance Computing, Taiwan, and (4) National Science Council of Taiwan under Grant No. NSC-096-2628-E-110-005-MY2. One of us, M.C.L., wants to acknowledge the supports from Taiwan National Science Council for the NSC Distin-guished Visiting Professorship and from Taiwan Semiconductor Manufacturing Co. for a TSMC Distinguished Professorship at National Chiao Tung University, Hsinchu, Taiwan.
Supporting Information Available: Tables S1-S8:
Opti-mized geometries in Cartesian for the snapshots of the Cl2and HCl dissociation on the W(111) surface at specific times which are calculated by the VASP program. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
(1) Cha, S. C.; Wo¨lpert, P. Mater. Corros. 2002, 53, 886.
(2) Perry, S. S.; Galloway, H. C.; Cao, Paul.; Mitchell, E. J.R.; Koeck, D. C.; Smith, C. L.; Lim, M. S. Appl. Surf. Sci. 2001, 180, 6.
(3) Chen, B.-H.; Zhang, H.; Chooi, S. Y. M.; Chan, L.; Xu, Y.; Ye, J. H. Ind. Eng. Chem. Res. 2003, 42, 6096.
(4) Kim, S. Y.; Kim, H.; Kwan, H. S. Mater. Corros. 2006, 57, 835. (5) Michaelides, A.; Hu, P.; Lee, M. H.; Alavi, A.; King, D. A. Phys.
ReV. Lett. 2003, 90, 246103.
(6) Chen, L.; Sholl, D. S.; Johnson, J. K. J. Phys. Chem. B 2006, 110, 1344.
(7) Choe, S. J.; Kang, H. J.; Park, D. H.; Huh, D. S.; Lee, S. B. Bull.
Korean Chem. Soc. 2004, 25, 1314.
(8) Hukka, T. I.; Pakkanen, T. A.; D’Evelyn, M. P. J. Phys. Chem.
1995, 99, 4710.
(9) Svanberg, M.; Pettersson, J. B. C.; Bolton, K. J. Phys. Chem. A
2000, 104, 5787.
(10) Mantz, Y. A.; Geiger, F. M.; Molina, L. T.; Molina, M. J.; Trout, B. L. J. Phys. Chem. A 2001, 105, 7037.
(11) Alavi, S.; Sorescu, D. C.; Thompson, D. L. J. Phys. Chem. B 2003,
107, 186.
(12) Sa´nchez-Castillo, A.; Cocoletzi, G. H.; Takeuchi, N. Surf. Sci. 2002,
521, 95.
(13) Kresse, G.; Hafner, J. Phys. ReV. B 1993, 47, 558. (14) Kresse, G.; Hafner, J. Phys. ReV. B 1994, 49, 14251. (15) Kresse, G.; Furthmuller, J. Comput. Mater. Sci. 1996, 6, 15. (16) Kresse, G.; Hafner, J. Phys. ReV. B 1996, 54, 11169. (17) Blo¨chl, P. E. Phys. ReV. B 1994, 50, 17953. (18) Kresse, G.; Joubert, D. Phys. ReV. B 1999, 59, 1758. (19) White, J. A.; Bird, D. M. Phys. ReV. B 1994, 50, 4954. (20) Perdew, J. P.; Chevary, J. A.; Vosko, S. H.; Jackson, K. A.; Pederson, M. R.; Singh, D. J.; Fiolhais, C. Phys. ReV. B 1992, 46, 6671. (21) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. ReV. Lett. 1996, 77, 3865.
(22) Zhang, Y.; Yang, W. Phys. ReV. Lett. 1998, 80, 890. (23) Monkhorst, H. J.; Pack, J. D. Phys. ReV. B 1976, 13, 5188. (24) (a) Chen, H.-T.; Musaev, D. G.; Lin, M. C. J. Phys. Chem. C 2007,
111, 11117. (b) Chen, H.-T.; Musaev, D. G.; Lin, M. C. J. Phys. Chem. C
2007, 112, 3341.
(25) Ulitsky, A.; Elber, R. J. Chem. Phys. 1990, 92, 1510.
(26) Mills, G.; Jo´nsson, H.; Schenter, G. K. Surf. Sci. 1995, 324, 305. (27) Henkelman, G.; Uberuaga, B. P.; Jo´nsson, H. J. Chem. Phys. 2000,
113, 9901.
(28) (a) Baer, T.; Hase, W. L. Unimolecular Reaction Dynamics. Theory
and Experiments; Oxford University Press: Oxford, 1996. (b) Klippenstein,
S. J.; Wagner, A. F.; Dunbar, R. C.; Wardlaw, D. M.; Robertson, S. H. Variflex, 1999.
(29) Villars, P.; Calvert, L. D. Pearson’s Handbook of Crystallographic
Data for Intermetallic Phase, 2nd ed.; ASM International: Materials Park,
OH, 1991.
(30) Huber, K. P.; Herzberg, G. Molecular Spectra and Molecular
Structure. IV.; Constants of Diatomic Molecules; Van Nostrand Reinhold
Co.: New York, 1979.
(31) Shimanouchi, T. Tables of Molecular Vibrational Frequencies; Consolidated Volume 1, NSRDS NBS-39; National Bureau of Standards: Washington, DC, 1972.
(32) Hammer, B.; Hansen, L. B.; Nørskov, J. K. Phys. ReV. B 1999, 59, 7413.
(33) Bader, R. F. W. Atoms in Molecules-A Quantum Theory; Oxford University Press: Oxford, 1990.
(34) Rettner, C. T.; Ashfold, M. N. R. Dynamics of Gas-Surface
Interaction; Springer Verlag: Berlin, 1991; Chapter 5.