DFTB Parameters for the Periodic Table: Part 1, Electronic Structure
Mohammad Wahiduzzaman,
†,‡Augusto F. Oliveira,
‡,†Pier Philipsen,
‡Lyuben Zhechkov,
†Erik van Lenthe,
‡Henryk A. Witek,
§,∥and Thomas Heine*
,††School of Engineering and Science, Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany
‡Scientific Computing & Modelling NV, Theoretical Chemistry, Vrije Universiteit, De Boelelaan 1083, 1081 HV Amsterdam, The
Netherlands
§Department of Applied Chemistry, National Chiao Tung University, Hsinchu 30010, Taiwan ∥Institute of Molecular Science, National Chiao Tung University, Hsinchu 30010, Taiwan
*
S Supporting InformationABSTRACT: A parametrization scheme for the electronic part of the density-functional based tight-binding (DFTB) method that covers the periodic table is presented. A semiautomatic parametrization scheme has been developed that uses Kohn−Sham energies and band structure curvatures of real and fictitious homoatomic crystal structures as reference data. A confinement potential is used to tighten the Kohn−Sham orbitals, which includes two free parameters that are used to optimize the performance of the method. The method is tested on more than 100 systems and shows excellent overall performance.
1. INTRODUCTION
Density-functional based tight binding (DFTB) is an approximation to density-functional theory (DFT).1,2 It has been proposed in the 1980s by Seifert and co-workers3for the Xα method and was transferred later to DFT.4,5 The method
made use of an optimized atomic orbital minimal basis set and approximated the effective Kohn−Sham (KS) potential in a rigorous two-center approximation. Thus, terms of three or more centers are not present in the KS matrix, allowing the a priori calculation and tabulation of all matrix elements, employing the so-called Slater−Koster technique.6 Because of the algorithmic simplicity of the method, it has been straightforward to implement it early on massively parallel high-performance computers;7,8 all computationally intensive operations of DFTB are available in standard linear algebra packages and thus highly optimized for virtually all computer architectures.
DFTB has been successfully employed in studying a series of applications, most notably carbon-based systems,5,9−20but also boron nitrides,21,22 silicon and silicon carbide,23−25 and other systems.26,27 Properties arising as expectation values of the DFTB-approximated KS orbitals are available in a straightfor-ward way, including NMR,13,15,28 STM images,29,30 and vibrational spectroscopy and related thermal properties.31,32
DFTB showed an excellent overall performance and was a significant contribution to computational materials science as it made electronic structure calculations possible for nanoscale systems at a very early time. However, its accuracy concerning structures and energies was not always competitive to highly parametrized force fields and self-consistent semiempirical models, in particular for bioorganic systems. Hence, Elstner and co-workers have extended DFTB in order to include a
self-consistent charge (SCC) correction, accounting for valence electron density redistribution due to interatomic interactions. This reformulated theory is based on the superposition of atom-centered electron densities, applying the two-particle approximation to densities rather than to potential terms. The total energy is derived as second-order expansion of the KS energy with respect to the electron density, which is constructed as a sum of pseudoatomic electron densities.33 This method, commonly referred to as SCC-DFTB, has been very successful, most notably in the field of computational biology, where quantitative accuracy of structural and energetic parameters has been achieved.8,34,35
The better description of charge fluctuations in the system allowed for the time-dependent SCC-DFTB formulation,36−38 as well as a more accurate description, of the vibrational properties (IR and Raman spectra).39−41
In the same way as DFT, (SCC-)DFTB lacks London dispersion interactions (LDI). These can be treated in a similar fashion as in DFT by adding an empirical two-body force- field-like term with r−6long-range asymptotic and appropriate short-range behaviors.42,43
Other major refinements of DFTB are spin-polarized DFTB,44 LDA+U-like Hamiltonians for strongly correlated systems,45,46and order DFTB (DFTB3), where the third-order contributions of the electronicfluctuations, in the form of induced atomic Mulliken charges, are taken into account.47,48
The combination of SCC and LDI made the results of DFTB much more realistic, and applications in thefield of inorganic nanomaterials,49−55 metal−organic frameworks (MOFs),56−58
Received: June 11, 2013
Published: August 5, 2013
covalent organic frameworks (COFs),59−63 carbon-based systems,64−66and biomolecules67−71have been reported.
There have been several recent reviews on DFTB to which we refer the interested reader.72−74 Despite all the progress achieved with the help of the DFTB method in all its variations and the availability of the method in most of today’s computer codes, it is important to note that a straightforward general-ization of the method, offering parameters throughout the periodic table, has not been successful to date. There have been various attempts to automatically generate parts of the DFTB parameters,75−77 but we note that the parameters that are actually used in computer simulations are typically hand-tuned and, unfortunately, in many cases, limited to a small set of elements.
In this and a forthcoming work, we will present a scheme that produces optimized parameters throughout the periodic table. This work concentrates on the generation of electronic parameters, that is, those that are needed to generate the KS and overlap matrix elements and thus to generate band structures and all related properties. In a forthcoming contribution, we will provide the remaining terms that are needed for the total energy (also known as repulsive potentials), thus completing a DFTB parameter set for the periodic table.
This article is organized as follows: First, we review the relevant part of the DFTB theory. Then, we motivate and rationalize our computational approach and present the automated parametrization scheme. After reporting our construction recipe for the calculation of the electronic parameters throughout the periodic table, we discuss the transferability of the approach and finally compare selected band structures with DFT reference calculations. This article is accompanied with a comprehensive Supporting Information, including the band structures of all materials selected for training and validation calculations.
2. REVIEW OF DFTB
The Kohn−Sham equations can be formulated as
ψ ψ ε ψ
̂ = ̂ + =
F i (T Veff) i i i (1)
with the effective potential Veff=Veff[n] being a functional of the electronic density n(r). In DFTB, Veff is expressed by a series
including only atom-centered terms. Two philosophies are found in the literature. In the potential superposition, introduced by Seifert and co-workers,3−5 Veff is written as series of atom-centered potential contributions VAof the atoms A at positions RA: Veff = ∑AVA(r − R
A). In the density superposition,
introduced by Elstner et al.,33the electronic density is expanded in a similar way, superposing atom-centered densities nA: Veff[n] = Veff [∑AnA(r − RA)]. Both expansions are formally
exact when pursued to the infinite order, and one could write the atomic contributions, for example, in a series of spherical harmonics centered at the respective positions of the nuclei of the atoms.
In the DFTB approximation, the individual elements of these series are replaced by atomic trial potentials or trial densities, respectively. Those contributions are constructed by calcu-lations of spherical pseudoatoms that are subject to a confinement potential Vconf· Vconf is acting in two roles. It reduces the spatial extension of the atomic orbitals and, consequently, atomic densities and potentials, and it acts as a tunable parameter. The trial potentials V0Aor densities n0A are
obtained by self-consistently solving the KS equations of a closed-shell pseudoatom of type A, with the electron density constrained to spherical symmetry
These atomic calculations also yield the confined atomic orbitalsϕvA. The linear combination of atomic orbitals (LCAO)
approach is now used to expand the KS orbitals
∑
ψ = ϕ − ν ν ν ν c r r R ( ) ( ) i i A A( ) (3)where A(ν) denotes center A to which atomic orbital ϕvA
belongs. DFTB approximates this series by restricting it to include only the atomic valence orbitals plus, in some cases, the first unoccupied shell. As basis functions, the method uses the solutions of the pseudoatomic calculations ϕvA of eq 2. Thus,
the overlap matrix S is defined as Sμν=⟨ϕμ|ϕν⟩. For the
zero-order KS matrix F0, two further approximations are necessary.
First, we apply the two-center approximation, that is, only those potential (or density) contributions that are centered at the same positions as the basis functions are considered
ϕ ϕ ϕ ϕ ϕ ϕ = ⟨ | ̂| ⟩ = ⟨ | ̂ + | ⟩ ≈ ⟨ | ̂ + | ⟩ μν μ ν μ ν μ ν μ ν F0 F T Veff T VeffA( ) ( )B (4) with = − + − VeffAB V0A(r RA) V0B(r RB) (5)
for the potential superposition, and
= − + −
VeffAB Veff[n0A(r RA) n0B(r RB)] (6) for the density superposition. Second, for the diagonal elements we use Fμμ0 = <ϕ
μ|F̂|ϕμ> = εμfree, with εμfree referring to the KS
energy of orbitalϕμof the free spherical atom, that is, with Vconf
= 0. This latter choice ensures correct dissociation energies.5 Because of the orthogonality of the atomic orbitals, all other one-center off-diagonal elements are zero, and we can summarize the zero-order DFTB KS matrix as
ε μ ν ϕ ϕ μ ν = = ⟨ | ̂ + | ⟩ ∈ ∈ ≠ μν μ μ ν ⎧ ⎨ ⎪⎪ ⎩ ⎪ ⎪ F T V A B A B , , { }, { }, 0, otherwise AB 0 free eff (7)
The orbital term of the total energy of the system is conveniently written as tr(P·F0), where the elements of the
density matrix P are defined as
∑
= μν μ ν P f c c i i i i (8)with orbital occupations fi. The SCC correction33accounts for
the second order terms with respect to the chargefluctuations in the system. Those modify the KS matrix to
∑
γ γ = + + μν μν μν ξ ξμ ξν ξ F F 1S q 2 ( ) 0 (9)where γ is an effective matrix including the Coulomb and exchange correlation interactions (for details see ref 33), and qξ
denote charges, typically Mulliken charges, of the atoms. If applied to atoms with charges qξ, the SCC correction shifts the
KS matrix elements up (for electron gain) or down (for electron loss) at the respective centers; hence, it balances the charge flow through the system depending on the atomic hardnesses. The SCC correction can be formulated for shells, being advantageous, e.g., for transition metals with notably different hardnesses for s, p shells on one hand, and the d shell on the other hand. In this case, the indexξ denotes the shell index, and qξ = nξ0 − nξ is the electric charge of the shell, computed as the shell population difference with respect to the free atom.
The DFTB total energy results from the orbital term corrected by the second-order term and a so-called repulsive energy Erep, which accounts for the internuclear repulsion and the double counting terms, and includes ad hoc corrections for the approximations made
The parametrization of the repulsive energy will be subject of a forthcoming work. Here, we concentrate on a high-quality parametrization of the electronic parameters, yielding the KS orbitals, and hence orbital energies and all other quantities that can be expressed as expectation values of the KS orbitals.
As commonly done in the DFTB community, we will employ the confinement potential as parameter to tune the DFTB electronic structure. There are various ways to describe the confinement potential. Traditionally, it has been defined as harmonic potential Vconf= (r/r0)2, with r
0being an empirical
parameter, typically having a value of about twice the covalent radius of the atom.78,79In this work, we generalize this potential to = σ ⎛ ⎝ ⎜ ⎞ ⎠ ⎟ V r r conf 0 (11)
with r0andσ being free, positive, real parameters that are used to optimize the DFTB performance to yield high-quality KS orbitals ψi. A DFTB performance comparable to the one presented in this work has been achieved, with a similar parametrization strategy using a Woods−Saxon confinement potential together with a fully relativistic atomic program, by Nishimura in the Irle and Witek groups.80,81
3. PARAMETRIZATION DETAILS
3.1. General Concept. The format of present-day DFTB parameters and the corresponding literature generate the impression that DFTB involves a diatomic parametrization, thus requiring the optimization of N(N + 1)/2 parameter sets to cover a number N of chemical elements. To put it into perspective, 5050 parameter sets would then be required to cover 100 elements. It is rather obvious that this amount of parameter sets is difficult to generate and to maintain, and this is certainly the principal reason why a DFTB for the periodic table has not been available so far. In principle, DFTB requires only a set of atomic parameters, that is, each parameter set depends on one atom type only. This is particularly true for the electronic part of DFTB, given in the so-called Slater−Koster tables,6 and this may even be transferred to the so-called repulsive potential, as discussed by Mirtschink and Seifert.82The adoption of a single atom-type strategy reduces the number of
parameter sets to be optimized to N, that is, one parameter set per chemical element, a number that is much easier to control. We will employ this strategy and describe it in detail in this section.
First, we discuss our selection of reference structures and methods and define the computational details for the generation of reference data. Then, we give the numerical details of the quantum-mechanical calculations leading to the DFTB pseudoatoms and consequently DFTB parameters and motivate their choice. We report the semiautomatic para-metrization procedure and the scoring functions that are used to optimize the DFTB parameters before we discuss the transferability to heteroatomic systems and measures to overcome shortcomings.
3.2. Reference Structures and Methods. DFTB parameters are supposed to be transferable between different systems. Therefore, it is important to include interactions between atoms in different bonding situations during the parametrization. As we want to have a simplified para-metrization strategy, we prefer homoatomic systems. A straightforward way to achieve this is to concentrate on solids and to include crystals of different coordination numbers, thus probing different bonding environments. An attractive para-metrization target will be the band structure, as it simultaneously samples the effect of a large number of bond distances on elements of the overlap and KS matrices. The bands are associated with the individual KS energies and thus the KS orbitals. The curvature of the bands gives important data for applications such as effective masses, and thus electron and hole mobilities, and is the first term needed for the calculation of vibrational frequencies (phonons).
We have employed an extensive database of crystal structures extracted from the work of Nishimura.80 This database comprises experimental lattices as well as lattices optimized with a PBE-PAW method. For the sake of better transferability of our parameters, we wanted to use as many reference systems as possible. We use three or more crystal systems for most of the atom types. The crystal systems used as reference in the parametrization of each element are discriminated in the Supporting Information (SI).
The DFT reference is always the all electron calculation with a numerical TZP basis set (available in ADF/BAND83,84), the generalized gradient approximation (GGA) functional sug-gested by Perdew, Burke, and Ernzerhof in 1996 (PBE),85and the scalar-relativistic correction (ZORA).86 The quadratic scheme proposed by Wiesenekker and Baerends87 with high precision settings has been used for numerical integration over the Brillouin zone.
The DFTB calculations of band structures have been carried out using the ADF/DFTB88program.
3.3. Generation of DFTB Pseudoatoms and DFTB Parameters. All necessary information for defining the DFTB parameters is obtained by solving the KS equations for the pseudoatoms of spherical symmetry as given in eq 7. For consistency, we employ the same density-functional as for the reference calculations (PBE) and also include the scalar-relativistic corrections (ZORA) for all elements.
The choice of basis functions was less straightforward. Intensive tests have shown that it is practically impossible to describe the pseudoatoms being subjected to different confine-ment potentials Vconf using standard analytical Slater-type or
Gaussian-type basis sets, even if they have excellent quality within DFT, and especially for confinement potentials with
Table 1. Electronic Configurations and Confinement Potentials for the Elements Considered in the Present Worka
element valence shell r0 σ εd εp εs Ud Up Us
H 1s1 1.6 2.2 − − −0.238603 − − 0.419731 He 1s2 1.4 11.4 − − −0.579318 − − 0.742961 Li 2s12p0 5.0 8.2 − −0.040054 −0.105624 − 0.131681 0.174131 Be 2s22p0 3.4 13.2 − −0.074172 −0.206152 − 0.224651 0.270796 B 2s22p1 3.0 10.4 − −0.132547 −0.347026 − 0.296157 0.333879 C 2s22p2 3.2 8.2 − −0.194236 −0.505337 − 0.364696 0.399218 N 2s22p3 3.4 13.4 − −0.260544 −0.682915 − 0.430903 0.464356 O 2s22p4 3.1 12.4 − −0.331865 −0.880592 − 0.495405 0.528922 F 2s22p5 2.7 10.6 − −0.408337 −1.098828 − 0.558631 0.592918 Ne 2s22p6 3.2 15.4 − −0.490009 −1.337930 − 0.620878 0.656414 Na 3s13p0 5.9 12.6 − −0.027320 −0.100836 − 0.087777 0.165505 Mg 3s23p0 5.0 6.2 − −0.048877 −0.172918 − 0.150727 0.224983 Al 3s23p13d0 5.9 12.4 0.116761 −0.099666 −0.284903 0.186573 0.203216 0.261285 Si 3s23p23d0 4.4 12.8 0.113134 −0.149976 −0.397349 0.196667 0.247841 0.300005 P 3s23p33d0 4.0 9.6 0.121111 −0.202363 −0.513346 0.206304 0.289262 0.338175 S 3s23p43d0 3.9 4.6 0.134677 −0.257553 −0.634144 0.212922 0.328724 0.375610 Cl 3s23p53d0 3.8 9.0 0.150683 −0.315848 −0.760399 0.214242 0.366885 0.412418 Ar 3s23p63d0 4.5 15.2 0.167583 −0.377389 −0.892514 0.207908 0.404106 0.448703 K 3d04s14p0 6.5 15.8 0.030121 −0.029573 −0.085219 0.171297 0.081938 0.136368 Ca 3d04s24p0 4.9 13.6 −0.070887 −0.051543 −0.138404 0.299447 0.128252 0.177196 Sc 3d14s24p0 5.1 13.6 −0.118911 −0.053913 −0.153708 0.322610 0.137969 0.189558 Ti 3d24s24p0 4.2 12.0 −0.156603 −0.053877 −0.164133 0.351019 0.144515 0.201341 V 3d34s24p0 4.3 13.0 −0.189894 −0.053055 −0.172774 0.376535 0.149029 0.211913 Cr 3d54s14p0 4.7 3.6 −0.107113 −0.036319 −0.147221 0.312190 0.123012 0.200284 Mn 3d54s24p0 3.6 11.6 −0.248949 −0.050354 −0.187649 0.422038 0.155087 0.230740 Fe 3d64s24p0 3.7 11.2 −0.275927 −0.048699 −0.194440 0.442914 0.156593 0.239398 Co 3d74s24p0 3.3 11.0 −0.301635 −0.046909 −0.200975 0.462884 0.157219 0.247710 Ni 3d94s14p0 3.7 2.2 −0.170792 −0.027659 −0.165046 0.401436 0.106180 0.235429 Cu 3d104s14p0 5.2 2.2 −0.185263 −0.025621 −0.169347 0.420670 0.097312 0.243169 Zn 3d104s24p0 4.6 2.2 −0.372826 −0.040997 −0.219658 0.518772 0.153852 0.271212 Ga 4s24p14d0 5.9 8.8 0.043096 −0.094773 −0.328789 0.051561 0.205025 0.279898 Ge 4s24p24d0 4.5 13.4 0.062123 −0.143136 −0.431044 0.101337 0.240251 0.304342 As 4s24p34d0 4.4 5.6 0.078654 −0.190887 −0.532564 0.127856 0.271613 0.330013 Se 4s24p44d0 4.5 3.8 0.104896 −0.239256 −0.635202 0.165858 0.300507 0.355433 Br 4s24p54d0 4.3 6.4 0.126121 −0.288792 −0.739820 0.189059 0.327745 0.380376 Kr 4s24p64d0 4.8 15.6 0.140945 −0.339778 −0.846921 0.200972 0.353804 0.404852 Rb 4d05s15p0 9.1 16.8 0.030672 −0.027523 −0.081999 0.180808 0.073660 0.130512 Sr 4d05s25p0 6.9 14.8 −0.041363 −0.047197 −0.129570 0.234583 0.115222 0.164724 Y 4d15s25p0 5.7 13.6 −0.092562 −0.052925 −0.150723 0.239393 0.127903 0.176814 Zr 4d25s25p0 5.2 14.0 −0.132380 −0.053976 −0.163093 0.269067 0.136205 0.189428 Nb 4d35s25p0 5.2 15.0 −0.170468 −0.053629 −0.172061 0.294607 0.141661 0.200280 Mo 4d45s25p0 4.3 11.6 −0.207857 −0.052675 −0.179215 0.317562 0.145599 0.209759 Tc 4d55s25p0 4.1 12.0 −0.244973 −0.051408 −0.185260 0.338742 0.148561 0.218221 Ru 4d75s15p0 4.1 3.8 −0.191289 −0.033507 −0.155713 0.329726 0.117901 0.212289 Rh 4d85s15p0 4.0 3.4 −0.218418 −0.031248 −0.157939 0.350167 0.113146 0.219321 Pd 4d95s15p0 4.4 2.8 −0.245882 −0.029100 −0.159936 0.369605 0.107666 0.225725 Ag 4d105s15p0 6.5 2.0 −0.273681 −0.027061 −0.161777 0.388238 0.099994 0.231628 Cd 4d105s25p0 5.4 2.0 −0.431379 −0.043481 −0.207892 0.430023 0.150506 0.251776 In 5s25p15d0 4.8 13.2 0.135383 −0.092539 −0.301650 0.156519 0.189913 0.257192 Sn 5s25p25d0 4.7 13.4 0.125834 −0.135732 −0.387547 0.171708 0.217398 0.275163 Sb 5s25p35d0 5.2 3.0 0.118556 −0.177383 −0.471377 0.184848 0.241589 0.294185 Te 5s25p45d0 5.2 3.0 0.114419 −0.218721 −0.555062 0.195946 0.263623 0.313028 I 5s25p55d0 6.2 2.0 0.112860 −0.260330 −0.639523 0.206534 0.284168 0.331460 Xe 5s25p65d0 5.2 16.2 0.111715 −0.302522 −0.725297 0.211949 0.303641 0.349484 Cs 5d06s16p0 10.6 13.6 −0.007997 −0.027142 −0.076658 0.159261 0.069450 0.120590 Ba 5d06s26p0 7.7 12.0 −0.074037 −0.045680 −0.118676 0.199559 0.105176 0.149382 La 5d16s26p0 7.4 8.6 −0.113716 −0.049659 −0.135171 0.220941 0.115479 0.160718 Lu 5d16s26p0 5.9 16.4 −0.064434 −0.049388 −0.171078 0.220882 0.126480 0.187365 Hf 5d26s26p0 5.2 14.8 −0.098991 −0.051266 −0.187557 0.249246 0.135605 0.200526 Ta 5d36s26p0 4.8 13.8 −0.132163 −0.051078 −0.199813 0.273105 0.141193 0.212539 W 5d46s26p0 4.2 8.6 −0.164874 −0.049978 −0.209733 0.294154 0.144425 0.223288
high exponents σ, which are very steep. This holds for the orbital basis set and alsoif employedfor the fitting functions to describe the electron density. To avoid numerical problems we employ here numerical atomic orbitals (NAOs), which are exact eigenfunctions of the DFT Hamiltonian for a spherical atom, obtained with the Herman−Skillman proce-dure.89
Various strategies have been suggested in the literature to optimize Vconf, motivated by different reasons,90−92 and alternative analytical forms of Vconf have been suggested.93,94 We have chosen an analytical expression for the confinement potential Vconf= (r/r0)σ, with free parameters r0 and σ. This
choice is numerically simple and can be taken as generalization of Vconfas it is used by most groups today, who take the same
parameter r0but integer values (2 or 4) forσ. Thus, this choice allows the DFTB community to easily adapt existing para-metrization tools.
With these details, we calculate the pseudoatomic potential V0, density n0, and atomic orbitals ϕμ, depending on the
parameters r0andσ. They are used to compute the elements of overlap and KS matrices using eqs 7 and 9 given in the previous section. It is convenient and common to precalculate the overlap and KS matrix elements between two atoms as function of the interatomic distance and to store them in a parameter file, the so-called Slater−Koster table.6
For each element, we carry out a further atomic calculation with Vconf= 0, yielding the orbital energiesεμof the free atoms. The Hubbard parameters Ul, required to obtain the SCC matrix γ,33 are calculated for
each atomic shell as the second derivative of the atomic energy with respect to the electronic occupation of the l orbital in the unconfined atom.
At this point, it is important to discuss what orbitals are to be included in the DFTB approach. DFTB is a nonorthogonal tight-binding scheme, so some attention must be given to the related orthogonality issues. Within the potential superposition (eq 5), one can interpret the orthogonalization terms as pseudopotential contributions. Those have been identified to be small and can be neglected.73Therefore, it is reasonable to disregard the core orbitals in DFTB. In DFT, it is necessary to employ a large series of unoccupied orbitals into the variational space in order to be able to obtain high-quality KS orbitals, energies, and properties. In DFTB, however, this variational space is expected to be represented in the pseudoatomic orbitals, which have been optimized during the parametrization, and that a minimum valence basis is sufficient. In cases where
explicit polarization is important, one shell of polarization functions has been included.90,91 The explicit choice for each element is given in Table 1. Typical choices are, for example, to include the next p shell in alkali metals or the unoccupied d shell for elements such as Si and P.
Before we turn to the optimization procedure of the DFTB parameters, we would like to motivate the importance of the choice of the numerical atomic orbitals. Figure 1 gives the
electronic band structure of Al(fcc), calculated with our optimized DFTB confinement (r0= 5.9 bohr and σ = 12.4),
in comparison with a DFT benchmark calculation. The use of analytical basis functions, even the enormous QZ4P basis set, for calculating the DFTB pseudoatom has been found insufficient for reproducing the DFT reference bands in the valence region (below the Fermi levelεF). As shown in Figure 1, the deviations are especially pronounced in the s band. Only the numerical atomic orbitals match our reference calculations closely. Hence, to reproduce the KS matrix elements with Slater-type orbitals, special basis sets would need to be constructed to handle steep confinement potentials. We have not pursued this route, as the NAOs already represent the basis set limit for this problem.
3.4. Optimization of DFTB Parameters in a Semi-automated Procedure. It remains to determine the free parameters r0andσ in such way that they lead to high-quality and transferable results. This means that the DFTB perform-Table 1. continued
element valence shell r0 σ εd εp εs Ud Up Us
Re 5d56s26p0 4.2 13.0 −0.197477 −0.048416 −0.218183 0.313288 0.146247 0.233028 Os 5d66s26p0 4.0 8.0 −0.230140 −0.046602 −0.225640 0.331031 0.146335 0.241981 Ir 5d76s26p0 3.9 12.6 −0.262953 −0.044644 −0.232400 0.347715 0.145121 0.250317 Pt 5d86s26p0 3.8 12.8 −0.295967 −0.042604 −0.238659 0.363569 0.143184 0.258165 Au 5d106s16p0 4.8 2.0 −0.252966 −0.028258 −0.211421 0.361156 0.090767 0.255962 Hg 5d106s26p0 6.7 2.0 −0.362705 −0.038408 −0.250189 0.393392 0.134398 0.272767 Tl 6s26p16d0 7.3 2.2 0.081292 −0.087069 −0.350442 0.119520 0.185496 0.267448 Pb 6s26p26d0 5.7 3.0 0.072602 −0.128479 −0.442037 0.128603 0.209811 0.280804 Bi 6s26p36d0 5.8 2.6 0.073863 −0.167900 −0.531518 0.142210 0.231243 0.296301 Po 6s26p46d0 5.5 2.2 0.081795 −0.206503 −0.620946 0.158136 0.250546 0.311976 Ra 6d07s27p0 7.0 14.0 −0.047857 −0.037077 −0.120543 0.167752 0.093584 0.151368 Th 6d27s27p0 6.2 4.4 −0.113604 −0.045825 −0.161992 0.211980 0.114896 0.174221
aConfinement radii r
0 are given in bohr, whereas orbital energiesεland Hubbard parameters Ulare given in hartree. When applying the SCC
correction per atom rather than per atomic shell, we use the Hubbard parameter of the occupied shell with highest l number.
Figure 1.Electronic band structures of the Al(fcc) crystal obtained with DFT (PBE/TZP) and DFTB, employing QZ4P, TZP, and numerical atomic orbitals (NAOs) in the calculation of the pseudoatoms. All further numerical parameters are identical in the three DFTB calculations.
ance shall be similarly good for the different crystal systems that have been chosen as reference systems for each element. The quality assessment is associated to the band structure differences to the reference values and thus contains a significant amount of information.
In principle, a careful eye observation of the band structures or projected density of states would be the best way to judge the quality of the DFTB parameters. However, this is most inconvenient in terms of time and manual effort and introduces a subjective component that is difficult to control. The main challenge of this work is tofind an optimization scheme that is as much automated as possible. As our target quantities are band structures, we defined two scoring functions η(ε) and η(κ). η(ε) evaluates the quality of the band structure energies. It is defined as
∑
η ε = |ε −ε | N ( ) 1 i N i i k k k , , DFT , DFTB (12)where the sum runs over the valence bands, indicated with i and a set of k points from the selected path through the Brillouin zone, yielding a total of N elements. A similar expression can be written for the curvature of the bands
∑
η κ = |κ −κ | N ( ) 1 . i N i i k k k , , DFT , DFTB (13)For some insulating systems, for example, Ge(dia), the lowest conduction band is also included in the scoring functions. For details on the number of bands used in the scoring functions for each reference crystal, we refer the reader to Table S1.1 on the SI.
The parametrization of the individual atom types moves along the following procedure:
1. We generate parameter sets by varying the parameters r0
andσ in a brute-force scheme. In detail, we change r0in
steps of 0.1 rcov, starting from the covalent radius rcovof the element up to 10 rcov. The values of rcov are taken
from the work of Cordero et al.95 The exponent σ is varied from 2 to 17 in steps of 0.2. Slater−Koster tables are prepared and saved for analysis.
2. For each atom type we have selected several crystal systems. For each of the crystal systems, we compute and score the band structures following eqs 12 and 13. We first rank the best parameters in terms of the band energies (η(ε)). It is interesting to note that a rather
large variation of parameters produces excellent band structures for many systems. We pick a set of the best performing sets, typically about 50.
3. We re-rank the best performing sets in terms of band curvatures (η(κ)).
4. Finally, the sets of ranked parameters of different crystal systems are progressively compared until the closest intersection point is found (i.e., those parameters that are performing well in all crystal systems with the lowest possible energy and curvature deviation). This step is comparable to drawing a Venn diagram, where only one single element is common between all sets. If there are no common parameters found, we extend the best-performing set in point 2 and start over.
This procedure yields values for r0andσ that produce
high-quality band energies and band curvatures for different crystal systems.
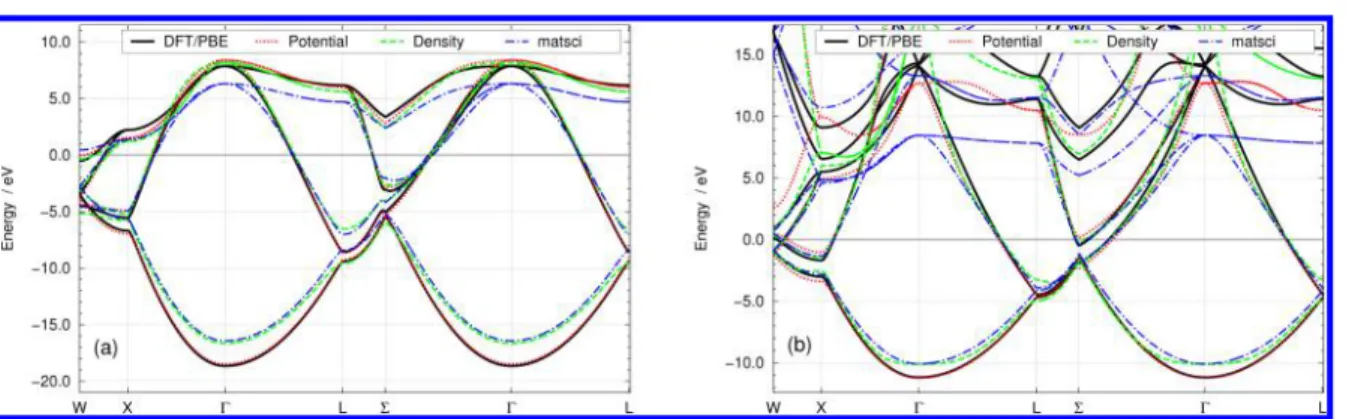
We have carried out this procedure for DFTB formulated in the density superposition as well as for the potential superposition scheme (eqs 5 and 6). It turned out that the potential superposition scheme showed, overall, superior results in terms of band energies and curvatures, as exemplified in Figure 2. We will therefore consistently apply this scheme to generate all parameters that will be presented below.
In addition to the procedure described above, different electronic configurations have been tested for transition metal elements. Although the ground state configuration should be used to ensure correct dissociation energies, as discussed in Section 2, alternative configurations, in which an electron is reassigned from the s to the d orbital or vice versa, may improve the description of electronic band structures. Hence, we have chosen to use the electronic configuration that resulted in the best band structure around the Fermi energy.
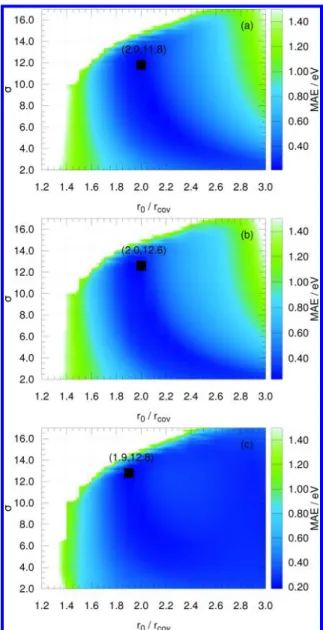
3.5. Securing Transferability. Already during the para-metrization procedure of homoatomic materials, we have generated sufficient data to discuss the transferability of DFTB. If a large number of parameter sets scores well in the description of each crystal type selected per element, a good transferability is expected. On the contrary, if very few parameter sets score well, transferability may turn out to be problematic and compromises would have to be made when selecting the best parameter set for a certain element. In Figures 3 and 4 we show an exceptionally good (phosphorus) and an exceptionally bad (carbon) example. It is surprising and interesting to note that carbon is one of the least transferable
Figure 2.Electronic band structures calculated for fcc crystals of (a) carbon and (b) aluminum, using DFTB parameters optimized with the potential superposition (potential) and density superposition (density) approaches. The bands have been shifted according to the Fermi energyεF. In the
present work, the potential superposition has generally performed better than the density superposition in terms of band energies and band curvatures. For reference, the band structures calculated with the matsci-0-396parameter set are also shown.
elements that we have encountered because a significant portion of the success of DFTB is related to its excellent performance for carbon-based systems. The original choice of Porezag et al.5for Vconf, with r0/rcov≈ 1.8 and σ = 2, is clearly outside the best range for carbon systems (Figure 4). This finding indicates that it is important to compromise between different benchmark systems. It further shows how inaccuracies in the electronic parameters may be compensated by the additional terms of the total energy (eq 10).
Because only homoatomic crystals have been used as references for our DFTB parametrization, the performance of our parameters with heteroatomic systems would be a true evidence of transferability. In the majority of the cases, we have obtained very good qualitative agreement between DFTB and DFT calculations of band energies and band curvatures of binary crystals. For some systems, however, poor results have been achieved. More specifically, systems containing elements
that do not naturally occur in solid phase, i.e., N, O, F, Cl, and Br. In order to overcome this problem, we have reoptimized the parameters of these elements using a series of binary compounds as reference. Upon reoptimization, significant improvement has been achieved (Figure 5). Table S1.1 of the Supporting Information shows a detailed list of the crystal systems used as reference for the parametrization of each element, as well as thefinal band energy and curvature scores for all systems considered in this work.
4. RESULTS AND DISCUSSION
Table 1 gives the selected DFTB valence shells, the optimized parameters r0andσ to express Vconf, the orbital energiesε of the unconfined pseudoatoms, and the Hubbard parameters U for all elements of the periodic table from H to Th, with completelyfilled f levels. With these parameters it is possible to compute all elements of the overlap and Hamiltonian matrices for DFTB calculations. It should be reminded that it is important to use an extremely extended basis set, preferentially numerical atomic orbitals.
For several elements, we have obtained a value of 2.0 forσ, which is the lowest limit in our optimization range. Hence, in these cases, we have reoptimized the confinement potential using 1.0 as the lower limit. Nevertheless, the results have not changed, confirming that 2.0 is truly the lower limit of σ.
It is interesting to notice that opposite to previous assumptions the optimal value of r0 is not related to the covalent radii by a simple constant. In fact, the values of both r0 andσ considerably differ from element to element. In Figure 6 we compare the bond lengths (De) in the reference crystals to the optimal values of r0. A trend can be observed between this values. Furthermore, it can be observed that the covalent radii do not reflect the bond lengths of the crystal systems.
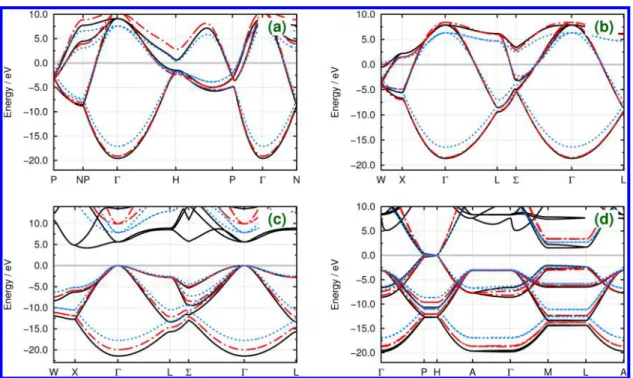
The corresponding band structures of the benchmark systems are compared to our DFT reference calculations and to results obtained using the matsci-0-3 parameter set50,51,96 available at DFTB.org.97 In the matsci-0-3 parameter set the confinement potential has been tuned manually, and the pseudoatomic calculations have been carried out with convenient Slater-type basis functions and the local density approximation.98 Not surprisingly, our new set of DFTB parameters, entitled QUASINANO2013.1 (quantum-mechan-ical simulations for the nanoscale, labeled according to the title of the funding project), performs very well for the vast majority of all homoatomic systems in the periodic table. Three examples have been selected for discussion here: C, Si, and Au. Despite the transferability issue discussed before, very good qualitative results have been obtained for all carbon systems (Figure 7), although the band gap in C(dia) has been compromised. In the case of Si (Figure 8), our parameters outperform the matsci-0-3 set in both the valence and conduction bands close to the Fermi level. The matsci-0-3 set does not include Au parameters; however, a very good agreement between our parameters and DFT can been observed in Figure 9. It is important to note that the conduction bands of DFTB will always differ from DFT. There are bands missing due to the choice of minimum basis set in DFTB, and the higher-energy conduction bands have not been included in the calculation of the scoring functions. This deviation from DFT is not relevant, as the high-energy part of the band structure does not influence the electronic ground state of the system.
Figure 3.Mean absolute error (MAE) of the electronic band energies for phosphorus in (a) bcc, (b) fcc, and (c) simple cubic arrangements, plotted as function of the confinement radius (r0) and exponent (σ).
The optimal performance regions (blue) within all three systems overlap nicely, meaning an excellent transferability of the DFTB parameters can be obtained for phosphorus. The best points are marked on the plots.
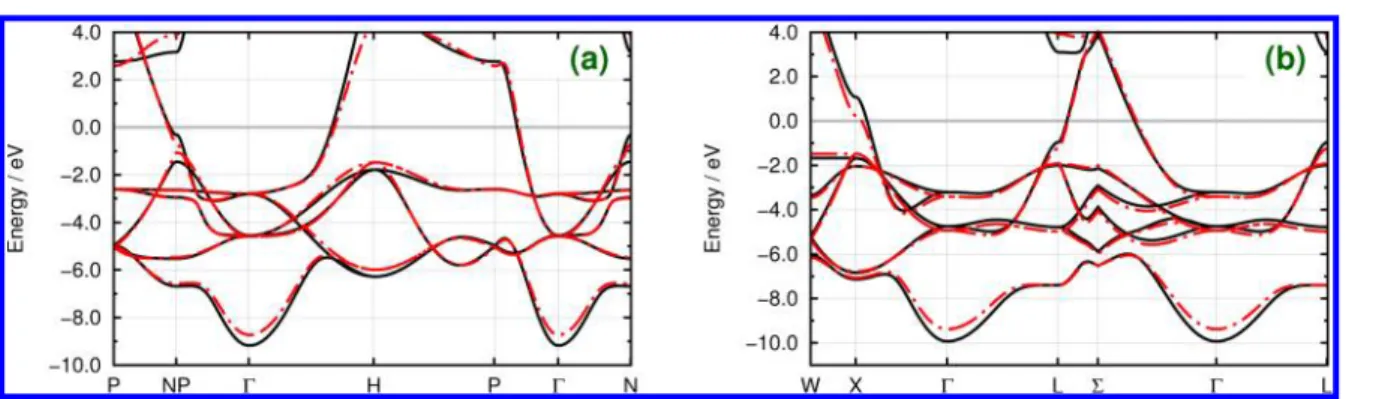
We have further tested our DFTB parameters for binary compounds. Four crystals that have not been used as reference in the parametrization have been chosen, namely, ScC (rock salt type) and TiC (rock salt type) (Figure 10), GeSi (zinc blende type), and SnSi (zinc blende type) (Figure 11). All these examples show that DFTB bands obtained with our parameters are in good agreement with DFT reference bands. With the exception of a few transition metal compounds, we observed results of similar quality for many other systems (SI), which confirms the transferability of the parameters.
Another test worth mentioning refers to the mechanical deformation of MoS2 monolayers. The change in electronic conductivity associated to mechanical strain in transition-metal dichalcogenides has been investigated in our group,99using full DFT calculations. We have repeated the band structure calculations (results not shown) of the MoS2 monolayer subject to compression of up to 15% and have observed the same trends with respect to the position and value of the
Figure 4.Mean absolute error (MAE) of the electronic band energies for carbon crystals in (a) bcc, (b) fcc, (c) diamond, and (d) graphite arrangements, plotted as function of the confinement radius (r0) and exponent (σ). The regions with optimal performance (blue) of diamond do not
overlap with other systems, indicating limited transferability of the DFTB parameters for carbon. The best points are marked on the plots.
Figure 5.Electronic band structures of (a) ZnO (zinc blende type) and (b) LiBr (rock salt type) after reoptimization of O and Br parameters by including these crystal structures in the set of reference systems. DFT reference bands are plotted in solid black lines, whereas the bands obtained with our DFTB parameters are shown in dashed−dotted red lines. The band energies have been shifted according to the Fermi energy εF.
Figure 6.Equilibrium distances (De) and optimized values of r0, both
electronic band gap. On the basis of these results, our DFTB parameters have been applied in a recent investigation of the electromechanical properties of MoS2nanotubes.100
Many more examples can be found in the Supporting Information. Our parameters are already applicable for band structures, electron mobilities, and quantum conductance calculations. The applicability of the parameter set is currently being extended by inclusion of the repulsion energy term Erep.
5. CONCLUSION
We have presented a semiautomated parametrization scheme for the density-functional based tight-binding method that yields parameters throughout the periodic table. Thus, electronic structure calculations of solids and finite systems are now possible for practically any material using the DFTB approximation.
Figure 7.Electronic band structures of carbon crystals in (a) bcc, (b) fcc, (c) diamond, and (d) graphite arrangements. Solid black lines show the DFT reference bands, whereas dashed−dotted red lines show the results with our DFTB parameters and dotted blue lines correspond to DFTB calculations using the matsci-0-396parameter set. Band energies have been shifted according to the Fermi energyεF.
Figure 8.Electronic band structures of silicon crystals in (a) bcc, (b) fcc, (c) simple cubic, and (d) diamond arrangements. Solid black lines show the DFT reference bands, whereas dashed−dotted red lines show the results with our DFTB parameters, and dotted blue lines correspond to DFTB calculations using the matsci-0-396parameter set. Band energies have been shifted according to the Fermi energyεF.
The parametrization scheme takes reference calculations, obtained with a standard density-functional (PBE), as bench-marks and analyzes the electronic band energies and curvatures for selecting the best possible DFTB parameters.
We have presented the optimized parameters for our first global parameter set, QUASINANO2013.1, and validated it for a set of test systems. Within the accuracy range of DFTB, a very good performance has been observed for the new QUASINA-NO2013.1 parameters, in most cases outperforming previous parameters that have been regularly employed. We note that a proper choice of basis functions is essential for obtaining correct DFTB parameters.
It should be noted that the calculation of other important properties, such as binding energies, reaction paths, and bond angles, requires the parametrization of repulsive potentials, which will be the focus of a forthcoming contribution. Once this parametrization is complete, a more detailed analysis of the performance and transferability of our DFTB parameters and
further comparison to other parameters already available will be possible.
The article is accompanied with extensive Supporting Information, which brings detailed results of benchmark and validation calculations for more than 100 systems. The QUASINANO2013.1 parameter set can be produced with the current version of the ADF software.
■
ASSOCIATED CONTENT*
S Supporting InformationDetailed information on the optimized DFTB parameters, electronic band structure plots, and geometrical data of all crystal structures considered in this work. This material is available free of charge via the Internet at http://pubs.acs.org.
■
AUTHOR INFORMATIONCorresponding Author
*E-mail: t.heine@jacobs-university.de.
Figure 9.Electronic band structures of gold crystals in (a) bcc and (b) fcc arrangements. Solid black lines show the DFT reference bands, whereas dashed−dotted red lines show the results with our DFTB parameters. Band energies have been shifted according to the Fermi energy εF.
Figure 10.Electronic band structures of (a) ScC (rock salt type) and (b) TiC (rock salt type). Solid black lines show the DFT reference bands, whereas dashed−dotted red lines show the results with our DFTB parameters. Band energies have been shifted according to the Fermi energy εF.
Figure 11.Electronic band structures of (a) GeSi (zinc blende type) and (b) SnSi (zinc blende type). Solid black lines show the DFT reference bands, whereas dashed−dotted red lines show the results with our DFTB parameters. Band energies have been shifted according to the Fermi energy εF.
Notes
The authors declare no competingfinancial interest.
■
ACKNOWLEDGMENTSThe authors thank the European Commission for financial support for the Marie Curie Industrial-Academic Partnership Pathways project QUASINANO (REA-FP7-PEOPLE-2009-IAPP/251149). Stan van Gisbergen is thanked for his support. We also thank Marcus Elstner, Gotthard Seifert, Stephan Irle, Chien-Pin Chou, Yoshifumi Nishimura, Stefano Borini, and Agnieszka Kuc for enlightening and inspiring discussions.
■
REFERENCES(1) Hohenberg, P.; Kohn, W. Phys. Rev. 1964, 136, B864−B871. (2) Kohn, W.; Sham, L. J. Phys. Rev. 1965, 140, A1133−A1138. (3) Seifert, G.; Eschrig, H.; Bieger, W. Z. Phys. Chem. 1986, 267, 529−539.
(4) Seifert, G.; Porezag, D.; Frauenheim, T. Int. J. Quantum Chem. 1996, 58, 185−192.
(5) Porezag, D.; Frauenheim, T.; Köhler, T.; Seifert, G.; Kaschner, R. Phys. Rev. B 1995, 51, 12947−12957.
(6) Slater, J. C.; Koster, G. F. Phys. Rev. 1954, 94, 1498−1524. (7) Haugk, M.; Elsner, J.; Heine, T.; Frauenheim, T.; Seifert, G. Comput. Mater. Sci. 1999, 13, 239−251.
(8) Elstner, M.; Frauenheim, T.; Kaxiras, E.; Seifert, G.; Suhai, S. Phys. Rev. B 2000, 217, 357−376.
(9) Fowler, P. W.; Heine, T.; Manolopoulos, D. E.; Mitchell, D.; Orlandi, G.; Schmidt, R.; Seifert, G.; Zerbetto, F. J. Phys. Chem. 1996, 100, 6984−6991.
(10) Fowler, P. W.; Heine, T.; Mitchell, D.; Orlandi, G.; Schmidt, R.; Seifert, G.; Zerbetto, F. J. Chem. Soc., Faraday Trans. 1996, 92, 2203. (11) Heine, T.; Fowler, P.; Seifert, G. Solid State Commun. 1999, 111, 19−22.
(12) Albertazzi, E.; Domene, C.; Fowler, P. W.; Heine, T.; Seifert, G.; Van Alsenoy, C.; Zerbetto, F. Phys. Chem. Chem. Phys. 1999, 1, 2913− 2918.
(13) Heine, T.; Seifert, G.; Fowler, P. W.; Zerbetto, F. J. Phys. Chem. A 1999, 103, 8738−8746.
(14) Heine, T.; Zerbetto, F.; Seifert, G.; Fowler, P. W. J. Phys. Chem. A 2001, 105, 1140−1143.
(15) Heine, T.; Zerbetto, F.; Seifert, G.; Fowler, P. W. J. Phys. Chem. A 2000, 104, 3865−3868.
(16) Seifert, G.; Enyashin, A.; Heine, T. Phys. Rev. B 2005, 72, 012102.
(17) Ivanovskaya, V. V.; Ranjan, N.; Heine, T.; Merino, G.; Seifert, G. Small 2005, 1, 399−402.
(18) Kuc, A.; Zhechkov, L.; Patchkovskii, S.; Seifert, G.; Heine, T. Nano Lett. 2007, 7, 1−5.
(19) Lukose, B.; Wahiduzzaman, M.; Kuc, A.; Heine, T. J. Phys. Chem. C 2012, 116, 22878−22884.
(20) Heine, T.; Vietze, K.; Seifert, G. Magn. Reson. Chem. 2004, 42, S199−201.
(21) Widany, J.; Frauenheim, T.; Köhler, T.; Sternberg, M.; Porezag, D.; Jungnickel, G.; Seifert, G. Phys. Rev. B 1996, 53, 4443−4452.
(22) Fowler, P. W.; Heine, T.; Mitchell, D.; Schmidt, R.; Seifert, G. J. Chem. Soc., Faraday Trans. 1996, 92, 2197.
(23) Frauenheim, T.; Weich, F.; Köhler, T.; Uhlmann, S.; Porezag, D.; Seifert, G. Phys. Rev. B 1995, 52, 11492−11501.
(24) Gutierrez, R.; Frauenheim, T.; Köhler, T.; Seifert, G. J. Mater. Chem. 1996, 6, 1657.
(25) Astala, R.; Kaukonen, M.; Nieminen, R.; Heine, T. Phys. Rev. B 2000, 61, 2973−2980.
(26) Goyal, P.; Elstner, M.; Cui, Q. J. Phys. Chem. B 2011, 115, 6790−805.
(27) Choi, T. H.; Liang, R.; Maupin, C. M.; Voth, G. A. J. Phys. Chem. B 2013, 117, 5165−79.
(28) Heine, T.; Bühl, M.; Fowler, P.; Seifert, G. Chem. Phys. Lett. 2000, 316, 373−380.
(29) Sternberg, M.; Frauenheim, T.; Zimmermann-Edling, W.; Busmann, H.-G. Surf. Sci. 1997, 370, 232−244.
(30) Haugk, M.; Elsner, J.; Frauenheim, T.; Staab, T.; Latham, C.; Jones, R.; Leipner, H.; Heine, T.; Seifert, G.; Sternberg, M. Phys. Status Solidi B 2000, 217, 473−511.
(31) Fowler, P. W.; Heine, T.; Zerbetto, F. J. Phys. Chem. A 2000, 104, 9625−9629.
(32) Heine, T.; Zerbetto, F. Chem. Phys. Lett. 2002, 358, 359−367. (33) Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Phys. Rev. B 1998, 58, 7260− 7268.
(34) Liu, H.; Elstner, M.; Kaxiras, E.; Frauenheim, T.; Hermans, J.; Yang, W. Proteins 2001, 44, 484−9.
(35) Sugihara, M.; Buss, V.; Entel, P.; Elstner, M.; Frauenheim, T. Biochemistry 2002, 41, 15259−15266.
(36) Niehaus, T.; Suhai, S.; Della Sala, F.; Lugli, P.; Elstner, M.; Seifert, G.; Frauenheim, T. Phys. Rev. B 2001, 63, 085108.
(37) Frauenheim, T.; Seifert, G.; Elstner, M.; Niehaus, T.; Köhler, C.; Amkreutz, M.; Sternberg, M.; Hajnal, Z.; Carlo, A. D.; Suhai, S. J. Phys.: Condens. Matter 2002, 14, 3015−3047.
(38) Frenzel, J.; Joswig, J.-O.; Seifert, G. J. Phys. Chem. C 2007, 111, 10761−10770.
(39) Abdali, S.; Niehaus, T. A.; Jalkanen, K. J.; Cao, X.; Nafie, L. A.; Frauenheim, T.; Suhai, S.; Bohr, H. Phys. Chem. Chem. Phys. 2003, 5, 1295−1300.
(40) Witek, H. A.; Irle, S.; Zheng, G.; de Jong, W. A.; Morokuma, K. J. Chem. Phys. 2006, 125, 214706.
(41) Li, W.; Irle, S.; Witek, H. A. ACS Nano 2010, 4, 4475−86. (42) Elstner, M.; Hobza, P.; Frauenheim, T.; Suhai, S.; Kaxiras, E. J. Chem. Phys. 2001, 114, 5149.
(43) Zhechkov, L.; Heine, T.; Patchkovskii, S.; Seifert, G.; Duarte, H. A. J. Chem. Theory Comput. 2005, 1, 841−847.
(44) Köhler, C.; Seifert, G.; Frauenheim, T. Chem. Phys. 2005, 309, 23−31.
(45) Sanna, S.; Hourahine, B.; Gallauner, T.; Frauenheim, T. J. Phys. Chem. A 2007, 111, 5665−70.
(46) Hourahine, B.; Sanna, S.; Aradi, B.; Köhler, C.; Niehaus, T.; Frauenheim, T. J. Phys. Chem. A 2007, 111, 5671−7.
(47) Gaus, M.; Cui, Q.; Elstner, M. J. Chem. Theory Comput. 2011, 7, 931−948.
(48) Gaus, M.; Goez, A.; Elstner, M. J. Chem. Theory Comput. 2013, 9, 338−354.
(49) Guimarães, L.; Pinto, Y. N.; Lourenço, M. P.; Duarte, H. A. Phys. Chem. Chem. Phys. 2013, 15, 4303−9.
(50) Guimarães, L.; Enyashin, A. N.; Frenzel, J.; Heine, T.; Duarte, H. A.; Seifert, G. ACS Nano 2007, 1, 362−8.
(51) Frenzel, J.; Oliveira, A. F.; Duarte, H. A.; Heine, T.; Seifert, G. Z. Anorg. Allg. Chem. 2005, 631, 1267−1271.
(52) Vörös, M.; Gali, A.; Kaxiras, E.; Frauenheim, T.; Knaup, J. M. Phys. Status Solidi B 2012, 249, 360−367.
(53) Saha, S.; Pal, S.; Sarkar, P.; Rosa, A. L.; Frauenheim, T. J. Comput. Chem. 2012, 33, 1165−1178.
(54) Dolgonos, G.; Aradi, B.; Moreira, N. H.; Frauenheim, T. J. Chem. Theory Comput. 2010, 6, 266−278.
(55) Sarkar, S.; Pal, S.; Sarkar, P.; Rosa, A. L.; Frauenheim, T. J. Chem. Theory Comput. 2011, 7, 2262−2276.
(56) Kuc, A.; Enyashin, A.; Seifert, G. J. Phys. Chem. B 2007, 111, 8179−86.
(57) Lukose, B.; Supronowicz, B.; St. Petkov, P.; Frenzel, J.; Kuc, A. B.; Seifert, G.; Vayssilov, G. N.; Heine, T. Phys. Status Solidi B 2012, 249, 335−342.
(58) Liu, J.; Lukose, B.; Shekhah, O.; Arslan, H. K.; Weidler, P.; Gliemann, H.; Bräse, S.; Grosjean, S.; Godt, A.; Feng, X.; Müllen, K.; Magdau, I.-B.; Heine, T.; Wöll, C. Sci. Rep. 2012, 2, 921.
(59) Ding, X.; Chen, L.; Honsho, Y.; Feng, X.; Saengsawang, O.; Guo, J.; Saeki, A.; Seki, S.; Irle, S.; Nagase, S.; Parasuk, V.; Jiang, D. J. Am. Chem. Soc. 2011, 133, 14510−14513.
(60) Kandambeth, S.; Mallick, A.; Lukose, B.; Mane, M. V.; Heine, T.; Banerjee, R. J. Am. Chem. Soc. 2012, 134, 19524−7.
(61) Lukose, B.; Kuc, A.; Heine, T. J. Mol. Model. 2013, 19, 2143−8. (62) Chen, X.; Addicoat, M.; Irle, S.; Nagai, A.; Jiang, D. J. Am. Chem. Soc. 2013, 135, 546−549.
(63) Biswal, B. P.; Chandra, S.; Kandambeth, S.; Lukose, B.; Heine, T.; Banerjee, R. J. Am. Chem. Soc. 2013, 135, 5328−31.
(64) Nishimoto, Y.; Wang, Z.; Morokuma, K.; Irle, S. Phys. Status Solidi B 2012, 249, 324−334.
(65) Zheng, G.; Irle, S.; Morokuma, K. Chem. Phys. Lett. 2005, 412, 210−216.
(66) Page, A. J.; Chou, C.-P.; Pham, B. Q.; Witek, H. A.; Irle, S.; Morokuma, K. Phys. Chem. Chem. Phys. 2013, 15, 3725−35.
(67) Elstner, M. Theor. Chem. Acc. 2005, 116, 316−325.
(68) Hou, G.; Zhu, X.; Elstner, M.; Cui, Q. J. Chem. Theory Comput. 2012, 8, 4293−4304.
(69) Woiczikowski, P. B.; Kubar, T.; Gutiérrez, R.; Cuniberti, G.; Elstner, M. J. Chem. Phys. 2010, 133, 035103.
(70) Frähmcke, J. S.; Wanko, M.; Elstner, M. J. Phys. Chem. B 2012, 116, 3313−21.
(71) Goyal, P.; Ghosh, N.; Phatak, P.; Clemens, M.; Gaus, M.; Elstner, M.; Cui, Q. J. Am. Chem. Soc. 2011, 133, 14981−97.
(72) Seifert, G.; Joswig, J.-O. WIREs Comput. Mol. Sci. 2012, 2, 456− 465.
(73) Oliveira, A. F.; Seifert, G.; Heine, T.; Duarte, H. a. J. Braz. Chem. Soc. 2009, 20, 1193−1205.
(74) Elstner, M.; Frauenheim, T.; McKelvey, J.; Seifert, G. J. Phys. Chem. A 2007, 111, 5607−5608.
(75) Knaup, J. M.; Hourahine, B.; Frauenheim, T. J. Phys. Chem. A 2007, 111, 5637−41.
(76) Gaus, M.; Chou, C.-P.; Witek, H.; Elstner, M. J. Phys. Chem. A 2009, 113, 11866−81.
(77) Bodrog, Z.; Aradi, B.; Frauenheim, T. J. Chem. Theory Comput. 2011, 7, 2654−2664.
(78) Eschrig, H.; Bergert, I. Phys. Status Solidi B 1978, 90, 621−628. (79) Eschrig, H. Optimized LCAO Method and the Electronic Structure of Extended Systems; Springer: Berlin, 1989.
(80) Nishimura, Y. Development of the Density-Functional Tight-Binding Method and Its Application in Computational Nanomaterial Science. Ph.D. Thesis, Nagoya University, Japan, 2013.
(81) Irle, S. personal communication.
(82) Mirtschink, A. Berechnungen von Bindungsenergien zweiato-miger Moleküle der Elemente der ersten und zweiten Periode im Rahmen der DFTB-Methode. M.Sc. Thesis, University of Technology Dresden, Germany, 2009.
(83) te Velde, G.; Baerends, E. J. Phys. Rev. B 1991, 44, 7888−7903. (84) Philipsen, P. H. T.; te Velde, G.; Baerends, E. J.; Berger, J. A.; de Boeij, P. L.; Franchini, M.; Groeneveld, J. A.; Kadantsev, E. S.; Klooster, R.; Kootstra, F.; Romaniello, P.; Skachkov, D. G.; Snijders, J. G.; Wiesenekker, G.; Ziegler, T. BAND2013, Scientific Computing & Modeling, 2013. http://www.scm.com.
(85) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865−3868.
(86) van Lenthe, E.; Ehlers, A.; Baerends, E.-J. J. Chem. Phys. 1999, 110, 8943.
(87) Wiesenekker, G.; Baerends, E. J. J. Phys.: Condens. Matter 1991, 3, 6721−6742.
(88) Borini, S.; Philipsen, P.; Yakovlev, A.; McCormack, D.; Patchkovskii, S.; Heine, T. ADF DFTB, Scientific Computing & Modeling, 2013. http://www.scm.com.
(89) Herman, F.; Skillman, S. Atomic Structure Calculations; Prentice-Hall, Inc., Englewood Cliffs, NJ, 1963.
(90) Kaschner, R.; Frauenheim, T. J. Comput.-Aided Mater. Des. 1997, 4, 53−62.
(91) Niehaus, T. J. Mol. Struc.: THEOCHEM 2001, 541, 185−194. (92) Zheng, G.; Witek, H. A.; Bobadova-Parvanova, P.; Irle, S.; Musaev, D. G.; Prabhakar, R.; Morokuma, K.; Lundberg, M.; Elstner, M.; Köhler, C.; Frauenheim, T. J. Chem. Theory Comput. 2007, 3, 1349−1367.
(93) Witek, H. A.; Köhler, C.; Frauenheim, T.; Morokuma, K.; Elstner, M. J. Phys. Chem. A 2007, 111, 5712−9.
(94) Junquera, J.; Paz, O.; Sánchez-Portal, D.; Artacho, E. Phys. Rev. B 2001, 64, 235111.
(95) Cordero, B.; Gómez, V.; Platero-Prats, A. E.; Revés, M.; Echeverra, J.; Cremades, E.; Barragán, F.; Alvarez, S. Dalton Trans. 2008, 2832−8.
(96) Frenzel, J.; Oliveira, A. F.; Jardillier, N.; Heine, T.; Seifert, G. Semi-Relativistic, Self-Consistent Charge Slater−Koster Tables for Density-Functional Based Tight-Binding (DFTB) for Materials Science Simulations. http://www.dftb.org/parameters/download/ matsci/matsci_0_3.
(97) DFTB Web site. http://www.dftb.org.
(98) Ceperley, D. M.; Alder, B. J. Phys. Rev. Lett. 1980, 45, 566−569. (99) Ghorbani-Asl, M.; Borini, S.; Kuc, A.; Heine, T. Phys. Rev. B 2013, 87, 235434.
(100) Ghorbani-Asl, M.; Zibouche, N.; Wahiduzzaman, M.; Oliveira, A. F.; Kuc, A.; Heine, T. Sci. Rep. 2013, submitted.