The Identification and Location of Succinyl Residues and the
Characterization of the Interior Arabinan Region Allow for a
Model of the Complete Primary Structure of
Mycobacterium tuberculosis Mycolyl Arabinogalactan
*
Received for publication, January 9, 2008, and in revised form, February 26, 2008Published, JBC Papers in Press, February 26, 2008, DOI 10.1074/jbc.M800222200
Suresh Bhamidi‡, Michael S. Scherman‡, Christopher D. Rithner§, Jessica E. Prenni¶, Delphi Chatterjee‡,
Kay-Hooi Khoo储, and Michael R. McNeil‡1
From the Departments of‡Microbiology,§Chemistry, and the¶Proteomics and Metabolomics Facility, Colorado State University, Fort Collins, Colorado 80523 and the储Institute of Biochemical Sciences, National Taiwan University, Taipei 106, Taiwan, and the Institute of Biological Chemistry, Academia Sinica, Taipei 115, Taiwan
The complex cell wall of Mycobacterium tuberculosis is the hallmark of acid fast bacteria and is responsible for much of its physiological characteristics. Hence, much effort has been made to determine its primary structure. Such studies have been ham-pered by its extreme complexity. Also, its insolubility leads to difficulties determining the presence or absence of base labile groups. We have used an endogenous arabinase to solubilize the arabinan region of the cell wall and have shown using mass spec-trometry and NMR that succinyl esters are present on O2 of the inner-branched 1,3,5-␣-D-arabinofuranosyl residues. In
addi-tion, an inner arabinan region of 14 linear␣-1,5 arabinofurano-syl residues has been identified. These and earlier results now allow the presentation of a model of the entire primary structure of the mycobacterial mycolyl arabinogalactan highlighted by three arabinan chains of 31 residues each.
The primary structure of the cell wall core of mycobacteria has been an object of study for many years. The fundamental concept that it consists of peptidoglycan attached to mycolic acids via the polysaccharide arabinogalactan (AG)2was
deter-mined some time ago (1). Also, early studies revealed a
chemo-type IV peptidoglycan similar to that of Escherichia coli with both diaminopimelic acid (DAP)-Ala and DAP-DAP cross-linking (2). Until very recently, little additional study of the peptidoglycan of M. tuberculosis has occurred. These recent studies have focused on substituents on the peptide carboxyl groups (3) and on whether the peptidoglycan chains are parallel or perpendicular to the plasma membrane (4). The structure of the mycolic acids was determined in detail some time ago (5); however, notably lacking in these studies was the structure of the arabinogalactan polymer, although the esterification of the mycolic acids at C5 of some arabinofuranosyl residues was established (6, 7). Also some linkage and ring form information on the arabinosyl and galactosyl residues was determined by methylation analysis (8 –10).
The primary structure of AG is very complex and is not com-posed of a simple repeating unit as is the case for most bacterial polysaccharides. Elucidation of the structure was fundamen-tally advanced by the isolation and characterization of per-O-alkylated oligosaccharide alditols (11). This analysis revealed a linear galactan to which arabinan side chains were attached and also allowed elucidation of the detailed structure of the hexa-arabinoside non-reducing end of the arabinan. The data were consistent with many different structures of the complete ara-binan, and two possibilities were proposed for further study (11). During the same time period, the linker disaccharide (12) at the reducing end of AG and the exact location of the mycolic acids at the non-reducing end of the arabinan were determined (13). A follow-up study extended the structure of the arabinan at the non-reducing end from the hexa-arabinoside to include the last 17–22 arabinosyl residues (14), but until very recently, additional progress on the structure of AG has been lacking. Now three fundamental advances have occurred, which allow the structure to be further refined. First, AG has been found to be nonessential in the related Corynebacteria genus, which per-mits structural studies of the AG produced by mutants of AG biosynthetic enzymes (15, 16). In this regard, elegant studies by Besra et al. (16) have shown that in Corynebacterium
glutami-cum, three arabinan chains are attached to the galactan. These researchers knocked out the gene of the arabinosyl transferase, Emb, that extends the arabinan after a different arabinosyl transferase, AftA (15) attaches the first arabinosyl unit.
Methy-*This work was supported, in whole or in part, by National Institutes of Health Grant AI-33706 from NIAID (to M. R. M.) and Grant AI-37139 (to D. C.). This work was also supported by United States Public Health Service Grant NSC 96-2628-B-001-014-MY3 (to K. K.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article is dedicated to Patrick J. Brennan in honor of his 70th birthday and in recognition of his unprecedented accomplishments of defining the structures of mycobacterial cell surface molecules and of elucidating mycobacterial biosynthetic pathways.
1To whom correspondence should be addressed: Dept. of Microbiology, Immunology, & Pathology Colorado State University, Fort Collins, CO 80523-1682. Tel.: 970-491-1784; Fax: 970-491-1815; E-mail: mmcneil@ colostate.edu.
2The abbreviations used are: AG, arabinogalactan; DAP, diaminopimelic acid; MALDI-TOF MS, matrix-assisted laser desorption/ionization-time of flight mass spectrometry; GalNH2, galactosamine; mAGP, mycolyl arabinogalac-tan peptidoglycan complex; GC/MS, gas chromatography/mass spectrom-etry; gHMBC, two-dimensional gradient-enhanced proton-detected mul-tiple bond correlation NMR; HSQC, two-dimensional proton-detected heteronuclear single quantum coherence NMR; DPFGSE TOCSY, one-di-mensional double-pulsed field gradient spin echo total correlation spec-troscopy NMR; LAM, lipoarabinomannan; amu, atomic mass units.
at National Taiwan University on June 19, 2009
www.jbc.org
lation of the cell wall core of the emb knock-out strain yielded galactan substituted with what turned out to be three terminal Araf units (attached by the still active arabinosyl transferase AftA) in the form of a methyl rhamnoside unit at the reducing end. Mass spectrometry revealed that the single Araf units are attached at positions 8, 10, and 12 of the galactan; this showed the presence and location of three arabinan chains per galactan. Additionally, in recent times, an endogenous arabinase, which releases large oligo-arabinosides, has been purified suf-ficiently for structural studies (17, 18). Use of this enzyme, com-bined with the third major recent advance, matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS), allowed the sequencing of large oligo-ar-abinosides (17). These studies revealed that in M. tuberculosis, a non N-acetylated galactosamine residue (GalNH2) (19) is located on the interior branched arabinosyl residue (17). Two fundamental structural issues remain: the presence and loca-tion of alkali labile groups on AG, and the structure of the inte-rior region of the arabinan. These two issues are addressed in this report, leading to a model of the complete primary struc-ture of AG.
EXPERIMENTAL PROCEDURES
Bacterial Cultures—M. tuberculosis (H37Rv) was grown in GAS medium and M. smegmatis (⌬embC mutant, Ref. 20) was grown in 7H9 medium (with 0.2% glycerol and 50g/ml kana-mycin), both at 37 °C. Bacteria were harvested in late log phase.
Preparation of AG—Harvested cells were disrupted mechan-ically using a French press at 1500 psi followed by centrifuga-tion at 21,000⫻ g for 40 min at 4 °C. The pellet thus obtained was resuspended in 30 ml of Milli-Q water containing 2% SDS and stirred gently at room temperature overnight followed by centrifugation (25,000⫻ g). The pellet was resuspended in 30 ml of Milli-Q water containing 2% SDS and stirred gently at 100 °C for 1 h. This was followed by centrifuging at 25,000⫻ g for 30 min. The pellet was repeatedly washed with water (three times) each time followed by centrifugation as above. After three water washes, the pellet was resuspended in 30 ml of 80% acetone/Milli-Q water and centrifuged at 25,000⫻ g for 20 min to remove SDS. This was again followed by two water washes and, after freeze-drying, the material thus obtained was used as mycolyl arabinogalactan peptidoglycan complex (mAGP) for further experiments.
Endogenous Arabinase Treatment and Purification of Arabinans—The mAGPs (of both M. tuberculosis H37Rv and
M. smegmatis (⌬emb C mutant) were treated with partially
purified endogenous arabinase (21) at 37 °C, for 12 h, followed by extraction with chloroform/methanol/water (10:10:3). Sur-prisingly, the chloroform/methanol/water (10:10:3) was shown to be a good solvent system for solubilizing the resulting oligo-arabinans. This fraction was dried and treated with chloro-form/methanol/water (8:4:3), and the arabinan in these cir-cumstances was in the aqueous phase. Then the aqueous fraction from this Folch extract was passed through a P-2 col-umn (Biogel, 1⫻ 25 cm column) eluted with 50 mMsodium
acetate buffer, pH 5, and the fractions were analyzed for arabi-nose content by GC/MS of the alditol acetate derivatives. In the case of the arabinan purified from M. tuberculosis cell wall, it
was found that the arabinans with a GalNH2eluted slightly
earlier than arabinan without this sugar and thus could be sep-arated. The purified arabinan containing fractions were again passed through a P-2 column (1⫻ 15 cm column) and were eluted this time with water for the purpose of desalting. The purified fractions from P-2 column were used directly for MALDI-TOF MS or exchanged with D2O for NMR analysis.
NMR Analysis—NMR spectra were recorded on a Varian Inova 500 MHz spectrometer equipped with a 5 mm, gradient-enhanced HCN probe (Varian, Palo Alto, CA). During one-dimensional and two-one-dimensional experiments, data were recorded at 298 K. All data were processed using Varian VnmrJ 1.1D software. One-dimensional double-pulsed field gradient spin echo total correlation spectroscopy (22) (1D-DPFGSE TOCSY) NMR spectra were acquired with four different mix-ing times of 0, 30, 80, and 150 ms. The signal at 4.92 ppm that we have assigned to the H2 proton was selected in these experi-ments. Two-dimensional proton-detected heteronuclear single quantum coherence (HSQC) NMR spectra were acquired in phase-sensitive mode with 256 increments (complex, States-TPPI) at sixteen signal-averaging transients per increment. The total cycle time per transient including relaxation delay was 1.7 s. The spectral windows were 7K Hz in the proton directly detected dimension and 17K Hz in the carbon indirectly detected dimension. Forward linear prediction was used in the indirect dimension, and zero-filling was applied to both dimen-sions. The data were weighted with cosine-squared functions before Fourier transformation. The final two-dimensional spectrum was 2K complex points in F2 by 1K complex points in F1. Two-dimensional gradient-enhanced proton-detected multiple bond correlation (gHMBC) NMR spectra were acquired in absolute value mode with 512 increments and 64 signal-averaging transients per increment. The total cycle time per transient including relaxation delay was 1.9 s. The spectral windows were 7K Hz in the proton directly detected dimension and 30K Hz in the carbon indirectly detected dimension. For-ward linear prediction was used in the indirect dimension, and zero-filling was applied to both dimensions. The data were weighted with sinebell-squared functions before Fourier trans-formation. The final two-dimensional spectrum was 4K real points in F2 by 1K real points in F1.
GC/MS Analysis of Succinates—Succinates were detected and quantified by GC/MS of their octyl succinate derivatives obtained from 1 mg of mAGP after octanolysis with 3MHCl in 1-octanol (100l) at 120 °C for 30 min as reported earlier (23). The octyl derivatives were dissolved in hexanes prior to injec-tion on a DB-5 column at an initial temperature of 60 °C held for 1 min. The temperature was increased to 330 °C at a rate of 30 °C/min. The retention times and mass spectra were com-pared with those of standard succinic acid and were quantified by comparison with the areas of the internal standards, malonic acid and glutaric acid (as octyl esters), which were also sub-jected to octanolysis per the procedure.
MALDI-TOF MS—Samples (5g) were peracetylated with 100l of acetic anhydride in the presence of 50 l of pyridine at room temperature overnight. After removal of the solvents, they were extracted three times using 1:1 chloroform/water, the final chloroform layer was dried, and the residue dissolved in
at National Taiwan University on June 19, 2009
www.jbc.org
10l of chloroform prior to MALD-TOF MS. 1 l of the acety-lated AG sample in chloroform was mixed with 1l of matrix, dihydroxybenzoic acid (10 mg/ml in 50% acetonitrile in 0.1% trifluoroacetic acid) and allowed to air-dry on the MALDI tar-get plate. The acetylated AG samples were analyzed by an Ultraflex-TOF/TOF mass spectrometer (Bruker Daltonics, Bil-lerica, MA) in positive ion, reflector mode using a 25 kV accel-erating voltage. External calibration was performed using an 8 standard calibration mixture on a spot adjacent to the sample. To observe the weaker high mass peaks (⬎4000 Da), the laser power was incrementally increased at the expense of the reso-lution of lower mass species. Thus the spectra were collected in suitable mass ranges corresponding to the laser power increases.
RESULTS
Succinyl Residues Are Present on AG Arabinan—The cell wall core, mAGP, was prepared from M. tuberculosis and from
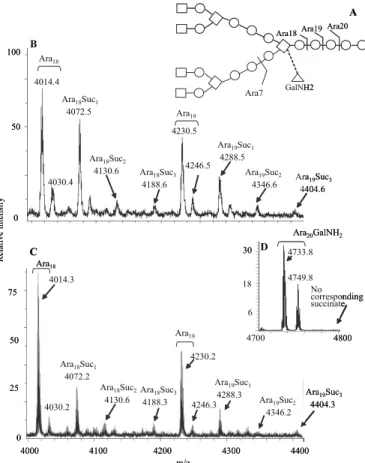
M. smegmatis embC⫺ (a lipoarabinomannan-free strain, Ref. 20) under non-basic and non-acidic conditions. These prepa-rations of mAGP were treated with partially purified endoge-nous ␣-1,5-endoarabinanase (21), and the solubilized oligo-arabinans were purified by sizing chromatography. In the case of M. tuberculosis arabinan, the galactosaminylated arabinan eluted slightly earlier than the non-galactosaminylated arabi-nan. The arabinan fractions were then acetylated and analyzed by MALDI-TOF MS. The arabinase, although capable of cleav-ing any␣-1,5 bond, has been shown to preferentially cleave in the positions indicated in Fig. 1A (17). The enzyme preparation lacks the ability to cleave the-terminal arabinosyl residues, and, indeed, no activity other than the endo-␣-1,5 activity has been detected (17) including any galactanase activity.3 The
resulting spectra showed the M⫹Na⫹(and M⫹K⫹) ions from
the expected Ara18product, previously found using soluble AG
(17), but importantly, also showed ions that were 58 amu heavier, consistent with some arabinans substituted with suc-cinyl esters (Fig. 1, B and C).
Careful examination of the MALDI-TOF MS spectra revealed that although one succinyl group was most common, up to three succinyl groups per released arabinan fragment could be found on oligo-arabinans from both M. smegmatis and M. tuberculosis (Fig. 1, B and C, respectively). Careful analysis of the arabinan fragments from M. tuberculosis substituted with a GalNH2led to the realiza-tion that none of these were succinylated, as shown in Fig. 1D.
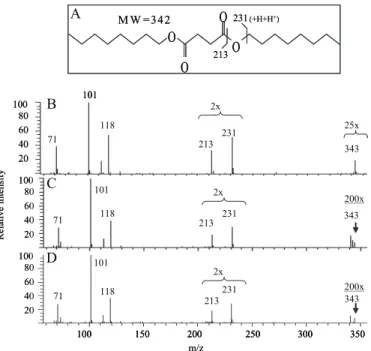
Direct analysis to confirm the presence of succinyl groups in AG was performed by octanolysis of the mAGP of M.
tubercu-losisand M. smegmatis to yield dioctyl esters of any succinyl groups present (Fig. 2A). GC/MS analysis (Fig. 2, C and D and the standard in Fig. 2B) confirmed the presence of succinate in mAGP from both species. Quantitation of the ratio of succinyl to arabinosyl groups showed a ratio of 1:41 (S.D.,⫾2.4 for four determinations) for M. tuberculosis H37Rv and 1:29 for
M. smegmatis(S.D.,⫾2.0 for two determinations). For a clinical isolate of M. tuberculosis (CSU20) the ratio was 1:30 (S.D.,⫾0.4 for two determinations). The significance of the variations is
not known, nor were studies to compare succinylation versus growth phase performed. However the data suggest that approximately one of the three arabinosyl chains contains one (or sometime more) succinyl groups.
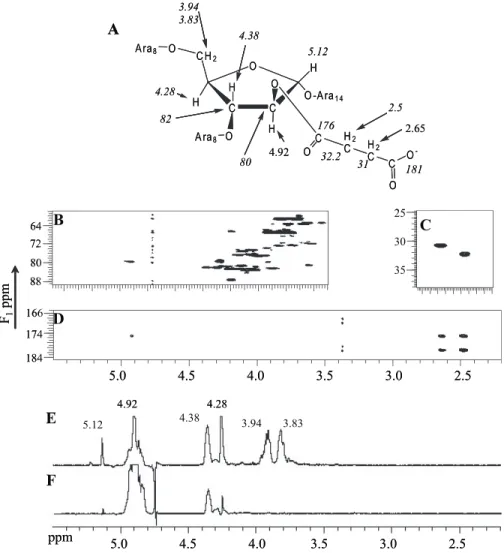
Location of the Succinyl Groups by NMR and Mass Spectrometry—The succinyl groups were located to the specific hydroxyl group on a specific arabinosyl residue by NMR and
MALDI-TOF MS analysis. One-dimensional1H NMR analysis
of the endogenous arabinase-released oligo-arabinans from the cell wall of M. tuberculosis (the non-galactosaminylated frac-tion from the sizing column) showed two multiplets at 2.5 and 2.65 ppm (labeled b and c in Fig. 3), which were assigned to the methylene groups of succinate, consistent with the MALDI-TOF MS and GC/MS data. Detailed NMR studies were then performed using HSQC and gHMBC to determine which arabi-nosyl residue and to which position on that arabiarabi-nosyl residue the succinyl groups were attached. This procedure followed in general terms previous work locating succinyl groups on lipoarabinomannan (24). The two-dimensional1H-13C HSQC
3X. Dong, D. Chatterjee, K. H. Khoo, S. Bhamidi, and M. R. McNeil, unpublished observations. 4014.4 4030.4 Ara18Suc1 4072.5 Ara18Suc2 4130.6 Ara18Suc3 4188.6 4230.5 4246.5 Ara19Suc1 4288.5 Ara19Suc2 4346.6 Ara19Suc3 4404.6 100 50 0 Relat ive intensit y A C Ara18 Ara19
Ara18Ara19 Ara20
GalNH2 Ara7 4800 4700 Ara20GalNH2 No corresponding succinate D 4733.8 4749.8 30 6 18 4014.3 4030.2 Ara18 Ara18Suc1 4072.2 Ara18Suc3 4188.3 Ara18Suc2 4130.6 4230.2 Ara19 4246.3 Ara19Suc1 4288.3 Ara19Suc2 4346.2 Ara19Suc3 4404.3 4000 4100 4200 4300 4400 m/z 50 0 25 75 4014.4 4030.4 Ara18Suc1 4072.5 Ara18Suc2 4130.6 Ara18Suc3 4188.6 4230.5 4246.5 Ara19Suc1 4288.5 Ara19Suc2 4346.6 Ara19Suc3 4404.6 100 50 0 Relat ive intensit y A B C Ara18 Ara19
Ara18Ara19 Ara20
GalNH2 Ara7 4800 4700 Ara20GalNH2 No corresponding succinate D 4733.8 4749.8 30 6 18 4014.3 4030.2 Ara18 Ara18Suc1 4072.2 Ara18Suc3 4188.3 Ara18Suc2 4130.6 4230.2 Ara19 4246.3 Ara19Suc1 4288.3 Ara19Suc2 4346.2 Ara19Suc3 4404.3 4000 4100 4200 4300 4400 m/z 50 0 25 75
FIGURE 1. The endogenous arabinase releases oligo-arabinosides
con-taining succinyl groups. A, preferred sites of action of the endogenous
ara-binase. When the GalNH2is present (dashed bond), the Ara20site is strongly preferred over the Ara19and Ara18site. B, MALDI-TOF mass spectrum of the arabinan fragments released from M. smegmatis mycolyl arabinogalactan between m/z 4000 and 4400. The non-succinylated and succinylated Ara18 and Ara19 oligo-arabinosides are labeled. Both M⫹Na⫹, and, at16 amu higher, M⫹ K⫹ions are seen. C, MALDI-TOF mass spectrum of the non-galac-tosaminylated arabinan fragments released from M. tuberculosis mycolyl arabinogalactan between m/z 4000 and 4400. The non-succinylated and suc-cinylated Ara18and Ara19oligo-arabinosides are labeled. D, MALDI-TOF mass spectrum of the galactosaminylated arabinan fragments released from M. tuber-culosis mycolyl arabinogalactan between m/z 4700 and 4800 showing the major Ara20GalNH2fragment (both as M⫹ Na⫹and M⫹ K⫹) and the lack of any corre-sponding succinylated version.
at National Taiwan University on June 19, 2009
www.jbc.org
spectrum allowed the identification of the succinyl methylene carbon resonances at 32.2 and 31 ppm, corresponding to the1H
signals at 2.5 and 2.65, respectively (Fig. 4C). The two-dimen-sional1H-13C gHMBC spectrum revealed two and three bond
couplings between the methylene proton resonances at 2.5 and
2.65 ppm to two carbonyl groups (Fig. 4D). The two carbonyl resonances at 176 and 181 ppm are assigned to the ester and carboxylic groups of the succinyl group, respectively, based on their chemical shifts (24). Further, the carbonyl ester resonance at 176 ppm showed a cross peak with a proton resonance at 4.92 ppm (Fig. 4D and peak a in Fig. 3). This proton was found to correlate with a non-anomeric carbon resonance at 80 ppm by HSQC (Fig. 4B), corresponding to the carbon substituted by the quantitatively major succinyl group. 1D-DPFGSE TOCSY was used to identify the entire spin system of the succinylated arabi-nosyl residue and thereby to identify the position of the succinyl group (Fig. 4E). Thus the peak at 4.92 ppm was selectively excited, and then four different spin-lock mixing times were used to examine the propagation of coherence through this signal spin-coupled network. We recognized that because of the weak H2/H1 coupling (24), this propagation would likely not be observed at shorter mixing times. The experiment showed that a single signal arose at 4.38 ppm for the shortest mixing time (Fig. 4F), suggesting that the proton at 4.92 ppm was on position 2. We would have expected two signals from the strong H3/H2 and H3/H4 coupling if the proton was at position 3 whereas it being at position 5 is not possible because position 5 is always substituted with an arabinosyl residue. At longer mixing times, signals at 4.28 (H4), 3.83 (H5), and 3.94 (H5⬘) appeared progressively as expected for the more remote couplings. Also a weak signal for H1 at 5.12 ppm arises at the longest mixing time. Hence, we conclude that the succinyl is on position 2 of an arabinosyl residue. The H3 signal at 4.38 was shown to be correlated to C3 at 82 ppm (Fig. 4B); this downfield position taken together with data from known standards revealed that C3 is substituted with a glycosyl residue (24), thereby showing that the arabinosyl is 3,5-linked. These assign-ments are illustrated in Fig. 4A. Mass spectrometry was then used to determine which of the two possible 3,5-linked arabi-nosyl residues was succinylated, the one in the interior region of the arabinan or the one near the non-reducing end.
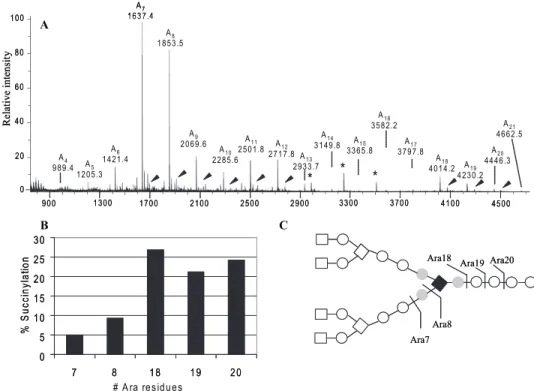
The analysis shown in Fig. 5A of the entire MALDI-TOF MS scan of the endogenous arabinase-released arabinan (from
M. tuberculosis) reveals that Ara7– 8 shows a significantly smaller degree of succinylation compared with Ara18 –21(Fig.
5B). This result is readily interpreted as the succinyl residues located interior of the non-reducing Ara7unit, i.e. on the inte-rior branched 3,5-Araf residue (Fig. 5C). Thus Ara7as released
by the enzyme in a single cleavage contains very few succinyl groups in contrast to the larger Ara18 –21 oligo-arabinosides. Ara7(and Ara8) can also be produced from the interior region
of the arabinan by multiple enzyme cleavages (the only way
Ara9 –12are produced), which explains why the small amount of
succinylation is seen for Ara7and Ara8, although our data does
not absolutely rule out some succinylation on the exterior 3,5-Araf residue. The lack of succinylation of Ara7combined with
the fact that Ara18may have up to 3 succinyl residues leads to
the identification of the quantitatively minor positions of suc-cinylation shown with the gray residues in Fig. 5C.
Structure of the Reducing End Region of the Arabinan —Pre-vious results have suggested that additional␣-1,5-arabinosyl residues are found connecting the Ara18motif to the galactan.
Thus in the mass spectra of arabinans produced by partial acid O O O O 213 231(+H+H+) 101 71 118 213 231 343 101 118 213 231 343 71 101 71 118 213231 343 100 150 200 250 300 350 Relat ive intensit y 20 40 60 80 100 20 40 60 80 100 20 40 60 80 100 2x 25x 200x 200x m/z A B C D M W = 34 2 2x 2x O O O O 213 231(+H+H+) 101 71 118 213 231 343 101 118 213 231 343 71 101 71 118 213231 343 100 150 200 250 300 350 101 71 118 213 231 343 101 118 213 231 343 71 101 71 118 213231 343 100 150 200 250 300 350 Relat ive intensit y 20 40 60 80 100 20 40 60 80 100 20 40 60 80 100 2x 25x 200x 200x m/z B C D M W = 34 2 2x 2x
FIGURE 2. Identification of the succinyl residues in mycobacterial mAGP. A, structure of the dioctyl derivative of succinic acid and depiction of the origin of the ions at m/z 213 and 231. B, mass spectrum of standard succinyl dioctyl ester. The M⫹H ion at m/z 343 is formed in the e.i. mode in the ion trap of the mass spectrometer. C, mass spectrum of succinyl dioctyl ester released by octanolysis of mAGP from M. tuberculosis. D, mass spectrum of succinyl dioctyl ester released by octanolysis of mAGP from M. smegmatis. In C and D, the ion in the molecular weight region is less pronounced because of less material in the ion trap than in B.
FIGURE 3. The one-dimensional NMR spectrum of the mixture of
endog-enous arabinase solubilized non-galactosaminylated oligo-arabinans from M. tuberculosis. The methylene hydrogens on the succinyl group
(labeled b and c) are evident at 2.5 and 2.65 ppm as is the upfield arabinosyl ring proton (from succinyl substitution) at 4.92 (labeled a).
at National Taiwan University on June 19, 2009
www.jbc.org
hydrolysis, arabinans containing up to 22 residues were found (14), and in MADI-TOF MS of arabinan released from base-solubilized AG from M. smegmatis up to Ara21was found. But
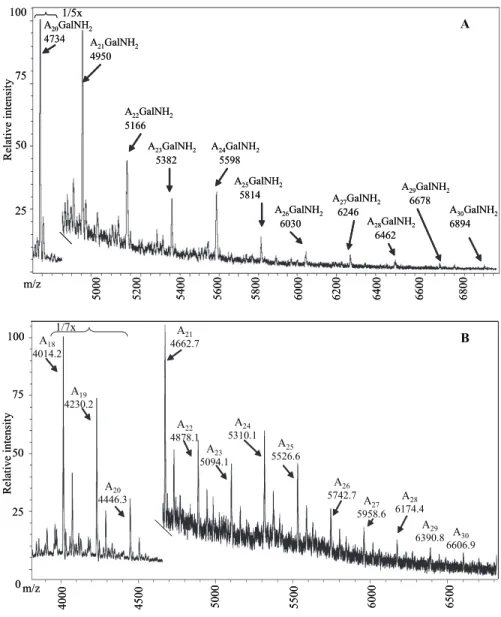
exactly how many arabinosyl residues are present before attachment to the galactan is unknown. However, the MALDI-TOF mass spectrum of the galactosaminylated arabinans released by endogenous arabinase turned out to be informative on this issue. What could be seen was a series of galactosami-nylated arabinosides from the major Ara20(GalNH2)1 up to
Ara30(GalNH2)1 (Fig. 6A). Initially, analogous series for the
non-GalNH2Ara fragments were not observed. However by
applying significantly higher laser power in the mass spectrom-eter, such fragments were also detected in this series, thereby showing that regardless of galactosaminylation, up to 31 arabi-nosyl residues (30 released and 1 “stub” on the galactan) are found for each arabinan (Fig. 6B). The evenness of both series shown in Fig. 6 (in contrast to the distribution seen in Fig. 5) precludes the presence of branched residues, and the linkage and NMR analysis of AG (11) requires that these interior arabi-nosyl residues be␣-5-linked. Thus the Ara17non-reducing end
of the arabinan is attached to the galactan via a linear␣-1,5-arabinan, with up to 14 units.
DISCUSSION
General Considerations—The arabinogalactan polysaccharide is clearly one of the more structurally complex polysaccharides in nature (25–27). Although the galactan of AG is made of a disaccharide-repeating unit, [36)--D
-Galf-(135)--D-Galf]15 the arabinan
most emphatically is not. Initial par-tial acid hydrolysis studies (11) pointed toward two fundamentally different motifs around the ␣-3,5-linked Araf residues: the first motif is evident in the non-reducing hepta-arabinoside (Fig. 1) where -D-Araf-(132)-␣-D-Araf- units are attached to both positions 3 and 5 and form the non-reducing end of the molecule. This result was con-firmed by physical isolation of the non-reducing end of arabinan (as a hexa-arabinoside) using an arabi-nase secreted by a Cellulomonas species (28). On the other hand, ␣-3,5-linked Araf residues are found in the interior where35)-␣-D-
Araf-residues are found on positions 3 and 5 (11). The two types of ␣-3,5-Araf residues were detected con-nected together by the generation of large fragments by mild acid hydrol-ysis (14); this structure (see Fig. 1) was again confirmed enzymatically (17) employing the endogenous␣-1,5 arabinase used in this study. Interestingly, some of the interior␣-3,5-Araf residues have been shown to be further substituted at position 2, with either a GalNH2
(17) or, as shown herein, with a succinyl residue.
The region interior of the inner ␣-3,5-Araf residue has remained a mystery. We have now shown that at least 14 linear ␣-1,5-arabinosyl residues make up this region. For the enzyme to cleave, it is assumed that at least one additional Araf must be present before the galactan, and hence our conclusion that at least 15 Araf units make up this interior region leading to 31 Araf residues in the complete arabinan. Our data will not dis-tinguish between the interior region consisting of precisely 15 arabinosyl residues or consisting of a mixture of linear chains up to this length.
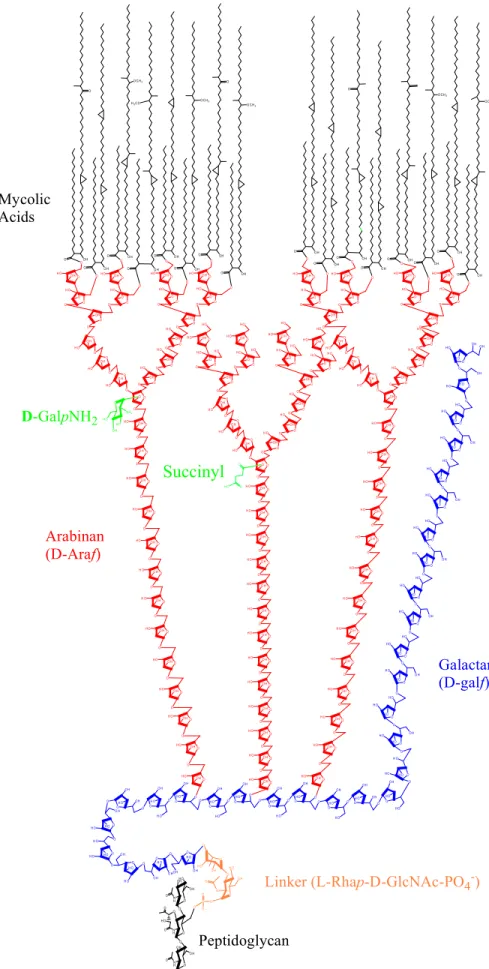
A Detailed Model of Arabinogalactan Structure—The results herein in combination with earlier work (11–14, 16) now allow for a detailed model of the primary structure of the mycolyl arabinogalactan complex of M. tuberculosis to be proposed (Fig. 7). Key features include the presence of an alternating 36)--D-Galf-(135)--D-Galf galactan with 30 residues in
5.0 4.5 4.0 3.5 3.0 2.5 ppm 5.12 4.92 4.38 4.28 3.94 3.83
E
F
A
5.0 4.5 4.0 3.5 3.0 2.5 184 174 166 88 80 72 64 F1 ppm 35 25 30B
D
C
Ara8 C O C O O C H2 O O -A ra1 4 A ra8 C O H2 C H2 C C O O -H H H 176 32.2 31 181 2.5 2.65 80 4.92 82 4.38 4.28 3.94 3.83 H 5.12 5.0 4.5 4.0 3.5 3.0 2.5 ppm 5.12 4.92 4.38 4.28 3.94 3.83E
F
A
5.0 4.5 4.0 3.5 3.0 2.5 184 174 166 88 80 72 64 F1 ppm 35 25 30B
D
C
Ara8 C O C O O C H2 O O -A ra1 4 A ra8 C O H2 C H2 C C O O -H H H 176 32.2 31 181 2.5 2.65 80 4.92 82 4.38 4.28 3.94 3.83 H 5.12FIGURE 4. Location of the succinyl group on O2 of an␣-3,5-Araf residue by NMR. A, summary of the chemical shifts of the relevant nuclei. B, HSQC two-dimensional NMR spectrum between 3.5 and 5.5 ppm (1H) and 58 and 89 ppm (13C). C, HSQC two-dimensional NMR spectrum between 2.2 and 2.8 ppm (1H) and 25 and 38 ppm (13C). D, gHMBC two-dimensional NMR between 2.2 and 5.5 ppm (1H) and 166 and 184 ppm (13C).
E, one-dimensional TOCSY from 2.5 to 5.5 ppm with a mixing time of a 150 ms. F, one-dimensional TOCSY from 2.5 to 5.5 ppm with a mixing time of 30 ms.
at National Taiwan University on June 19, 2009
www.jbc.org
total; 3 arabinans (16) each containing 31 Araf residues arranged in a precise structure and attached to O5 of galacto-furanosyl residues 8, 10, and 12 units from the galactan reduc-ing end; a specific linker unit of a rhamnosyl residue attached to an N-acetylglucosaminosyl-1-phosphate residue; and hence 125 glycosyl residues in total. Importantly, in the case of M.
tu-berculosis, and presumably in all slow growers, both a positive charge (protonated GalNH2as GalNH3⫹) and a negative charge (succinyl) are present in the middle regions of the arabinan, specifically at O2 of the inner 3,5-␣-D-Araf units. The mycolates
are attached to position 5 of both the non-reducing terminal -D-Araf units and the penultimate 2 linked␣-D-Araf units as
shown in the figure. The model is presented with two arabinan chains being fully mycolylated (i.e. eight mycolates) and thereby leaving the other chain fully unsubstituted. Previous data (13) showed that some of the non-reducing arms of the arabinan, i.e. the non-reducing terminal defined by Ara7(Fig. 1A), are fully
substituted with mycolates as shown in Fig. 7, and similarly that other non-reducing Ara7arms of the arabinan are fully unsub-stituted (13). The same data also showed the lack of partially substituted non-reducing arms and that ⬃two-thirds of the non-reducing Ara7units are mycolylated. The all or nothing mycolylation of any given arabinan chain shown in Fig. 7 (as opposed to random groups of four) is suggested by our present studies. Thus quantitation of the arabinase treatments described herein showed that⬃25% of the arabinosyl residues of mAGP are solubilized in a mycolate-free form corresponding
roughly to the one-third of the arabinan expected to be fully non-mycolylated.
Finally the succinyl residue is shown in the model (Fig. 7) to be on the non-mycolylated chain. This is because integration of the NMR spectrum of the soluble non-myco-lylated material suggested that the bulk of the succinylated arabinan was recovered from mAGP after enzyme treatment. It is also clear from the mass spectrum of this material (Fig. 1) that some of these non-mycolylated arabinan chains contain no succinyl residues and that some contain GalNH2, and this
diversity is not indicated in the model. It should be noted that the negative charge on the succinyl res-idues makes quantitation of the three arabinan forms by MS essen-tially impossible. The presence of the majority of the succinylated ara-binans in the soluble fraction sug-gests that the mycolylated arabinan chains are succinate-free, although this has not been demonstrated directly. Finally, because there is
approximately one GalNH2 per
entire AG molecule (19) it seems likely that mycolylated arabinan chains are both galactosami-nylated and nonsubstituted as shown in the model.
Limitations of the Model—In our first model of AG (11), we were careful to point out the limitations of our data and even illustrated two of the several possible arrangements that the limited structural data were consistent with. Now we are able to narrow the model to a much more specific presentation, which is, not surprisingly, different than either of the earlier possibil-ities suggested (Fig. 7). Nevertheless there remain limitations and uncertainties that need to be clearly delineated. Most importantly, the homogeneity or heterogeneity of the arabinan and galactan chain lengths is not well established. Thus galac-tans of different sizes averaging around 30 residues and inner arabinan of different sizes (the size of the non-reducing end is fixed) averaging around 14 residues is possible. Perhaps this issue will best be resolved if mass spectrometric analysis of the intact AG molecule can be performed.
Additionally, in the model (Fig. 7), the three major types of mycolic acids, alpha, keto, and methoxy, found in M.
tubercu-losis(29) are shown. These three classes themselves each con-sist of a large number of forms varying primarily on the number of CH2groups in various regions of the long meromycolate chain (29). At this point in time, we assume these various myco-lates are attached to the arabinan in a random fashion, but no data on this exists.
Finally, the distribution of galactosaminylated, succinylated, and non-substituted arabinan chains is not rigorously
estab-0 5 10 15 20 25 30 7 8 18 19 20 # A ra res idues % S u cc in yl a tio n Ara7 Ara19 Ara20 Ara18 Ara8 100 A5 1205.3 A4 989.4 A6 1421.4 A7 1637.4 A8 1853.5 A9 2069.6 A10 2285.6 A11 2501.8 A12 2717.8 A13 2933.7 A14 3149.8 A17 3797.8 A18 4014.2 A20 4446.3 A21 4662.5 900 1300 1700 2100 2500 2900 3300 3700 4100 4500 80 60 40 20 0 A16 3582.2 A B Relat ive inte nsit y A15 3365.8 A19 4230.2 * * * 0 5 10 15 20 25 30 7 8 18 19 20 % S u cc in yl a tio n Ara7 Ara19 Ara20 Ara18 C Ara8 100 A5 1205.3 A4 989.4 A6 1421.4 A7 1637.4 A8 1853.5 A9 2069.6 A10 2285.6 A11 2501.8 A12 2717.8 A13 2933.7 A14 3149.8 A17 3797.8 A18 4014.2 A20 4446.3 A21 4662.5 900 1300 1700 2100 2500 2900 3300 3700 4100 4500 80 60 40 20 0 A16 3582.2 A Relat ive inte nsit y A15 3365.8 A19 4230.2 * * *
FIGURE 5. Location of the succinyl groups in mycobacterial arabinan by mass spectrometry. A, full mass spectrum of the non-galactosaminylated arabinan enzyme-released fragments from M. tuberculosis mAGP after per-O-acetylation. The succinylated version of each fragment is indicated with arrows. The peaks marked with asterisks are of unknown origin. B, plot of % succinylation (as revealed by the M⫹Na⫹ions of the non-succinylated oligo-arabinan enzyme products and the non-succinylated versions at 58 amu higher m/z values) for Ara7– 8and Ara18 –20. The different degree of succinylation of the two classes of oligo-arabinosides combined with the NMR data shows that the major succinyl group is on the interior␣-3,5-branched arabinosyl residue. C, major (dark square) and minor (gray circles) positions of succinylation revealed by mass spectrometry and NMR analyses together (see text for further information).
at National Taiwan University on June 19, 2009
www.jbc.org
lished; what is clear is that all three modifications occur on the non-mycolylated arabinan chains, and succinylation of the mycolylated chains is diminished or absent.
Ramifications of the Mycolyl Arabinogalactan Primary Structure—Although the model of the primary structure of mycolyl arabinogalactan is presented as a primary structural model (analogous to an amino acid sequence) rather than a tertiary model (analogous to a protein structure), the bond lengths of the carbohydrate and the mycolic acids are approxi-mately at the same scale. This results in the realization that the “periplasmic” space between the mycolate layer and the pepti-doglycan can be fairly sizeable, depending on the conforma-tion(s) adapted by the polysaccharide. It also results in the rec-ognition of the hydrophilicity of the “periplasmic” space with the long galactan “tail” (Fig. 7) and extensive arabinan polymer. It seems likely that the charged groups, both negative and itive, affect the environment of the periplasmic space. The pos-sibility has previously been raised that the protonated GalNH2
(i.e. GalNH3⫹) interacts with anionic
substances such as phosphates of glycolipid anchors like those of lipoarabinomannan (LAM) (19). It is also possible that the GalNH3⫹
interacts with the negatively
charged succinyl residues leading to a more rigid and tightened AG structure. It is fairly certain that some aspects of cell wall biosynthe-sis occur in this region, and it seems likely that other important biologi-cal events occur in this region as well. Finally, the size of the AG poly-mer and its furanose nature allow considerable flexibility between the mycolate and peptidoglycan layer as has been previously pointed out (30), but would nevertheless keep the two layers connected within cer-tain spatial limits.
Comparison of the Arabinan Structure of AG with the Arabinan Structure of LAM—In addition to the cell wall core polysaccharide, AG, mycobacteria produce a cell wall-associated polysaccharide, LAM. The arabinan of LAM is very similar to that of AG in that the same linkages of Araf units are found and the same hepta-arabino-side non-reducing end as shown in Fig. 1A. However it has been known for some time that the non-reduc-ing end can also be a
penta-arabino-side, where the disaccharide
attached at O3 of the branched res-idue of the hepta-arabinoside is missing (31). Also the non-reducing end of the LAM arabinan is substi-tuted with␣-mannosyl residues rather than with mycolic acids (32). However, using the same M. smegmatis endogenous ara-binan, we have recently shown that even the interior regions of the LAM arabinan are fundamentally different than that of the AG arabinan (18). In LAM, the precise Ara18arrangement (Fig. 1) is not always present, but rather chains of various lengths extend from interior 3,5-Araf residues (18). Such differences may well reflect the function of the two molecules. Thus, in the case of AG, it is reasonable that the number of glycosyl residues separating the peptidoglycan and mycolate layers would be rea-sonably constant in order to form the distinct two-layered cell wall core. In contrast, the arabinan region of LAM is expected to be part of the extracellular milieu (with the lipid anchor likely in the mycolic acids layer), in which case precise arabinan lengths and branching would not be required or perhaps even desirable.
Biosynthetic Considerations—The complete definition of the arabinan structure of AG is likely to be an aid to deciphering the
6200 5400 A20GalNH2 4734 A 21GalNH2 4950 A22GalNH2 5166 A23GalNH2 5382 A24GalNH2 5598 A25GalNH2 5814 A26GalNH2 6030 A27GalNH2 6246 A28GalNH2 6462 A29GalNH2 6678 A30GalNH2 6894 5600 5800 6000 50 75 25 100 Relat ive intensit y 6400 6600 6800 5200 5000 A m/z 1/7x 4000 4500 5000 5500 6000 6500 100 0 25 50 75 A18 4014.2 A19 4230.2 A20 4446.3 A21 4662.7 A22 4878.1 A23 5094.1 A24 5310.1 A25 5526.6 A26 5742.7 A 27 5958.6 A28 6174.4 A29 6390.8 A30 6606.9 Rela ti ve intensit y B m/z 1/5x 6200 5400 A20GalNH2 4734 A 21GalNH2 4950 A22GalNH2 5166 A23GalNH2 5382 A24GalNH2 5598 A25GalNH2 5814 A26GalNH2 6030 A27GalNH2 6246 A28GalNH2 6462 A29GalNH2 6678 A30GalNH2 6894 5600 5800 6000 50 75 25 100 Relat ive intensit y 6400 6600 6800 5200 5000 m/z 1/7x 4000 4500 5000 5500 6000 6500 100 0 25 50 75 A18 4014.2 A19 4230.2 A20 4446.3 A21 4662.7 A22 4878.1 A23 5094.1 A24 5310.1 A25 5526.6 A26 5742.7 A 27 5958.6 A28 6174.4 A29 6390.8 A30 6606.9 Rela ti ve intensit y B m/z 1/5x
FIGURE 6. The MALDI-TOF mass spectrum of the higher molecular weight arabinan fragments released
from M. tuberculosis mycolyl arabinogalactan. A, galactosaminylated fraction, which eluted first on the
Biogel P2 column. B, non-galactosaminylated fraction which eluted later on the Biogel P2 column. The series of Ara20GalNH2to Ara30GalNH2is readily apparent in A and Ara18-Ara30in B; the succinylated oligo-arabinans in B are also apparent (not marked).
at National Taiwan University on June 19, 2009
www.jbc.org
O O O H O H O H O O O H O H O O H O O H O H O H O O O H O H O O O H O H O O H O O H O H O O H O H O O H O O H O H O O H O O H O H O O OH O H O H O O H O O H O H O O H O N H H O O H O P O -O O O O H H O O O H O O H H O O O H O O H H O O O O O H O H O O H H O O H O H O O O H O H O O H O O H O H O O H O O O H O H O O H O O H O H O O H O O H O O H O O O H OH N H O O O N H O O O O H O H N H O H O O O H O H O H O O O H O H O O H O H O O H H O O O O H H O O O H H O O O O H H O O O O H H O O O O H H O O O O O H H O O O O H H O O O OH H O O O O H H O O O H O O O H H O O O O H H O O O O H H O O O H O H O O O H H O O O O H H O O O H H O O O H H O O O O H H O H O O O O O H H O O H O O O O H H O O O H O O O H O O O H O O O H O H O O O H O O O O H O O O H O O OH O O H H O O O O H O O H O O H H O O O O H H O O O H H O O O H H O O OH H O O O H H O O O H H O O O H O O H H O O O O O O O H H O O O O H H O O O H H O O O O H H O O O O H H O O O O H H O O O O H O O O O O H H O O O O H O O O O O H H O O O OH O O O H O O O O H H O O O O H O O O O O H O O O H O H O O O H O O H H O OH H O O O O H H O O O O O O O O H H O O O O H H O O O H H O O O O H H O O O O H H O O O O H H O O O O H H O O O O OH O O H O O H O O O H H O H O O O H O O H O O O H H O H O O O H O O O H O O H O O O H H O H O O O H O O H O O O H H O H O O O H H O O O O H H O O OH H O O O O H H O O O O H H O O O O H H O O O O O H H O O O O H H O O O O H H O O O O H H O O O H H O O O O H H O O O O H O O O O H H O O O H O O H H O O O H O O O H H O O O O O H O O O O O H H O O O H O O O H O O O O O H O O H O O O H H O O H O H O O O O O O O O H O O O H O O H O O H O O O O H O O O O H H3C O O O H O C H3 O O H O O H O C H3 O OH O C H3 O O H O O H O O H O O O H O O H O O H O C H3 O O H O O H O C H3 Arabinan (D-Araf) Mycolic Acids Galactan (D-galf)
Linker (L-Rhap-D-GlcNAc-PO4-)
Peptidoglycan O O H O
Succinyl
O N H2 H O O H H O D-GalpNH2FIGURE 7. The structure of the mycolyl arabinogalactan and its attachment to peptidoglycan. The key features include the 31-unit long arabinan (red) of which two-thirds are substituted with mycolyl residues, one-third with succinyl and one-third with glucosaminosyl residues. Three chains are present per galactan (blue) chain. The GalNH2and the succinyl residues are shown in green. The remaining features and ambiguities of the structure are presented under “Discussion.”
at National Taiwan University on June 19, 2009
www.jbc.org
various arabinosyl transferases required for its synthesis. At present, three relevant arabinosyl transferases are known. The first is AftA, which adds a single arabinosyl residue to the galac-tan (15). Second is EmbB (and/or EmbA or an EmbA/EmbB dimer), which most likely is the␣-1,5-arabinosyl transferase that extends the arabinan from the single arabinosyl residue added by AftA (16, 18, 20). The Emb protein(s) thus may be responsible for the synthesis of the entire linear interior arabi-nan portion the structure of which was elucidated herein. How-ever, changes in Emb proteins also affect the structure of the arabinan at the non-reducing end in ways that are not yet understood (33). The third arabinosyl transferase AftB is a -Araf transferase responsible for the -Araf units at the non-reducing end of the AG (34). The structure immediately asks the question of how the interior chain lengths between various motifs are controlled. The C-terminal of the EmbB proteins may have a role in that processes for the interior␣-1,5-arabinan region. In addition other, yet to be identified, enzymes are clearly required for AG synthesis. Thus, a branching enzyme that puts a␣-Araf on position 3 of the 15th Araf unit from the reducing end (Fig. 7) is required, and further enzymes (in addi-tion to AftB) that precisely form the non-reducing structure are also necessary.
The observation suggesting a lack of succinylation on the mycolylated arabinan chains suggests that succinylation nega-tively controls mycolylation. However, the succinyl group is rather far from the site of mycolylation, and thus this possibility needs to be considered with care. In fact, the opposite might be true in that succinylation might follow mycolylation. Thus fas-cinating biosynthetic questions involving control of the length of␣-1,5 arabinan regions, the identification of specific arabino-syl transferases, and the control of mycolylation remain to be elucidated.
REFERENCES
1. Kanetsuna, F. (1968) Biochim. Biophys. Acta 158, 130 –143
2. Wietzerbin, J., Das, B. C., Petit, J. F., Lederer, E., Leyh Bouille, M., and Ghuysen, J. M. (1974) Biochemistry 13, 3471–3476
3. Mahapatra, S., Yagi, T., Belisle, J. T., Espinosa, B. J., Hill, P. J., McNeil, M. R., Brennan, P. J., and Crick, D. C. (2005) J. Bacteriol. 187, 2747–2757 4. Dmitriev, B. A., Ehlers, S., Rietschel, E. T., and Brennan, P. J. (2000) Int.
J. Med. Microbiol. 290,251–258
5. Minnikin, D. E. (1982) in The Biology of the Mycobacteria (Ratledge, C., and Stanford, J. L., eds) Vol. I, pp. 95–184, Academic Press, London 6. Azuma, I., Yamamura, Y., and Misaki, A. (1969) J. Bacteriol. 98, 331–333 7. Amar-Nagasch, C., and Vilkas, E. (1970) Bull. Soc. Chim. Biol. 52, 145–151 8. Misaki, A., Seto, N., and Azuma, I. (1974) J. Biochem. 76, 15–22 9. Vilkas, E., Amar, C., Markovits, J., Vliegenthart, J. G. F., and Kamerling,
J. P. (1973) Biochim. Biophys. Acta 297, 423– 435
10. Kotani, S., Kato, T., Kato, K., Matsuda, T., and Misaki, A. (1971) Biken J.
14,379 –387
11. Daffe, M., Brennan, P. J., and McNeil, M. (1990) J. Biol. Chem. 265, 6734 – 6743
12. McNeil, M., Daffe, M., and Brennan, P. J. (1990) J. Biol. Chem. 265, 18200 –18206
13. McNeil, M., Daffe, M., and Brennan, P. J. (1991) J. Biol. Chem. 266, 13217–13223
14. Besra, G. S., Khoo, K.-H., McNeil, M., Dell, A., Morris, H. R., and Brennan, P. J. (1995) Biochemistry 34, 4257– 4266
15. Alderwick, L. J., Seidel, M., Sahm, H., Besra, G. S., and Eggeling, L. (2006) J. Biol. Chem. 281,15653–15661
16. Alderwick, L. J., Radmacher, E., Seidel, M., Gande, R., Hitchen, P. G., Morris, H. R., Dell, A., Sahm, H., Eggeling, L., and Besra, G. S. (2005) J. Biol. Chem. 280,32362–32371
17. Lee, A., Wu, S. W., Scherman, M. S., Torrelles, J. B., Chatterjee, D., Mc-Neil, M. R., and Khoo, K. H. (2006) Biochemistry 45, 15817–15828 18. Shi, L. B., Berg, S., Lee, A., Spencer, J. S., Zhang, J., Vissa, V., McNeil, M. R.,
Khoo, K. H., and Chatterjee, D. (2006) J. Biol. Chem. 281, 19512–19526 19. Draper, P., Khoo, K. H., Chatterjee, D., Dell, A., and Morris, H. R. (1997)
Biochem. J. 327,519 –525
20. Zhang, N., Torrelles, J. B., McNeil, M. R., Escuyer, V. E., Khoo, K. H., Brennan, P. J., and Chatterjee, D. (2003) Mol. Microbiol. 50, 69 –76 21. Dong, X., Bhamidi, S., Scherman, M., Xin, Y., and McNeil, M. R. (2006)
Appl. Environ. Microbiol. 72,2601–2605
22. Stott, K., Stonehouse, J., Keeler, J., Hwang, T. L., and Shaka, A. J. (1995) J. Am. Chem. Soc. 117,4199 – 4200
23. Torrelles, J. B., Khoo, K. H., Sieling, P. A., Modlin, R. L., Zhang, N. N., Marques, A. M., Treumann, A., Rithner, C. D., Brennan, P. J., and Chat-terjee, D. (2004) J. Biol. Chem. 279, 41227– 41239
24. Delmas, C., Gilleron, M., Brando, T., Vercellone, A., Gheorghiu, M., Riviere, M., and Puzo, G. (1997) Glycobiology 7, 811– 817
25. McNeil, M. R. (1999) in Genetics of Bacterial Polysaccharides (Goldberg, J. B., ed), pp. 207–223, CRC Press, Boca Raton
26. McNeil, M., Besra, G. S., and Brennan, P. J. (1996) in Tuberculosis (Rom, W. N., and Garay, S. M., eds), pp. 171–186, Little, Brown, and Company, Boston
27. McNeil, M., and Brennan, P. J. (1991) Res. Microbiol. 142, 451– 463 28. McNeil, M. R., Robuck, K. G., Harter, M., and Brennan, P. J. (1994)
Gly-cobiology 4,165–174
29. Barry, C. E., III, Lee, R. E., Mdluli, K., Sampson, A. E., Schroeder, B. G., Slayden, R. A., and Yuan, Y. (1998) Prog. Lipid Res. 37, 143–179 30. Liu, J., Barry, C. E., Besra, G. S., and Nikaido, H. (1996) J. Biol. Chem. 271,
29545–29551
31. Chatterjee, D., Bozic, C. M., McNeil, M., and Brennan, P. J. (1991) J. Biol. Chem. 266,9652–9660
32. Chatterjee, D., Lowell, K., Rivoire, B., McNeil, M., and Brennan, P. J. (1992) J. Biol. Chem. 267,6234 – 6239
33. Escuyer, V. E., Lety, M. A., Torrelles, J. B., Khoo, K. H., Tang, J. B., Rithner, C. D., Frehel, C., McNeil, M. R., Brennan, P. J., and Chatterjee, D. (2001) J. Biol. Chem. 276,48854 – 48862
34. Seidel, M., Alderwick, L. J., Birch, H. L., Sahm, H., Eggeling, L., and Besra, G. S. (2007) J. Biol. Chem. 282, 14729 –14740
at National Taiwan University on June 19, 2009
www.jbc.org