Darling–Dennison resonance and Coriolis coupling in the bending overtones of the A
A u 1 state of acetylene, C 2 H 2
Anthony J. Merer, Nami Yamakita, Soji Tsuchiya, Adam H. Steeves, Hans A. Bechtel, and Robert W. Field
Citation: The Journal of Chemical Physics 129, 054304 (2008); doi: 10.1063/1.2939246
View online: http://dx.doi.org/10.1063/1.2939246
View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/129/5?ver=pdfcov Published by the AIP Publishing
Articles you may be interested in
The double Renner effect in the X A 2 and A A 2 electronic states of H O 2 J. Chem. Phys. 128, 114316 (2008); 10.1063/1.2827490
The vibrational structure of the X A 1 1 A B 1 1 and A B 1 1 B A 1 1 band systems of Ge H 2 Ge D 2 based on global potential energy surfaces
J. Chem. Phys. 126, 044313 (2007); 10.1063/1.2431653
A dispersed fluorescence and ab initio investigation of the X B 1 2 and A A 1 2 electronic states of the PH 2 molecule
J. Chem. Phys. 124, 094306 (2006); 10.1063/1.2168155
Absorption cross sections and correlation functions of the N H 2 A A 1 2 X B 1 2 Renner–Teller system J. Chem. Phys. 122, 234315 (2005); 10.1063/1.1929737
Resonances of CH 2 ( a A 1 1 ) and their roles in unimolecular and bimolecular reactions J. Chem. Phys. 122, 124308 (2005); 10.1063/1.1866094
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32
Darling–Dennison resonance and Coriolis coupling in the bending
overtones of the A
˜
1A
ustate of acetylene, C
2H
2Anthony J. Merer,1,2Nami Yamakita,3Soji Tsuchiya,4Adam H. Steeves,5Hans A. Bechtel,6 and Robert W. Field5,a兲
1
Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei 10617, Taiwan 2
Department of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z1, Canada 3
Department of Chemical and Biological Sciences, Japan Women’s University, Mejirodai, Bunkyo-ku, Tokyo 112-8681, Japan
4
Department of Applied Chemistry and Institute of Molecular Science, National Chiao Tung University, Hsinchu 30010, Taiwan
5Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA 6Advanced Light Source Division, Lawrence Berkeley National Laboratory, Berkeley,
California 94720, USA
共Received 27 March 2008; accepted 13 May 2008; published online 4 August 2008兲
Rotational analyses have been carried out for the overtones of the 4 共torsion兲 and 6 共in-plane cis-bend兲 vibrations of the A˜1A
u state of C2H2. Thev4+v6= 2 vibrational polyad was observed in high-sensitivity one-photon laser-induced fluorescence spectra and the v4+v6= 3 polyad was observed in IR-UV double resonance spectra via the ground state 3 共⌺+u兲 and 3+4 共⌸u兲
vibrational levels. The structures of these polyads are dominated by the effects of vibrational angular momentum: Vibrational levels of different symmetry interact via strong a-and b-axis Coriolis coupling, while levels of the same symmetry interact via Darling–Dennison resonance, where the interaction parameter has the exceptionally large value K4466= −51.68 cm−1. The K-structures of the polyads bear almost no resemblance to the normal asymmetric top patterns, and many local avoided crossings occur between close-lying levels with nominal K-values differing by one or more units. Least squares analysis shows that the coupling parameters change only slightly with vibrational excitation, which has allowed successful predictions of the structures of the higher polyads: A number of weak bands from thev4+v6= 4 and 5 polyads have been identified unambiguously. The state discovered by Scherer et al.关J. Chem. Phys. 85, 6315 共1986兲兴, which appears to interact with the K = 1 levels of the 33vibrational state at low J, is identified as the second highest of the five K = 1 members of the v4+v6= 4 polyad. After allowing for the Darling–Dennison resonance, the zero-order bending structure can be represented by4= 764.71,6= 772.50, x44= 0.19, x66= −4.23, and x46= 11.39 cm−1. The parameters x
46 and K4466 are both sums of contributions from the vibrational angular momentum and from the anharmonic force field. For x46these contributions are 14.12 and −2.73 cm−1, respectively, while the corresponding values for K
4466 are −28.24 and −23.44 cm−1. It is remarkable how severely the coupling of4and6distorts the overtone polyads, and also how in this case the effects of vibrational angular momentum outweigh those of anharmonicity in causing the distortion. © 2008 American Institute of Physics.
关DOI:10.1063/1.2939246兴
I. INTRODUCTION
The A˜ 1Au− X˜ 1⌺+g electronic transition of acetylene is
one of the most widely studied of all polyatomic spectra. The principal reason is that acetylene is the simplest compound containing a CwC triple bond, so that analysis of the spec-trum gives a clear picture of what happens on*← elec-tronic excitation of such a bond. As is well known, acetylene is linear in its ground electronic state, but, as was shown by Ingold and King1,2 and by Innes3 over 50 years ago, it be-comes trans-bent in its first singlet excited state A˜ 1Au. This
was one of the first demonstrations that a molecule can
change its point group on electronic excitation. In fact, it is more complicated than this. The first singlet excited state 共S1兲, which corresponds to a 1⌺−u state of the linear
mol-ecule, has potential minima corresponding to both cis- and
trans-bent isomers,4–11 but transitions from the ground state are only permitted to levels of the trans-isomer by the dipole selection rules. The S1 state of the cis-isomer, A˜ 1A2, has never been observed, though it is calculated to lie about 3000 cm−1higher than the A˜ 1Austate of the trans-isomer
7,11
or roughly 1000 cm−1 below the dissociation limit.12
Inter-esting dynamics will occur at the barrier to cis-trans-isomerization, and a search for the spectroscopic signatures of these dynamics has been one of the motivations for this work.
a兲Author to whom correspondence should be addressed. Tel.:共617兲253-1489.
FAX:共617兲253-7030. Electronic mail: [email protected].
THE JOURNAL OF CHEMICAL PHYSICS 129, 054304共2008兲
0021-9606/2008/129共5兲/054304/19/$23.00 129, 054304-1 © 2008 American Institute of Physics
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32
A second reason for interest in the A˜ 1Au− X˜ 1⌺+g transi-tion of acetylene is the wealth of detail it contains. The Franck–Condon pattern in absorption13,14 consists of a long progression in the trans-bending 共or “straightening”兲 vibra-tion 3, each member of which is the origin of a short pro-gression in the CuC stretching vibration2. This is consis-tent with the rotational analysis, which shows15 that the equilibrium HCC angle is 122.5°, and that the CuC bond length has increased from 1.208 to 1.375 Å, which is greater than that in C2H4. One of the results of the change of point group on electronic excitation, which was found in the spec-trum of acetylene for the first time, is “axis-switching,”16 where a small rotation of the principal inertial axis system upon excitation causes the appearance of unexpected
K
⬘
−ᐉ⬙
= 0 and ⫾2 subbands in an otherwise perpendicular 共K⬘
−ᐉ⬙
=⫾1兲 transition. Although predissociation sets in12 just below the 340band, its effects are minimal at first. Very extensive rotational analyses have therefore been possible, leading to a detailed description of the level structures of the 2 and 3 progressions and, from hot bands, of the ground state 4 共trans-bending兲 vibration.13,14,17With a detailed understanding of the upper state level structure available, the A˜ 1Au state has been a valuable step-ping stone for emission studies,18–23 and particularly for double resonance experiments. Among these are many com-prehensive studies of the high vibrational levels of the ground state using stimulated emission pumping24–27 and of the level structures of various Rydberg and valence states28–30and the acetylene cation.31,32
Interestingly, although the Franck–Condon active 共gerade兲 vibrational levels are fairly well understood, much less is known about the ungerade vibrations of the A˜ 1Au
state. IR-UV double resonance experiments via the ground state 33level33,34have allowed analyses of the three unger-ade fundamentals,4共au, torsion兲,5共bu, antisymmetric CH
stretch兲, and 6 共bu, in-plane cis-bend兲, together with the
combination 3151. The4and6 fundamentals of the A˜ 1A
u
state are almost degenerate,33 with wavenumbers of 764.9 and 768.3 cm−1, respectively; they are also extremely strongly coupled by Coriolis interactions. Further double resonance experiments via the ground state3 fundamental have allowed analyses35 of the combinations 3241, 3261, 3341, and 3361, where again the structure is massively dis-torted by Coriolis effects. The only other information about the ungerade vibrations comes from one-photon laser-induced fluorescence 共LIF兲 studies of the excitation spectrum36 where, with the help of supersonic jet-cooling, some weak bands in among the strong Franck–Condon progressions were identified as combinations involving overtones of the4and6vibrations.
The assignment of these weak combination bands36 sug-gested that many other bands involving the low-lying unger-ade vibrations 4 and6 should be observable given suffi-ciently sensitive experiments. These bands would be highly forbidden according to the Franck–Condon principle, but could obtain small amounts of intensity through anharmonic interactions of their upper levels with the Franck–Condon allowed levels. The purpose of the present paper is to
de-scribe the successful observation of a number of such bands belonging to overtones of the4and6vibrations with up to five vibrational quanta. The gerade members have been ob-served in one-photon LIF experiments, and the ungerade members in IR-UV double resonance experiments via the ground state3and3+4 levels.
The rotational and vibrational structures of these bands are highly unusual. Because the 4 and 6 vibrations have very nearly the same frequency, many of the features of a doubly degenerate vibration, with its associated vibrational angular momentum, appear in their overtone spectra. The strong Coriolis coupling of the4and6vibrations is one of these. Another is the strikingly large Darling–Dennison reso-nance that occurs between the overtones of 4 and 6, and which causes the vibrational levels to be grouped into what looks like the vibrational angular momentum level structure of a degenerate vibration. As far as we are aware this type of pattern has not been seen before in the bending vibrations of an asymmetric top molecule. The combination of a- and
b-axis Coriolis coupling with Darling–Dennison resonance
distorts the spectra very severely. The K-structure is totally disorganized, and local rotational perturbations occur in the
J-structure at many places where appropriate sets of levels
happen to lie close to each other. For K⬎0 all the members of an overtone polyad appear in the spectra, whatever their nominal vibrational symmetries. Specifically, the distinction between a and b irreducible representations is lost, although
g-u symmetry remains valid.
II. EXPERIMENTAL DETAILS
The c-axis polarization of the A˜ −X˜ transition implies the rotational selection rule K
⬘
−ᐉ⬙
=⫾1. Taking account of theg-u symmetry properties of the levels, it has been necessary
to carry out four sets of experiments in order to map the
K
⬘
= 0 − 2 structure of the A˜ state, as illustrated in Fig.1. In one-photon jet-cooled experiments from the ground vibra-tional level, which has ᐉ⬙
= 0, only the vibrationally geradeK
⬘
= 1 levels are accessible in the absence of Coriolis- or axis-switching-induced “forbidden” subbands. To get at the gerade K⬘
= 0 and 2 levels, it is necessary to use a warmed FIG. 1. Schematic energy level diagram showing how the K⬘−ᐉ⬙=⫾1 selection rule necessitates two experiments, both in one-photon excitation and in IR-UV double resonance, in order to observe the complete set of upper state levels with K⬘= 0 – 2.054304-2 Merer et al. J. Chem. Phys. 129, 054304共2008兲
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32
sample that has a sufficient population in the ground state4 fundamental, whereᐉ
⬙
= 1. Similarly, for the ungerade vibra-tional levels, observed in IR-UV double resonance experi-ments, only the ungerade K⬘
= 1 levels can be reached if the intermediate level is a ⌺+u 共ᐉ⬙
= 0兲 ground state vibrationallevel, such as the 3 fundamental; a ⌸u vibrational
inter-mediate 共ᐉ
⬙
= 1兲 is needed in order to reach the ungeradeK
⬘
= 0 and 2 levels.Laser-induced fluorescence spectra of neat acetylene have been recorded in an unskimmed pulsed jet expansion. The gas was expanded through a pulsed valve 共General Valve, Series 9兲 with a 0.5 mm orifice from a backing pressure of 200 kPa. The ultimate vacuum achieved in the apparatus was 2.7⫻10−5Pa, which rose to 6.7⫻10−3Pa under normal gas load.
The laser radiation was the frequency-doubled output of a Lambda Physik 3002E dye laser, pumped by the third har-monic of a Nd:YAG laser共Spectra-Physics DCR-3兲. A small portion of the dye laser power was passed through a heated gas cell containing130Te2vapor for calibration共⫾0.02 cm−1 accuracy兲, while the remainder was doubled in a -barium borate crystal, and sent to the molecular beam chamber. The laser radiation crossed the pulsed jet about 3 cm from the orifice.
Fluorescence from the excited acetylene was observed at right angles to both the laser beam and the jet axis. The fluorescence was collected by a lens system and detected by a Hamamatsu R331 photomultiplier after passing through a UG-5 or UG-11 colored glass filter.
To observe the K
⬘
= 0 and 2 levels belonging to gerade vibrational states it has been necessary to record hot band transitions from the ground state4fundamental. In order to induce hot bands in the jet spectra while maintaining reason-ably low rotational temperatures, the distance between the nozzle and the intersection of the laser with the pulse of molecules was reduced from ⬃30 to ⬃5 mm. Additionally, the relative timing of the pulsed valve and the laser was adjusted so that the laser radiation intersects the leading edge of the gas pulse, which is characterized by higher effective vibrational temperatures.The ungerade vibrational states have been observed by IR-UV double resonance, using the 3and 3+4 IR bands as intermediates. The infrared radiation was generated in a two-step difference frequency generation/optical parametric amplification process. A portion of the 1064 nm output of an injection-seeded Nd:YAG laser 共Spectra-Physics PRO-270兲 was mixed, in a lithium niobate 共LiNbO3兲 crystal, with the output of a dye laser 共Lambda Physik FL 2002兲 operating with either LDS 798 or LDS 751. The resulting infrared radiation was then passed through a second LiNbO3 crystal, which was pumped by the remainder of the 1064 nm beam. The amplified IR radiation had an energy of approximately 3 mJ/pulse and a spectral width of 0.15 cm−1, which is lim-ited by the resolution of the grating-tuned dye laser.
A small fraction of the IR beam was sent to a photoa-coustic cell containing 10 torr of acetylene gas. The observed photoacoustic signal was used to ensure that the IR fre-quency stayed in resonance with the desired vibrational tran-sition. The remaining IR radiation entered the chamber
through a CaF2window in the opposite direction to the UV laser. The relative timing of the two lasers was adjusted so that the beams were temporally overlapped, with the precise timing adjusted to maximize the observed double resonance fluorescence signals.
III. THEORY
The energy level pattern for the coupled4and6 bend-ing fundamentals of the A˜ 1Au state of C2H2 has been de-scribed by Utz et al.33 and is similar to that analyzed by Hegelund et al.37 in the infrared spectrum of the trans-bent molecule diimide N2H2. The overtones of 4 and 6, which are the subject of the present work on C2H2, require, in addition, considerations of anharmonicity and Darling–Dennison resonance.38 A summary of the relevant theory follows.
A. Matrix elements of the rotational and Coriolis operators
For an asymmetric top molecule such as C2H2 in its
A ˜ 1A
ustate, the general rotational Hamiltonian 39–41
simplifies to
Hrot= A共Ja− Ga兲2+ B共Jb− Gb兲2+ C共Jc− Gc兲2, 共1兲
where A, B, and C are the rotational constants, J is the total angular momentum, G is the vibrational angular momentum, and a, b, and c refer to the principal inertial axes. When the squares are expanded, this equation becomes
Hrot= AJa2+ BJb2+ CJc2− 2AJaGa− 2BJbGb − 2CJcGc+ AGa 2 + BGb 2 + CGc 2 . 共2兲
The first three terms are the familiar rigid rotator Hamiltonian, followed by the three first-order Coriolis terms, and finally three terms involving the squares of the components of the vibrational angular momentum. These components are defined42,43as
G␣= Qtr␣P, 共3兲
where Q and P are the vectors of vibrational normal coordinates and their conjugate momenta, and ␣ is a skew-symmetric matrix of Coriolis coupling constants. Multiplying this out, and considering just the vibrations 4and6,
G␣= Q446␣P6− Q646␣P4. 共4兲
For the A˜ 1Austate of C2H2the only nonvanishing coupling constants between the4and6vibrations are46aand46b, which are related by the sum rule,13,41
共46a兲2+共46b兲2= 1. 共5兲 The terms in Gccan therefore be ignored, since there is no c-axis coupling.
The matrix elements of the vibrational angular momen-tum operators follow from the matrix elements of the Q and P operators in a harmonic basis,40
054304-3 Bending overtones of the A˜ 1A
ustate of acetylene C2H2 J. Chem. Phys. 129, 054304共2008兲
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32
具v4+ 1v6兩G␣兩v4v6+ 1典 = − i46␣ប⍀关共v4+ 1兲共v6+ 1兲兴1/2, 共6兲 具v4v6+ 1兩G␣兩v4+ 1v6典 = i46␣ប⍀关共v4+ 1兲共v6+ 1兲兴1/2, where⍀ is Mills’ abbreviation,44
⍀ = 关共4/6兲1/2+共6/4兲1/2兴/2. 共7兲 Since the highest J values in either our jet-cooled spectra or our double resonance spectra are never more than 9, centrifugal distortion effects can be ignored. In a signed-k basis the matrix elements of the rigid rotator and first-order Coriolis terms are then
具v4v6Jk兩H兩v4v6Jk典 =
关
A −12共B + C兲兴
k2+21共B + C兲J共J + 1兲, 具v4v6Jk⫾ 2兩H兩v4v6Jk典 =14共B − C兲关J共J + 1兲 − k共k ⫾ 1兲兴1/2关J共J + 1兲 −共k ⫾ 1兲共k ⫾ 2兲兴1/2, 具v4+ 1v6Jk兩H兩v4v6+ 1Jk典 = 2iA46a⍀k关共v4+ 1兲共v6+ 1兲兴1/2, 具v4+ 1v6Jk⫾ 1兩H兩v4v6+ 1Jk典 = iB46b⍀关J共J + 1兲 − k共k ⫾ 1兲兴1/2关共v4+ 1兲共v6+ 1兲兴1/2, 具v4v6+ 1Jk兩H兩v4+ 1v6Jk典 = − 2iA46a⍀k关共v4+ 1兲共v6+ 1兲兴1/2, 具v4v6+ 1Jk⫾ 1兩H兩v4+ 1v6Jk典 = − iB46b⍀关J共J + 1兲 − k共k ⫾ 1兲兴1/2关共v4+ 1兲共v6+ 1兲兴1/2. 共8兲 The matrix elements of the terms in G␣2can be obtained by matrix multiplication from Eq. 共6兲. There are nine pos-sible expressions, but the only important ones are the diago-nal element and the elements that act within a given vibra-tional polyad, defining this as one where the levels have the same value ofv4+v6, 具v4v6兩AGa 2 + BGb 2兩v 4v6典 =关A共46a兲2+ B共46b兲2兴共2v4v6+v4+v6兲, 具v4v6兩AGa 2 + BGb 2兩v 4+ 2v6− 2典 = −关A共46a兲2+ B共46b兲2兴关共v4+ 1兲共v4+ 2兲v6共v6− 1兲兴1/2, 具v4v6兩AGa2+ BGb2兩v4− 2v6+ 2典 = −关A共46a兲2+ B共46b兲2兴关v4共v4− 1兲共v6+ 1兲共v6+ 2兲兴1/2. 共9兲 In deriving these elements the factors ofប2are absorbed into the rotational constants, and⍀2has been taken as exactly 1; terms involving 关共4/6兲1/2−共6/4兲1/2兴2 were ignored since 4 and6 are very nearly the same. The approximation of retaining only the matrix elements of G␣2 acting within agiven polyad is not expected to cause problems, since the closest interacting polyads must differ by two units of v4 +v6, as a result of the g − u symmetry properties of the levels, and will be separated by about 1500 cm−1.
It can be seen from the first line of Eq.共9兲that one of the effects of the vibrational angular momentum is to add a quantity关A共46a兲2+ B共46b兲2兴 to the vibrational frequencies4 and 6. With the values of the parameters taken from the least squares analysis of thev4+v6= 2 polyad, described be-low, this quantity is 7.06 cm−1.
The vibrational angular momentum also adds twice this quantity, i.e., 14.12 cm−1, to the anharmonicity parameter
x46. This is slightly larger than the observed value x46 = 11.39 cm−1 共described below兲, and implies that the anhar-monic force field contributes a mere −2.73 cm−1 to x46. The dominance of the angular momentum contribution to x46 is unusual and somewhat surprising. It also emphasizes that the role of vibrational angular momentum in generating what looks like anharmonicity should not be neglected.
B. Darling–Dennison resonance
The off-diagonal elements in Eq.共9兲 have the same vi-brational quantum number dependence as those responsible for Darling–Dennison resonance.38 This is a well-known effect45–47in the overtone spectroscopy of molecules such as H2O and the ground state of C2H2. Provided that certain definite relationships between the Darling–Dennison reso-nance parameter and the anharmonicity constants are satisfied,48the Darling–Dennison resonance converts the nor-mal mode energy level pattern of the low-lying stretching vibrational levels into a local mode pattern at higher energy.45 This represents how the vibrational structure changes from the low energy pattern, where the two bonds vibrate in phase, to the high energy pattern approaching dis-sociation, where just one of the two bonds breaks. The strong Darling–Dennison resonance involving nearly degenerate bending vibrations in an asymmetric top appears to be a new phenomenon. Some aspects of the resulting vibrational level structure resemble what is found for stretching vibrations, but there are also differences; research into the various effects is continuing.
Darling–Dennison resonance has been considered in some detail by Lehmann.49 He gives the matrix element as 具na+ 2nb− 2兩H兩nanb典 =14Kaabb关共na+ 1兲共na+ 2兲nb共nb− 1兲兴1/2, 共10兲 where Kaabb= 1 4aabb+
兺
␣ − B␣共ab ␣兲2共a+b兲2 ab + 1 8兺
k kaakbbk冉
1 4a2−k2 + 1 4b2−k2冊
− 1 2兺
k kab2 k k 2 −共a−b兲2 . 共11兲Although the notation is different, the term involving the ab␣ parameters in Eq.共11兲corresponds exactly to the
coef-054304-4 Merer et al. J. Chem. Phys. 129, 054304共2008兲
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32
ficient in the off-diagonal elements of Eq. 共9兲. Allowing for the factor 14 in Eq. 共10兲, this term contributes an amount −4关A共46a兲2+ B共46b兲2兴=−28.24 cm−1 to the parameter K
4466. Again this is a surprisingly large amount, which 共as shown below兲 outweighs the effects of the cubic and quartic anhar-monic potential constants. It is remarkable that the vibra-tional angular momentum should make such a large contri-bution to what is usually thought of as anharmonicity, both for the x46and K4466parameters.
C. Structures of the Hamiltonian matrices
A complication in using the elements of Eq. 共8兲 for a matrix calculation of the energy levels is that the first-order Coriolis terms are imaginary. This can be overcome by mul-tiplying the 兩v6典 harmonic oscillator basis functions by a phase factor共i兲v6. To implement this, the Hamiltonian
matri-ces for each J-value from Eq.共8兲are subjected to a similarity transformation, H
⬘
= S†HS. The S matrix consists of blocks for each vibrational level that take the sums and differences of the signed-k basis functions, converting them to an unsigned-K basis,50 but with all the elements in the blocks for the various vibrational levels multiplied by 共i兲v6. Thetransformation factorizes the matrix for each J-value into two submatrices, which can be given e and f symmetry labels. Further factorization is not possible because of the ⌬K= ⫾1 form of the b-axis Coriolis elements.
After the transformation the matrix elements are all real, but any element off-diagonal in the vibrational quantum numbers carries a negative sign. The energy matrices can then be constructed directly from Eqs. 共8兲 and共10兲, taking account of the signs and the vibrational symmetries. Writing the basis functions as 兩K⫾x典, where ⫾ indicates sum
or difference and x is the irreducible representation label a or b, the e matrix contains the functions 兩0+a典,兩1−a典,兩1+b典,兩2+a典,兩2−b典,兩3−a典,..., while the f matrix has the same structure but with the a and b labels reversed. As is well known,50 the具k=−1兩H兩k=1典 asymmetry element is added to or subtracted from the兩K=1典 diagonal element by the similarity transformation, and any element connecting a兩k=0典 basis function to a 兩k= ⫾1典 or 兩k= ⫾2典 basis func-tion gets multiplied by 21/2.
D. Selection rules. Coriolis coupling and axis-switching
In the absence of Coriolis and axis-switching16 effects, the rotational selection rules for the c-axis polarized A˜ 1Au
− X˜ 1⌺+g transition are K
⬘
−ᐉ⬙
=⫾1, ⌬J=0, ⫾1. BothCori-olis and axis-switching effects act to destroy the strictness of the first rule, giving rise to additional K
⬘
−ᐉ⬙
= 0 ,⫾2 sub-bands. Because of this it is difficult to distinguish the two effects, and, in fact, it is possible to rationalize axis-switching effects using the formalism of Coriolis coupling.15,51,52 In this paper the term axis-switching will be used to describe the forbidden subbands of the Franck– Condon allowed 3n and 213n progressions. These are easilyrecognized since the K-structures of the upper levels follow the normal asymmetric top energy level expressions, because there is no competing Coriolis coupling. The forbidden
sub-bands in the bending polyads are best described as Coriolis-induced since this is the principal mechanism for their appearance. These forbidden subbands are mostly fairly weak but a few are surprisingly strong, particularly when two sets of levels with zero-order K values differing by one unit happen to lie close to each other.
As for the vibrational selection rules, these are found to be obeyed strictly only for the K = 0 levels of the v4+v6 = even polyads, where K
⬘
= 0 levels with bgvibrationalsym-metry are not seen. For the v4+v6= odd polyads, the K
⬘
= 0 levels with both au and bu vibrational symmetry appear indouble resonance spectra via⌸uintermediate levels 共ᐉ
⬙
= 1兲.They can be distinguished by their different rotational selec-tion rules. For instance, in transiselec-tions from f-symmetry rota-tional levels of a⌸uintermediate level, K
⬘
= 0 levels with auvibrational symmetry 共which have e rotational symmetry兲 give only Q branches, while those with bu vibrational
sym-metry 共f rotational symmetry兲 give R and P branches; the pattern is reversed in transitions from e-symmetry interme-diate levels. The relative strengths of double resonance tran-sitions to K
⬘
= 0 levels with au and buvibrational symmetryare found to depend on which⌸u intermediate state is
cho-sen. Transitions to bulevels dominate when3+4is used as
the intermediate, though experiments with other polyads have shown that transitions to au levels dominate when
1+5 is used as the intermediate. It is not clear why this should be so. For K⫽0 levels the a-axis Coriolis mixing is so strong that every vibrational level appears in the spectrum, with the only restriction being that K
⬘
−ᐉ⬙
=⫾1 transitions are usually the most intense.For simplicity in what follows the distinction between K for the A˜ 1Austate andᐉ for the X˜1⌺+gstate will not always
be made. The two quantities describe the projection of the total angular momentum along the linear or near-linear inertial axis, and are essentially equivalent.
IV. RESULTS
The structures of the bending polyads are highly irregu-lar because of the interplay of the Coriolis coupling and the Darling–Dennison resonance. Both of these are very large effects, with the experimental Darling–Dennison parameter
K4466found to be about −50 cm−1, and the Coriolis param-eter 2Aa共which is the coefficient of K in the a-axis coupling
elements兲 about 18 cm−1. It is not possible to separate them, or to understand the structures of the polyads, without data from levels with several K-values. For instance, it is essential to have data from as many of the K = 0 stacks as possible, because these contain no a-axis Coriolis effects, and allow the vibrational origins to be established. Even so, these vibrational origins are not the fully deperturbed origins, because they represent the levels that result after the Darling–Dennison resonance has acted between harmonic basis levels of the same symmetry. The higher-K stacks suf-fer from both Darling–Dennison and Coriolis effects, but the effects can be separated because the a-axis Coriolis coupling depends linearly on K, whereas the Darling–Dennison reso-nance is independent of K. This means that a successful least squares analysis of a bending polyad requires data from 054304-5 Bending overtones of the A˜ 1A
ustate of acetylene C2H2 J. Chem. Phys. 129, 054304共2008兲
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32
K = 0, 1, and 2 stacks, at the minimum. It is of course
neces-sary to use the deperturbed band origins in order to calculate the higher-K levels correctly, since the Coriolis coupling and the Darling–Dennison resonance are both perturbations on the rigid rotator-harmonic oscillator basis.
A. The v4+ v6= 2 polyad„B2…
The first overtone polyad 共v4+v6= 2, or B2, where B means “bending”兲 lies near 43 700 cm−1. It consists of three vibrational levels: 42, 4161, and 62. In room temperature ab-sorption spectra it is completely buried under very intense hot band structure from the Franck–Condon allowed 324
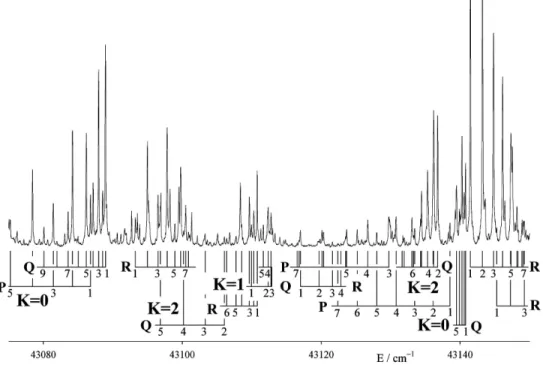
1 band, but in jet-cooled LIF experiments the hot bands are sufficiently reduced to make analysis possible. Two regions of structure can be recognized: a confused group of lines centered at 43 715 cm−1 and an elegantly simple K
⬘
−ᐉ⬙
= 1 − 0 subband at 43 796 cm−1. The 43 715 cm−1 group is illus-trated in Fig.2. In this figure the lines of an overlapping hot band have been coloured light gray.The bands shown in Fig.2represent transitions from the ground vibrational level of the molecule to the lower two of the three K
⬘
= 1 levels of the polyad, together with a Coriolis-induced K = 2 − 0 subband. One of the K = 1 − 0 subbands, with Q head at 43 712 cm−1, is easily assigned, and accounts for most of the strong lines. Somewhat surprisingly for a level so low in the vibrational manifold, it contains pertur-bations at J⬘
= 4e and 5f. Because of the unexpectedly large Coriolis perturbations and the severe blending, the remaining structure could not be assigned until the v4+v6= 3 共B3兲 polyad had been analyzed, and the Darling–Dennison reso-nance recognized. Calculations of the rotational structure then allowed the remaining lines, and the perturbations, to be assigned immediately.Experiments with a warmed beam gave the spectrum shown in Fig.3. The energy range illustrated lies below that of Fig.2 by the amount of the ground state4fundamental, and shows four of the K
⬘
= 0 and 2 stacks as hot bands from 4⬙
. The K⬘
= 0 subbands belong to the two overtones, namely 42 and 62, which have ag vibrational symmetry.
Although the 4 and6fundamentals lie only 3 cm−1 apart, the two overtones are 52 cm−1 apart, as a result of the Darling–Dennison resonance. The third K
⬘
= 0 hot band, going to the combination level 4161 共bg vibrational
symme-try兲, is not seen. It would be observable if it lay close enough to one of the K
⬘
= 1 levels to obtain some intensity by b-axis Coriolis coupling, but such is not the case here. It is calcu-lated to lie at 43 131 cm−1.The K
⬘
= 1 and 2 subbands near 43 110 cm−1 in Fig. 3are hot bands with the same upper states as the cold bands near 43 720 cm−1in Fig. 2. In Fig.2 the intensity is carried by the K
⬘
= 1 level, with the K⬘
= 2 level getting its intensity by b-axis Coriolis coupling; in Fig. 3 the roles of the two levels are reversed. Consistent with the line strengths for aK = 2 − 1 band, the P branches of both bands are expected to
be much weaker than the rest of the structure, and are not seen. The intense K = 2 − 1 hot band at 43 138 cm−1in Fig.3 goes to the middle K
⬘
= 2 level. The uppermost K = 2 − 1 hot band is calculated to lie at 43 258 cm−1, exactly at the posi-tion of the strong Franck–Condon allowed 310 band. It will be totally buried, and has not been searched for.The assignments of the K
⬘
= 0 and 2 hot bands near 43 140 cm−1 are confirmed by the observation of some very weak lines near 43 750 cm−1in the cold spectra, correspond-ing to Coriolis-induced K = 0-0 and 2-0 cold bands. These had been noted during the analysis of the cold spectra, but could not be assigned initially. The line assignments of the FIG. 2. Low frequency part of thev4+v6= 2共B2兲 polyad of the A˜1Austate of C2H2, observed in one-photon laser excitation. Three subbands are present,representing three interacting K-stacks, two with K⬘= 1 and one with K⬘= 2. The lines of an overlapping hot band共324
1,⌬-⌸兲 have been colored gray.
054304-6 Merer et al. J. Chem. Phys. 129, 054304共2008兲
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32
various subbands are given in Table I. In this table the sub-bands are labeled for convenience by Roman numerals in order of increasing energy for each K value.
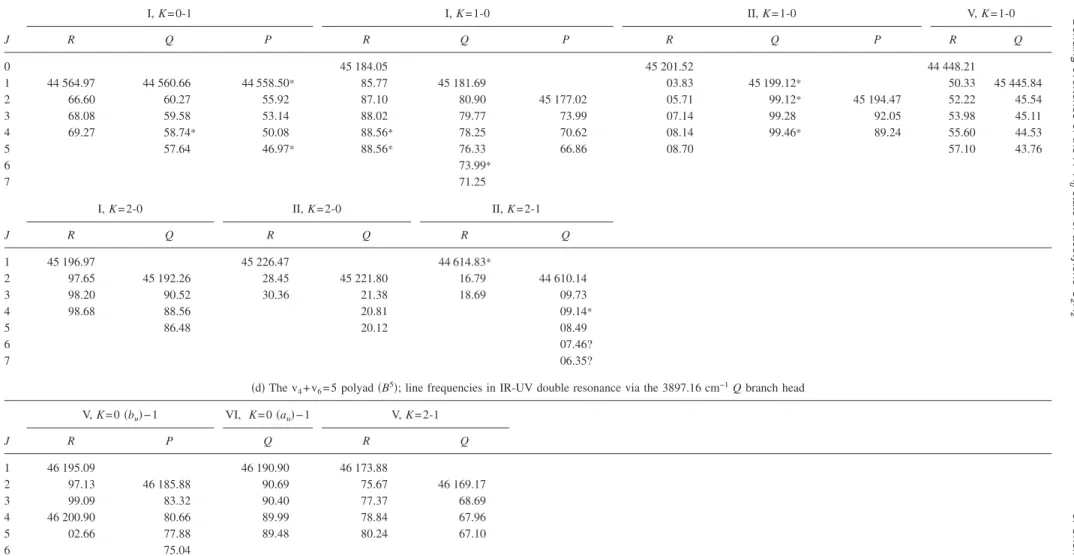
The level structure of the B2 polyad is illustrated in Fig.4. The left hand side关Fig.4共a兲兴 shows the K-structure. The two overtones 42 and 62 are almost degenerate in zero-order, with 42lying 2.1 cm−1above 62; however, as a result of the Darling–Dennison resonance, they give rise to two well-separated K = 0 levels, whose wave functions are very nearly the normalized sum and difference of 兩42典共0兲 and 兩62典共0兲. The K = 0 level of 4161lies above the mid-point of the two overtones because of the x46 term. For K = 1 the a-axis Coriolis coupling complicates the picture. The 4161level in-teracts essentially only with the upper of the mixed overtone levels, whose approximate wave function is the sum function 共2兲−1/2关兩42典共0兲+兩62典共0兲兴. The lower mixed overtone 共difference function兲 is almost unaffected. The result is that the two up-per K = 1 levels are pushed apart, with one of them dropping almost to the energy of the lower mixed overtone. For
K艌2 the Coriolis coupling between the upper mixed
over-tone and 4161 is so large that the Coriolis-coupled levels become the top and bottom levels with their K-value. It is possible to view these effects as an interference between the Coriolis and Darling–Dennison interactions, which allows the sign of the Darling–Dennison parameter to be deter-mined.
Figure 4共b兲 shows the observed and calculated
J-structure plotted against J共J+1兲. Only the low energy
lev-els are illustrated, so that the highest observed K = 1 level lies off the top of the figure. The most obvious irregularity is the very strong b-axis Coriolis perturbation between the lowest
K = 2 level and the second K = 1 level. The two levels are
almost exactly degenerate in zero-order, such that the split-ting between them rises to over 20 cm−1 at J = 7. The lower
level, nominally K = 2, is pushed down so hard that the R branch going to it共see Fig.2兲 degrades entirely to the red, in
contrast to the usual pattern. This level also cuts through the lowest K = 1 level, causing the small perturbations mentioned at the beginning of this section.
An unexpected feature in Fig.4共b兲is the presence of an unseen K = 3 level between the two uppermost K = 0 levels. It appears not to perturb the nearby K = 2 level, despite the possibility of b-axis Coriolis coupling 共following ⌬K= ⫾1 selection rules兲, but to interact strongly with the two K=0 levels. Detailed examination of the rotational energy matri-ces and their eigenvectors confirms that the K = 2 and K = 3 levels should not interact. The vibrational wave functions for these levels are, to good approximation,
K = 2: 共2兲−1/2关兩42典共0兲−兩62典共0兲兴,
共12兲
K = 3: 共2兲−1/2兩4161典共0兲+共1/2兲关兩42典共0兲+兩62典共0兲兴.
The b-axis Coriolis coupling between them would involve the matrix element
具4161兩共0兲G
b关兩42典共0兲−兩62典共0兲兴, 共13兲
where the two terms cancel exactly, to give zero. On the other hand, the⌬K= ⫾3 interactions between the K=3 level and the two K = 0 levels arise from vibrationally allowed cross-terms between the asymmetry and the b-axis Coriolis coupling.
With seven of the nine K-stacks with K = 0 – 2 assigned, there are enough data for a least squares fit to the upper state term values. A simple model was chosen for the rotational structure. The rotational constants A, 12共B+C兲, and B−C were varied just for the overtones 42 and 62, and it was assumed that the rotational constants for the combination FIG. 3. Hot bands of thev4+v6= 2共B2兲 polyad of the A˜1Austate of C2H2; the region shown lies to the red of that shown in Fig.2by the amount of the ground
state4⬙fundamental共g, 612 cm−1兲. Overlapping lines from bands with4⬙= 2 have been colored gray.
054304-7 Bending overtones of the A˜ 1A
ustate of acetylene C2H2 J. Chem. Phys. 129, 054304共2008兲
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32
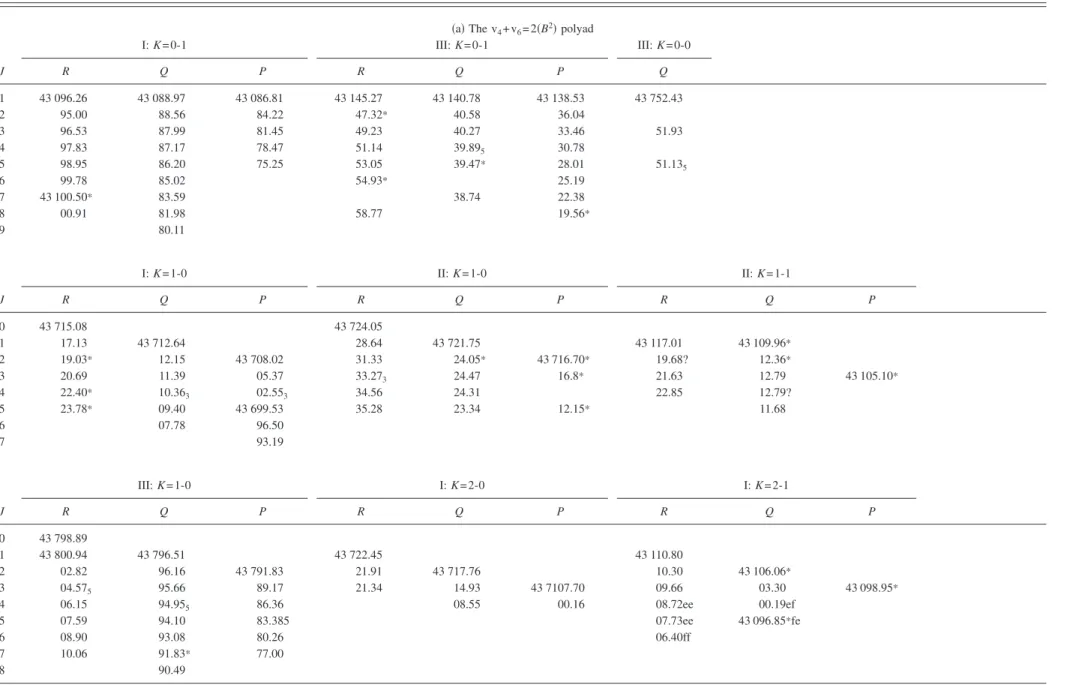
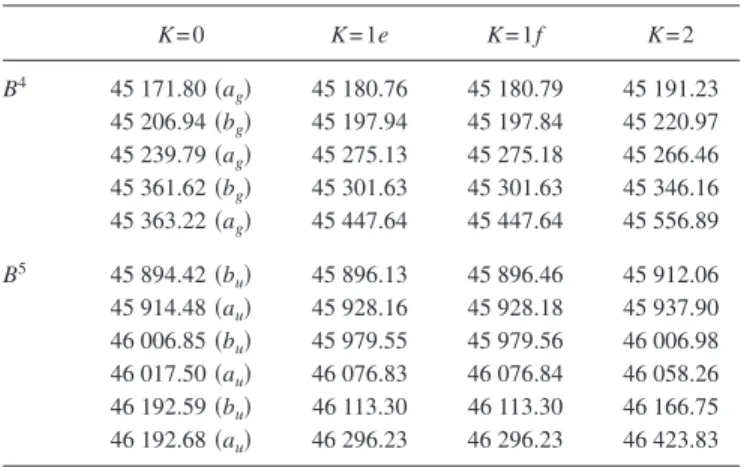
TABLE I. Assigned rotational lines of the pure bending polyads of the A˜1A
ustate of C2H2. The members of the upper state polyads are labeled with Roman numerals in order of increasing energy for each K-value.
Blended lines are marked with an asterisk共*兲.
共a兲 The v4+ v6= 2共B2兲 polyad
I: K = 0-1 III: K = 0-1 III: K = 0-0 J R Q P R Q P Q 1 43 096.26 43 088.97 43 086.81 43 145.27 43 140.78 43 138.53 43 752.43 2 95.00 88.56 84.22 47.32* 40.58 36.04 3 96.53 87.99 81.45 49.23 40.27 33.46 51.93 4 97.83 87.17 78.47 51.14 39.895 30.78 5 98.95 86.20 75.25 53.05 39.47* 28.01 51.135 6 99.78 85.02 54.93* 25.19 7 43 100.50* 83.59 38.74 22.38 8 00.91 81.98 58.77 19.56* 9 80.11 I: K = 1-0 II: K = 1-0 II: K = 1-1 J R Q P R Q P R Q P 0 43 715.08 43 724.05 1 17.13 43 712.64 28.64 43 721.75 43 117.01 43 109.96* 2 19.03* 12.15 43 708.02 31.33 24.05* 43 716.70* 19.68? 12.36* 3 20.69 11.39 05.37 33.273 24.47 16.8* 21.63 12.79 43 105.10* 4 22.40* 10.363 02.553 34.56 24.31 22.85 12.79? 5 23.78* 09.40 43 699.53 35.28 23.34 12.15* 11.68 6 07.78 96.50 7 93.19 III: K = 1-0 I: K = 2-0 I: K = 2-1 J R Q P R Q P R Q P 0 43 798.89 1 43 800.94 43 796.51 43 722.45 43 110.80 2 02.82 96.16 43 791.83 21.91 43 717.76 10.30 43 106.06* 3 04.575 95.66 89.17 21.34 14.93 43 7107.70 09.66 03.30 43 098.95* 4 06.15 94.955 86.36 08.55 00.16 08.72ee 00.19ef 5 07.59 94.10 83.385 07.73ee 43 096.85*fe 6 08.90 93.08 80.26 06.40ff 7 10.06 91.83* 77.00 8 90.49 054304-8 Merer et al. J. Chem. Phys. 129 , 054304 共2008 兲
TABLE I. 共Continued.兲 II: K = 2-0 II: K = 2-1 J R Q P Ree Rf f Qef Qfe Pee Pf f 1 43 753.155 43 141.48 2 54.915 43 748.44 43.21 43 136.77 3 56.48 47.845 43 741.39 44.80 36.205 43 129.75 4 57.82 47.045 38.43 46.11 35.31 35.41 26.69 5 59.00 46.08 35.28* 47.32* 34.29 34.44 23.68 23.53 6 60.01 48.30* 48.18 33.06 33.29 20.33? 20.13 7 60.72 49.12 48.91* 31.93 16.82 16.54 8 29.96 共b兲 The v4+ v6= 3共B3兲 polyad
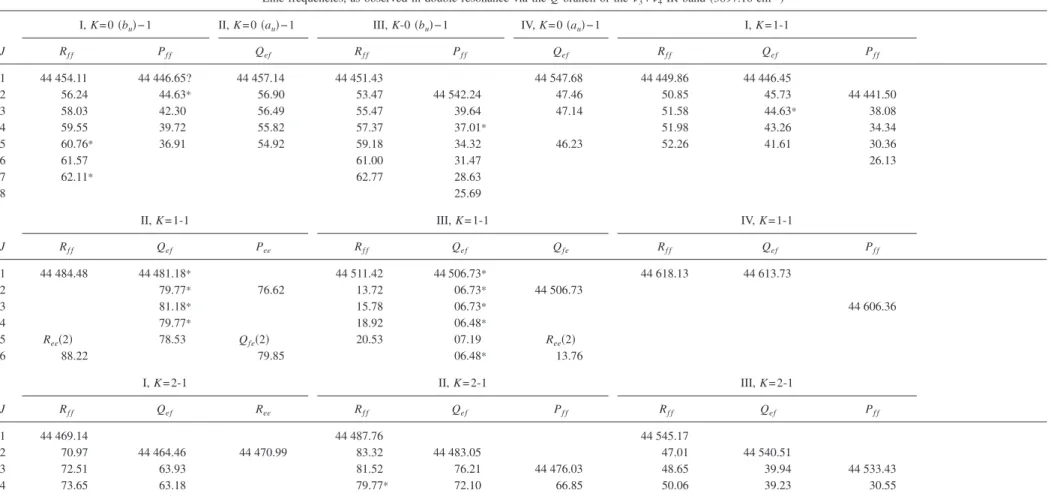
Line frequencies, as observed in double resonance via the Q branch of the3+4IR band共3897.16 cm−1兲
I, K = 0共bu兲−1 II, K = 0共au兲−1 III, K-0共bu兲−1 IV, K = 0共au兲−1 I, K = 1-1
J Rf f Pf f Qef Rf f Pf f Qef Rf f Qef Pf f 1 44 454.11 44 446.65? 44 457.14 44 451.43 44 547.68 44 449.86 44 446.45 2 56.24 44.63* 56.90 53.47 44 542.24 47.46 50.85 45.73 44 441.50 3 58.03 42.30 56.49 55.47 39.64 47.14 51.58 44.63* 38.08 4 59.55 39.72 55.82 57.37 37.01* 51.98 43.26 34.34 5 60.76* 36.91 54.92 59.18 34.32 46.23 52.26 41.61 30.36 6 61.57 61.00 31.47 26.13 7 62.11* 62.77 28.63 8 25.69
II, K = 1-1 III, K = 1-1 IV, K = 1-1
J Rf f Qef Pee Rf f Qef Qfe Rf f Qef Pf f 1 44 484.48 44 481.18* 44 511.42 44 506.73* 44 618.13 44 613.73 2 79.77* 76.62 13.72 06.73* 44 506.73 3 81.18* 15.78 06.73* 44 606.36 4 79.77* 18.92 06.48* 5 Ree共2兲 78.53 Qfe共2兲 20.53 07.19 Ree共2兲 6 88.22 79.85 06.48* 13.76 I, K = 2-1 II, K = 2-1 III, K = 2-1 J Rf f Qef Ree Rf f Qef Pf f Rf f Qef Pf f 1 44 469.14 44 487.76 44 545.17 2 70.97 44 464.46 44 470.99 83.32 44 483.05 47.01 44 540.51 3 72.51 63.93 81.52 76.21 44 476.03 48.65 39.94 44 533.43 4 73.65 63.18 79.77* 72.10 66.85 50.06 39.23 30.55 054304-9 Bending overtones of the A ˜ 1 A u state of acetylene C 2H 2 J. Chem. Phys. 129 , 054304 共2008 兲
TABLE I. 共Continued.兲 I, K = 2-1 II, K = 2-1 III, K = 2-1 J Rf f Qef Ree Rf f Qef Pf f Rf f Qef Pf f 5 73.94 62.10* 79.06 67.93 51.24 38.30 27.43 6 52.28 37.21 24.17 7 53.07 35.94 20.65 8 Qfe共2兲 Qfe共2兲 34.49 9 64.43 83.11 32.82 IV, K = 2-1 I, K = 3-1 II, K = 3-1 J Rf f Qef Pee Rf f Qef Pf f Rf f Qef Ree 1 44 707.97 2 09.82 44 703.26 44 497.06 44 515.18 44 415.23 3 11.46 02.75 44 696.23 99.12 44 489.99 17.04* 44 508.12 4 12.95 02.05 93.33 44 500.32 89.71 44 480.57 17.04* 07.63 5 14.30 01.21 90.29 00.89 88.50 77.95 18.13 05.28 6 15.46 00.22 01.10 86.70 04.07 7 16.46 44 699.01 02.65?
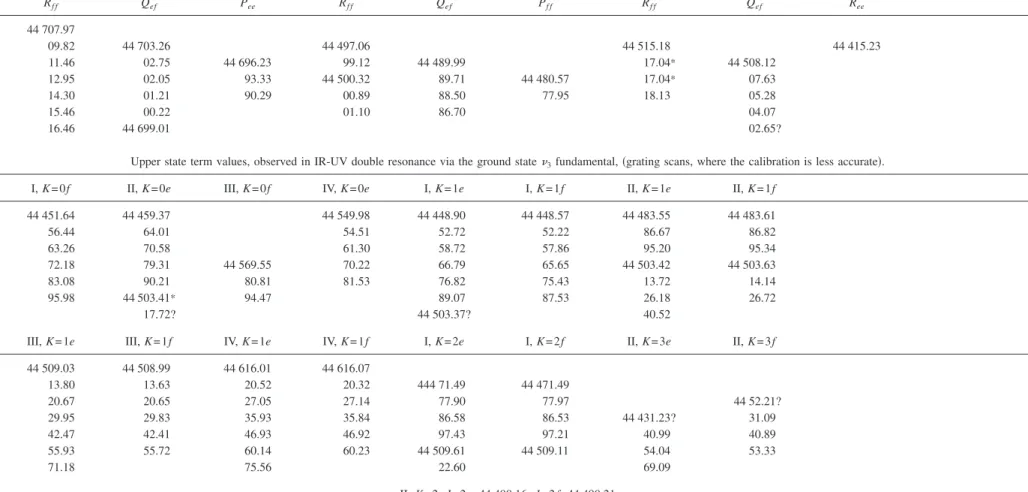
Upper state term values, observed in IR-UV double resonance via the ground state3fundamental,共grating scans, where the calibration is less accurate兲. J I, K = 0f II, K = 0e III, K = 0f IV, K = 0e I, K = 1e I, K = 1f II, K = 1e II, K = 1f
1 44 451.64 44 459.37 44 549.98 44 448.90 44 448.57 44 483.55 44 483.61 2 56.44 64.01 54.51 52.72 52.22 86.67 86.82 3 63.26 70.58 61.30 58.72 57.86 95.20 95.34 4 72.18 79.31 44 569.55 70.22 66.79 65.65 44 503.42 44 503.63 5 83.08 90.21 80.81 81.53 76.82 75.43 13.72 14.14 6 95.98 44 503.41* 94.47 89.07 87.53 26.18 26.72 7 17.72? 44 503.37? 40.52
J III, K = 1e III, K = 1f IV, K = 1e IV, K = 1f I, K = 2e I, K = 2f II, K = 3e II, K = 3f
1 44 509.03 44 508.99 44 616.01 44 616.07 2 13.80 13.63 20.52 20.32 444 71.49 44 471.49 3 20.67 20.65 27.05 27.14 77.90 77.97 44 52.21? 4 29.95 29.83 35.93 35.84 86.58 86.53 44 431.23? 31.09 5 42.47 42.41 46.93 46.92 97.43 97.21 40.99 40.89 6 55.93 55.72 60.14 60.23 44 509.61 44 509.11 54.04 53.33 7 71.18 75.56 22.60 69.09 II, K = 2, J = 2e: 44 490.16; J = 2f: 44 490.21 054304-10 Merer et al. J. Chem. Phys. 129 , 054304 共2008 兲
TABLE I. 共Continued.兲
共c兲 The v4+ v6= 4 polyad共B4兲; line frequencies in one-photon LIF
I, K = 0-1 I, K = 1-0 II, K = 1-0 V, K = 1-0 J R Q P R Q P R Q P R Q 0 45 184.05 45 201.52 44 448.21 1 44 564.97 44 560.66 44 558.50* 85.77 45 181.69 03.83 45 199.12* 50.33 45 445.84 2 66.60 60.27 55.92 87.10 80.90 45 177.02 05.71 99.12* 45 194.47 52.22 45.54 3 68.08 59.58 53.14 88.02 79.77 73.99 07.14 99.28 92.05 53.98 45.11 4 69.27 58.74* 50.08 88.56* 78.25 70.62 08.14 99.46* 89.24 55.60 44.53 5 57.64 46.97* 88.56* 76.33 66.86 08.70 57.10 43.76 6 73.99* 7 71.25 I, K = 2-0 II, K = 2-0 II, K = 2-1 J R Q R Q R Q 1 45 196.97 45 226.47 44 614.83* 2 97.65 45 192.26 28.45 45 221.80 16.79 44 610.14 3 98.20 90.52 30.36 21.38 18.69 09.73 4 98.68 88.56 20.81 09.14* 5 86.48 20.12 08.49 6 07.46? 7 06.35?
共d兲 The v4+ v6= 5 polyad共B5兲; line frequencies in IR-UV double resonance via the 3897.16 cm−1Q branch head
V, K = 0共bu兲−1 VI, K = 0共au兲−1 V, K = 2-1 J R P Q R Q 1 46 195.09 46 190.90 46 173.88 2 97.13 46 185.88 90.69 75.67 46 169.17 3 99.09 83.32 90.40 77.37 68.69 4 46 200.90 80.66 89.99 78.84 67.96 5 02.66 77.88 89.48 80.24 67.10 6 75.04 7 72.08 V, K = 2-1, P共2兲: 46 162.10 054304-1 1 Bending overtones of the A ˜ 1 A u state of acetylene C 2H 2 J. Chem. Phys. 129 , 054304 共2008 兲
level 4161were the averages of those for the two overtones. For the coupling terms, the two Coriolis parameters 2Aaand
Bb and the Darling–Dennison parameter K4466were varied initially, though it was later found that adding a centrifugal distortion correction to K4466 improved the fit considerably. This was taken as
K4466eff= K4466共0兲+ K4466,DK2. 共14兲 The vibrational parameters required some care. Since the
K
⬘
= 0 stack of the combination level 4161is not seen, there are only two observable band origins, corresponding to the overtones 42and 62, heavily mixed by the Darling–Dennison interaction. In the end it was decided to include the J = K = 0 energies of the two fundamentals 4 and 6 共from the work of Utz et al.33兲, and to adjust, by least squares,4,6, and two of the three anharmonicity parameters x44, x46, andx66. The value of x66 was then fixed at −4.226cm−1, as
ob-tained from combining the position of the 6 fundamental with the deperturbed origin of the 63 overtone关described in Sec. IV B兴. The fit is extremely good, with an rms error of 0.011 cm−1, which is comparable to the accuracy of the line measurements; the results are given in TableII.
The most surprising result is the large size of the param-eters x46and K4466. These are made up of contributions from the vibrational angular momentum and the anharmonic force field, and in both cases the vibrational angular momentum contribution is the larger. It is interesting to compare the anharmonic contributions with those calculated from the an-harmonic force field of Ref. 36. For the Darling–Dennison parameter K4466, where the experimental value, −51.68 cm−1, includes −28.24 cm−1 from the vibrational angular momen-tum 共meaning that the anharmonic contribution is −23.44 cm−1兲 a calculation using Eq.共11兲gives −16.6 cm−1. TABLE II. Rotational constants from least squares fitting of the B2polyad of the A˜1A
ustate of C2H2. Values are
in cm−1. The J = K = 0 levels lie at 43 700.85 cm−1共62, 1503.28 cm−1above T
00兲, 43 742.13 共4161, 1544.56兲 and
43 752.57共42, 1555.00兲. Derived Coriolis constants:a= 0.703
8,b= 0.7111;共a兲2+共b兲2= 1.001. The only
cor-relation coefficients with magnitudes over 0.9 are12共B+C兲 共62兲/1
2共B+C兲 共42兲 −0.996, A共62兲/A 共42兲 −0.981, and
4/x44−0.949. 6 772.497 ⫾ 0.032 A共62兲 13.356 ⫾ 0.128 4 764.709 0.075 1 2共B+C兲 共62兲 1.0806 0.0077 x66 −4.226 fixed 共B−C兲 共62兲 0.1262 0.0077 x46 11.385 0.080 A共42兲 12.857 0.138 x44 0.191 0.047 1 2共B+C兲 共42兲 1.0743 0.0080 2Aa 18.449 0.005 共B−C兲 共42兲 0.0530 0.0088 Bb 0.7980 0.0023 K 4466,D 0.0381 0.0281 K4466 −51.678 0.020 rms error= 0.0112 cm−1
FIG. 4.共a兲 K-structure of the v4+v6= 2共B2兲 polyad. Observed levels are shown in black, calculated levels in gray. 共b兲 J-structure of the eight lowest K-stacks
of the B2polyad, with observed levels shown as black dots, and calculated structure as gray lines. A quantity 1.05 J共J+1兲 has been subtracted in order to
expand the scale.
054304-12 Merer et al. J. Chem. Phys. 129, 054304共2008兲
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32
Similarly, for x46, where the anharmonic contribution is −2.73 cm−1, a calculation using the symmetry-allowed terms from Mills’ perturbation theory expression,53
x46= 1 44466− 1 4
兺
k 44kk66/k 共15兲gives +1.08 cm−1. At the same time the experimental anhar-monicity parameters x44 and x66in Table IIare quite small, which suggests that the pure bending motions are compara-tively harmonic, once allowance is made for the vibrational angular momentum.
As for the Coriolis coupling parameters, these are almost unchanged from the values found in the fundamentals,33 where 2Aa= 18.47 cm−1and Bb= 0.787 cm−1. It is interest-ing to see how accurately the zeta sum rule关Eq.共5兲兴 holds in the B2polyad. The derived values of46a
and46bare given in TableII; the sum of their squares is 1.001, compared to the theoretical value of 1.
The variation in the A rotational constants withv4andv6 appears to be much smaller for the overtones共TableII兲 than
for the fundamentals,33 where A共41兲=11.36共8兲 cm−1 and
A共61兲=14.59共13兲 cm−1. However, we note that the average of A共41兲 and A共61兲 is close to the value in the zero-point level, 13.057共5兲 cm−1.13
It is known from the N2H2 spectrum37that there is almost 100% correlation between the
A constants of two strongly a-axis Coriolis-coupled levels,
such that only their sum is well-determined; this may also be affecting the determinations for the C2H2 fundamentals. In the overtones the correlation is less severe because there are more vibrational levels to provide data.
An interesting minor point is that the 共deperturbed兲 asymmetry parameter B − C is much smaller in the 42 level than it is in the 62 level. This is consistent with the C2H2
molecule becoming nonplanar on average as the torsional vibration is excited. The rationale is as follows. Since the inertial b and c axes interchange when C2H2is twisted from
trans-bent to cis-bent, there must be a point near a twisting
angle of 90° where it is accidentally a symmetric top, with
B − C = 0. Therefore excitation of the torsional vibration must
reduce B − C. The prototype molecule for this effect is H2S2, which is 90° twisted and accidentally almost exactly a sym-metric top.50,54
B. The v4+ v6= 3 polyad„B3…
The B3 polyad consists of four vibrational levels, 43, 4261, 4162 and 63; their symmetries are a
u, bu, au, and bu,
respectively. Two spectra of the polyad have been recorded by IR-UV double resonance. In one, the ground state3 fun-damental共ᐉ=0兲 was used as the intermediate level in order to observe the K
⬘
= 1 levels; in the other the3+4 combina-tion level共ᐉ=1兲 was used to observe the K⬘
= 0 and 2 levels. Because of the strong b-axis Coriolis coupling, some of theK
⬘
levels appear in both spectra.The low energy part of the polyad is illustrated in Fig.5, as seen following IR pumping of the Q branch of the 3 +4 band at 3897.16 cm−1. This branch is very compact, so that when the IR laser is tuned to its head the first five lines are excited simultaneously, populating the J = 1f – 5f rota-tional levels. This allows the complete double resonance spectrum to be recorded in one scan, though it loses the state-selectivity of pumping individual rotational lines. Nev-ertheless the line assignments could be made straightfor-wardly, using lower state combination differences. To assist with the assignments a scan was also taken via the P共3兲 line of the band, which populates the J = 2e levels. This was valu-FIG. 5. Low energy part of thev4+v6= 3共B3兲 polyad of the A˜1Austate of C2H2, as observed in IR-UV double resonance via the Q共1兲–Q共5兲 lines of the
ground state3+4combination band共3897.16 cm−1兲.
054304-13 Bending overtones of the A˜ 1A
ustate of acetylene C2H2 J. Chem. Phys. 129, 054304共2008兲
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32
able in distinguishing the K
⬘
= 0 subbands, since the branch structures depend on the parity of the intermediate levels, as described above.The strongest features in Fig. 5 are two close-lying
K
⬘
= 2 subbands. The upper states interact with each other, and with nearby K⬘
= 1 and 3 levels, inducing extra subbands. At the low energy side are two K⬘
= 0 subbands separated by 8 cm−1; they form a pair with buand ausymmetries. There isalso another weak Coriolis-induced K
⬘
= 1 band.The central part of the B3polyad is illustrated in Fig.6. It contains the other two K
⬘
= 0 subbands, again as a bu/aupair, but this time separated by only 0.7 cm−1. Also present are another strong K
⬘
= 2 subband and weaker Coriolis-induced K⬘
= 1 and 3 bands. The remainder of the polyad, not illustrated, consists of a K⬘
= 1 subband at 44 614 cm−1and aK
⬘
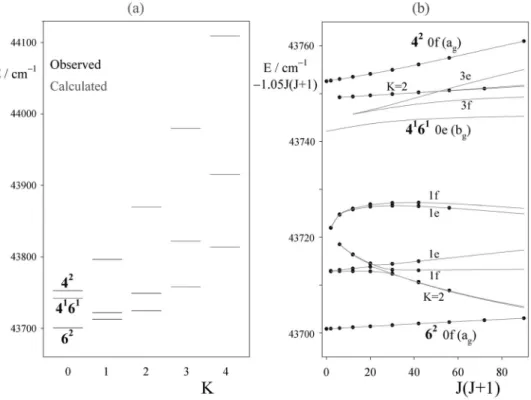
= 2 subband at 44 703 cm−1. These latter lie well above the rest of the polyad, and are not perturbed.The K-and J-structures of the B3polyad are illustrated in Fig.7. The left hand side关Fig.7共a兲兴 shows clearly how the
Darling–Dennison resonance groups the four K = 0 levels into two bu/au pairs, separated by about 100 cm−1. As expected
from the near degeneracy of the4and6fundamentals, the four zero-order basis levels lie quite close to each other, with the overtones below the combinations by an amount 2x46. However, their separations are much smaller than the Darling–Dennison matrix element. The result is that one level of each symmetry is pushed up, and the other down, by the amount of the resonance matrix element, which in this case is 50 cm−1. The patterns for higher K values are not so simple, though in first approximation the Coriolis coupling between the combination levels 4162and 4261pushes one of the levels far above the others.
The J-structure patterns 关Fig. 7共b兲兴 show a number of
local avoided crossings caused by the b-axis coupling ele-ments. These follow ⌬K= ⫾1 selection rules in the rigid rotator basis, and therefore act to destroy the goodness of K as a label. At the same time the Darling–Dennison resonance and the a-axis coupling, though diagonal in K, scramble the harmonic oscillator basis levels, so that the resulting patterns are often quite surprising. An example is given by the lowest two K = 2 levels. The upper of these two, which begins near 44 480 cm−1, is almost degenerate with the second K = 1 level, and gets pushed down strongly by b-axis coupling with it. As in the B2 polyad the R branch going to it degrades entirely to the red 共see Fig.5兲. At J=6 this K=2 level
un-dergoes an avoided crossing with the lowest K = 2, after which it goes on to perturb the two K = 0 levels at the bottom of the pattern. Very clearly the K quantum number loses all meaning, as was noted also by Utz et al.33in their analysis of the fundamentals, though it is retained here as a convenient label. Another unexpected avoided crossing occurs between the K = 1 and 3 levels near 44 510 cm−1.
Fourteen K
⬘
stacks have been identified, representing all the stacks with K = 0 – 2 together with two K⬘
= 3 stacks. This has allowed a detailed least squares treatment, of which the results are given in TableIII. As might be expected from the density of perturbations, the main problem encountered was that of matching the eigenvalues of the Hamiltonian to the observed upper state term values. After some experimenta-tion, the tactic adopted was to transform the Hamiltonian matrix in several steps. In the first step the Darling–Dennison resonance and the a-axis Coriolis coupling elements were diagonalized, after which the full Hamiltonian matrix was transformed by the resulting eigenvectors. Since the ele-ments eliminated were diagonal in K, the transformed basis states preserved the values of K. In the second step the asym-FIG. 6. Central part of the B3polyad; this spectrum is a continuation of Fig.5to higher energy. The two K⬘= 0 subbands appear to be a single subband withR, Q, and P branches, but closer examination shows that the Q branch共K=0, au兲 is shifted up by 0.73 cm−1relative to the R and P branches共K=0, bu兲.
054304-14 Merer et al. J. Chem. Phys. 129, 054304共2008兲
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32
metry elements 共⌬K= ⫾2兲 were diagonalized, such that the next set of transformed functions still retained the odd- or even-K character of the original basis functions. Finally the doubly transformed Hamiltonian matrix was diagonalized, eliminating the b-axis coupling, and the eigenvalues sorted according to their eigenvector coefficients. Even so, it was found that the sorting was not always accurate at the most severe of the avoided crossings, so that the stacks with a given nominal K-value were then placed in ascending order. Again, a simple model was used for the rotational energy, where only the A, 12共B+C兲, and B−C constants for the overtones 43and 63were varied, with those for the com-binations being interpolated between those of the overtones. As in the B2 polyad, the coupling terms used were K
4466,
2Aa, and Bb, though the first two were allowed centrifugal
distortion corrections as in Eq.共14兲. No attempt was made to write the band origins in terms of anharmonicity parameters, instead the four deperturbed origins were taken as adjustable parameters.
The final fit was not quite as good as that for the B2 polyad, but there are other factors to consider. The first is the possibility of Fermi resonance between the B3polyad and the 21B1polyad, which lies about 100 cm−1below. In fact, with the strong a-axis coupling in both polyads the K-structure of 21B1 catches up rapidly to that of B3, such that its upper
K = 2 level lies only 40 cm−1 below the lowest K = 2 of B3. An attempt was made to allow for interpolyad interactions by extending the rotational matrices to include this Fermi reso-FIG. 7.共a兲 K-structure of the B3polyad of the A˜1A
ustate of C2H2, with observed levels shown in black and calculated levels in gray. Vibrational assignments
are marked for the K = 0 levels, as given by their eigenvectors; for K艌1 levels the harmonic basis functions are so mixed that it is meaningless to give assignments.共b兲 J-structure of the 13 lowest energy K-stacks. Observed levels are shown as dots, calculated as gray lines. Two major avoided crossings occur: one between the K = 1 and 3 levels near 44 510 cm−1and the other involving the second K = 2 level, which interacts with the lowest K = 2 and the second K = 1
levels.
TABLE III. Rotational constants from least squares fitting of the B3polyad of the A˜1A
ustate of C2H2. Values
are in cm−1. The band origins共T
0兲 are given relative to T00共A˜1Au兲=42 197.57 cm−1, from Ref.13. The J = K
= 0 levels lie at 44 457.26共43兲, 44 547.04 共4261兲, 44 547.77 共4162兲, and 44 449.15 cm−1共63兲. Derived Coriolis
constants: a= 0.703
0, b= 0.7083; 共a兲2+共b兲2= 0.996. The parameter A⌬ raises the A constant of 4261
共and lowers that of 4162兲 relative to its value as interpolated between those for 43and 63. The only correlation
coefficients with magnitudes over 0.9 are 12共B+C兲 共63兲/1
2共B+C兲 共43兲 −0.948, A 共63兲/A 共43兲 −0.978, and 2Aa/A ⌬−0.934. T0共43兲 2295.098 ⫾ 0.102 A共63兲 13.000 ⫾ 0.050 T0共4261兲 2321.592 0.068 1 2共B+C兲 共63兲 1.0870 0.0028 T0共4162兲 2314.791 0.087 共B−C兲 共63兲 0.1406 0.0072 T0共63兲 2279.470 0.086 A共43兲 13.121 0.051 K4466共0兲 −51.019 0.009 12共B+C兲 共43兲 1.0685 0.0030 2Aa 18.363 0.009 共B−C兲 共43兲 0.0798 0.0102 Bb 0.8024 0.0029 K 4466,D 0.224 0.008 2Aa D −0.0228 0.00170 A⌬ −0.398 0.050 rms error= 0.0282 cm−1
054304-15 Bending overtones of the A˜ 1A
ustate of acetylene C2H2 J. Chem. Phys. 129, 054304共2008兲
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 140.113.38.11 On: Wed, 30 Apr 2014 06:16:32