國 立 交 通 大 學
生物資訊研究所
碩 士 論 文
使用重疊基因建構原核生物的基因體樹
Reconstructing Genome Trees of Prokaryotes Using
Overlapping Genes
研究生:鄭智先
指導教授:盧錦隆 博士
中華民國 九十八 年 六 月
使用重疊基因建構原核生物的基因體樹
Reconstructing Genome Trees of Prokaryotes Using
Overlapping Genes
研究生:鄭智先
Student:Chih-Hsien Cheng
指導教授:盧錦隆 博士
Advisor:Dr. Chin Lung Lu
國 立 交 通 大 學
生 物 資 訊 研 究 所
碩 士 論 文
A Thesis Submitted to Institute of Bioinformatics College of Biological Science and Technology
National Chiao Tung University in partial Fulfillment of the Requirements for the Degree of Master in
Biological Science and Technology June 2009
Hsinchu, Taiwan, Republic of China
i
使用重疊基因建構原核生物的基因體樹
學生:鄭智先
指導教授:盧錦隆 博士
國立交通大學生物科技系生物資訊碩士班
摘要
重疊基因被定義為在染色體位置相鄰且編碼序列內容會部分或全 部重疊的兩個基因。事實上,重疊基因在微生物的基因體上是非常普遍 的,而且他們比非重疊基因在演化上是更具有保留性。基於上述的特性, 我們之前已發展出一個網路伺服器的工具稱為OGtree,其可以讓使用者 根據兩兩原核生物基因體間的重疊基因距離來建構原核生物的基因體 樹。類似於基因內容與基因次序的研究,我們結合重疊基因內容(即兩 個基因體之間共有的直向同源重疊基因對的平均數)與次序(即兩個基 因體之間平均的重疊基因斷點距離)定義出兩個基因體之間的重疊基因 距離。但在利用斷點距離來定義重疊基因距離時有一個缺點,即無法將 其應用在多染色體的基因體並計算出他們的重疊基因距離。除此之外, 對於某些親緣關係較遠的物種,在他們之間能夠找到的直向同源重疊基 因可能很少,以致於沒有足夠的直向同源重疊基因可適當地衡量出他們 兩兩之間的重疊基因距離。 因此,在這篇論文中,我們定義了一個新的重疊基因距離,它是根 據較有生物正確性的基因重組(例如:翻轉、移位與易位)而不是斷點所ii 定義出來的,而且它能同時應用在單一染色體與多染色體的基因體上。 除此之外,我們也擴展了基因的範圍使之同時包含其編碼序列與調控區, 如此我們可以將兩個鄰近基因發生編碼序列重疊或調控區重疊都視是 一對重疊基因。這是因為不同基因若在調控區域發生重疊現象,或多或 少會影響這些基因的調控。根據上述的改變,我們將OGtree改版為一個 新的網路伺服器叫做OGtree2.0,並且利用二十一條蛋白細菌染色體去 建構其演化樹並用其結果來衡量OGtree2.0的正確性。最後,我們的實 驗結果顯示OGtree2.0的確比之前的版本OGtree以及另一個相似的工具 BPhyOG要來得好,因為OGtree2.0所建構出的演化樹,其蛋白細菌之間 的親緣關係與被生物學家所接受的是參考樹一致的。
iii
Reconstructing Genome Trees of Prokaryotes Using Overlapping
Genes
Student: Chih-Hsien Cheng
Advisor: Dr. Chin Lung Lu
Institute of Bioinformatics
Department of Biological Science and Technology
National Chiao Tung University
ABSTRACT
Overlapping genes (OGs) are defined as adjacent genes whose coding sequences overlap partially or entirely. In fact, they are ubiquitous in microbial genomes and more conserved between species than non-overlapping genes. Based on this property, we have previously implemented a web server, named OGtree, that allows the user to reconstruct genome trees of some prokaryotes according to their pairwise OG distances. By analogy to the analyses of gene content and gene order, the OG distance between two genomes we defined was based on a measure of combining OG content (i.e., the normalized number of shared orthologous OG pairs) and OG order (i.e. the normalized OG breakpoint distance) in their whole genomes. A shortcoming of using the concept of breakpoints to define the OG distance is its inability to analyze the OG distance of multi-chromosomal genomes. In addition, the amount of orthologous
iv
overlapping coding sequences between some distantly related prokaryotic genomes may be limited so that it is hard to find enough orthologous OGs to properly evaluate their pairwise OG distances.
In this study, we therefore define a new OG order distance that is based on more biologically accurate rearrangements (e.g., reversals, transpositions and translocations) rather than breakpoints and that is applicable to both uni-chromosomal and multi-chromosomal genomes. In addition, we expand the term ”gene” to include both its coding sequence and regulatory regions so that two adjacent genes whose coding sequences or regulatory regions overlap with each other are considered as a pair of overlapping genes. This is because overlapping of regulatory regions of distinct genes suggests that the regulation of expression for these genes should be more or less interrelated. Based on these modifications, we have reimplemented our OGtree as a new web server OGtree2.0 and have also evaluated its accuracy of genome tree reconstruction on a testing dataset consisting of 21 Proteobacteria genomes. Our experimental results have finally shown that our current OGtree2.0 indeed outperforms its previous version OGtree, as well as another similar server BPhyOG, significantly in the quality of genome tree reconstruction, because the phylogenetic tree obtained by OGtree2.0 is greatly congruent with the reference tree that coincides with the taxonomy accepted by biologists for these Proteobacteria.
v
誌謝
首先要感謝我們的家人跟女朋友,有你們不斷的支持與鼓勵,我才 能完成碩士學位。 感謝學姊在研究上給了我很多建議,常常提供各種優惠打折的消息。 謝謝明原學長在程式方面的幫忙,祝你與小護士進展順利。感謝研究夥 伴忠翰,藉由跟你頻繁地討論,研究主題才能慢慢成形。特別感謝你在 程式上給予我的協助,還有敬佩你願意留下來勇氣。感謝志偉、慶恩在 生活上的相互幫忙,你們殲滅僵屍的勇氣值得我學習。謝謝學妹芸蓁傳 承我的教育事業,孩子的教育不能等,下一棒就交給妳接下去了!謝謝學 弟昆澤常常陪我打球聊”智先話題”。還有給在魔獸世界裡的晟宸,希 望你早日回頭。最後,要謝謝指導教授盧錦隆老師。在您的指導之下,對於研究工 作有深刻的體會。很感謝您對於我報考博士班時的協助,也許結果不如 預期,但是您的幫忙,智先永記於心。恭喜您升等為教授,期待您升格 當丈夫的那天!
vi
Contents
Chinese Absract ... i ABSTRACT ... iii Acknowledgement ... v Contents…...………...viList of Figures ... vii
1 Introduction ... 1
2 Preliminaries ... 5
2.1 Overlapping genes ... 5
2.2 Orthologous Genes and Horizontal Gene Transfer ... 6
2.3 Bidirectional Best Hit ... 8
2.4 INPARANOID ... 9 2.5 Rearrangement Distance ...10 3 Methods ...12 3.1 Overlapping-Gene Distance ...12 3.2 Algorithm ...13 4 Implementation ...17 4.1 Input of OGtree2.0 ...17 4.2 Output of OGtree2.0 ...20
5 Result and Discussion ...23
6 Conclusion ...34
vii
List of Figures
1.1 The region of a gene is expanded to include both of its coding sequence and regulatory regions………...4 2.1 Genes A and B and C and D are overlapping genes, however A and B
are partially overlap and C and D are completely overlap…..………..6 2.2 Genes A1 and A2 are said to be paralogous genes if they are derived from a duplication event. Genes A1x and A1y are called orthologous genes if they are derived from a speciation event……….…....7 2.3 Effect of HGT on orthology. Gene XB in species B is acquired by HGT from gene XC in species C………...8 2.4 Gene a in genome Gi and gene b in genome Gj is called bidirectional
best-hit, if gene a is the most similar to gene b than any other gene in genome Gj, and vice versa………...9
3.1 The flowchart of our method…...……….17 4.1 OGtree2.0 web interface…...…...……….…..18 4.2 An example of OG distance matrix for 21 γ-Proteobacteria………...21 5.1 Phyolgenetic tree obtained from a trimmed alignment of 60
concatenated homologous proteins using maximum likelihood method, which was adapted from [18].………..………...25 5.2 Phylogenetic tree obtained from 16s rRNAs using the neighbor
joining method……….………..….26 5.3 Phylogenetic tree constructed using BPhyOG [6, 7]………..27 5.4 Genome tree obtained using OGtree with UPGMA method
[13]………..…28 5.5 Genome tree obtained using OGtree2 with NJ method.………...…..31 5.6 Genome tree obtained using OGtree2 with FM method.………31
viii
5.7 Genome tree obtained using OGtree2 with UPGMA method.………32
Chapter 1
Introduction
With the emergence of high-throughput sequencing techniques in the past decade, the amount of fully sequenced genomes from prokaryotes has increased enormously. The increasing availability of such complete prokaryotic genomes enables researchers to reconstruct their genome trees based on the whole genomic information of organisms rather than based on individual genes or a small number of genes. In addition to sequence-based phylogenomic approaches, methods based on whole genomes, like those based on gene content (i.e., the presence and absence of genes) [1,2] and gene orders (i.e., the presence and absence of gene pairs) [3–5], can be used to construct more precise and robust phylogenetic trees that are less influenced by anomalous events. As was pointed out in [6, 7], however, the genome trees constructed only based on gene content or gene order may not be suitable for microbial genomes, because gene content (respectively, gene order) might have changed too little (respectively, too much) for biologists to perform adequate analyses of evolutionary distances between closely (respectively, distantly) related genomes. More recently, to address these problems, Luo et al. [6, 7] have proposed an alternative way to reconstruct genome trees of bacteria based on the presence and absence of overlapping genes in their complete genomes.
The overlapping genes (OGs), defined as adjacent genes whose coding sequences partially or entirely overlap, are ubiquitous in microbial genomes. It has been observed that approximately a third of all genes in all the microbial genomes sequenced to date are overlapping and there is a strong relationship between the total number of genes and the number of OGs [8, 9]. In addition, OGs are more conserved between species than non-overlapping genes [10–12], because a mutation in the overlapping region causes changes in both genes and therefore natural selection against such mutations should be stronger. Based on these properties, Luo
et al. [6, 7] have reported that overlapping genes can serve as better
phylogenetic characters than non-overlapping genes for reconstructing the evolutionary relationships among bacterial genomes.
For the phylogenetic reconstruction of bacterial genomes, Luo et al. [6] defined the orthologous overlapping gene pairs between two different genomes to be pairs of genes that overlap in one genome and have orthologous counterparts that overlap in the other genome. In an analogous method to that used in the analysis of gene content, they defined a new distance measure between two genomes based on the normalized number of their shared orthologous OG pairs. Based on this definition, they utilized current distance-based approaches of building tree, such as Neighbor-Joining (NJ) and Unweighted Pair-Group Method using Arithmetic Averages (UPGMA), to construct the genome trees of many completely sequenced bacterial genomes. In addition, Luo et al. [7] have further maintained an database server, called BPhyOG (http://cmb.bnu.edu.cn/BPhyOG/), which allows the user to browse the genome trees of some bacterial genomes that were calculated in advance on the basis of shared orthologous OG pairs. However, their genome trees are not greatly consistent with those produced by traditional phylogenetic approaches based on 16S rRNAs and concatenation of multiple proteins.
It has been widely accepted that during evolutionary process, species genomes are subject to genome rearrangements, such as reversals, transpositions and translocations, that alter the order and orientation of genes on the genomes, inevitably leading to that the orders of orthologous genes, as well as the ones of orthologous OG pairs certainly, even between two closely related species may not be conserved. This suggests that not only OG content but also orthologous OG order should be considered to reconstruct the genome trees of prokaryotic species using OGs. For this purpose, we have defined the overlapping-gene distance between two genomes based on a measure of combining OG content (i.e., the presence and absence of OGs) and OG order (i.e., the presence and absence of orthologous OG pairs) in their whole genomes [13]. We have also developed a web server named OGtree (http://140.113.239.131/OGtree/) for reconstructing the genome tree of prokaryotic genomes according to their pairwise OG distances. Our experimental results for a set of closely related Proteobacteria showed that our OGtree outperformed BPhyOG in the quality of reconstruction of their genome trees.
In this study, we further improve the accuracy of our OGtree by extending the genes retrieved from their complete genomes to include their regulatory regions and redefining the distance measure between two orthologous OG orders using genome rearrangements rather than breakpoints. The reasons for above extension and replacement are described as follows. For some distantly related prokaryotic genomes, the amount of their overlapping coding sequences is limited so that it is hard to find enough orthologous OGs to properly evaluate their pairwise OG distances and reconstruct an accurate and robust genome tree for these species. Actually, the term ”gene” defined in modern genomics should include not only its coding region, but also its regulatory regions, such as
promoter (at the 5’ upstream end of the coding region) and terminator (at the 3’ downstream end of the coding region) [14]. In addition, overlapping of regulatory regions of distinct genes should be of certain interest, because the regulation of expression for these genes is more or less interrelated [15]. In this study, therefore, we expand the region of a gene to include both its coding sequence and regulatory regions so that two adjacent genes whose coding sequences or regulatory regions overlap with each other are considered as a pair of overlapping genes. On the other hand, the orders of orthologous OG pairs between two prokaryotic genomes, as mentioned above, are often different due to genome rearrangements. The distance measure between two orthologous OG orders we previously defined was analogous to the breakpoint distance between two gene orders, which has been widely used as a rough measure of genomic distance [3]. In contrast to the genome rearrangement distance, however, the breakpoint distance does not correspond to an optimal series of events that accounts for the rearrangements of one genome with respect to another. In addition, it is still not clear how to adapt the breakpoint analysis to multi-chromosomal genomes [16]. In this study, therefore, we try to use the genome rearrangement distance involved with reversals, block-interchanges (i.e., generalized transpositions) and translocations [17] to re-define the distance of the orthologous OG orders between two prokaryotic genomes.
Figure 1.1: The region of a gene is expanded to include both of its coding
sequence and regulatory regions.
5’ 3’
DNA Promoter 5’UTR Coding region 3’UTR
Chapter 2
Preliminaries
In this chapter, we shall first introduce basic concept of overlapping genes, orthologous genes, horizontal gene transfer and rearrangement distance. We shall then describe two approaches, BBH and INPARANOID, for identifying putative orthologous genes.
2.1
Overlapping genes
The overlapping genes (OGs) are defined as adjacent genes whose coding sequences partially or entirely overlap, as shown in Figure 2.1 for an example. OGs are ubiquitous in microbial genomes, because approximately a third of all genes in all the microbial genomes sequenced to date are overlapping [8, 9]. In fact, there is a strong relationship between the total number of genes and the number of overlapping genes [8, 9]. In addition, it has been reported that OGs are more conserved between species than non-overlapping genes [10–12], because a mutation in the overlapping region causes changes in both genes and therefore natural selection against such mutations should be stronger. All these properties above may suggest that overlapping genes can serve as better phylogenetic characters than non-overlapping genes for reconstructing the
evolutionary relationships among bacterial genomes.
Figure 2.1: Genes A and B and genes C and D are two pairs of
overlapping genes, where A and B overlap partially and C and D overlap completely.
2.2 Orthologous Genes and Horizontal Gene
Transfer
Basically, orthologous genes are in different species that derived from a single gene in the last common ancestor of these species. By contrast, paralogous genes are duplicated within a genome. In general, orhologous genes have the same functions in the respective organisms; however, the biological functions of paralogous genes are distinct.
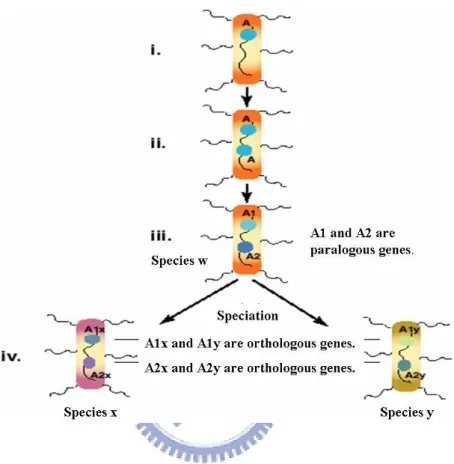
Figure 2.2 shows evolutionary process illustrating orhologous genes and paralogous genes relationships. (i) Initially, there is a gene called A in species w. (ii) Gene A is duplicated by producing two copies of A in the same chromosome. (iii) After that, the two copies diverge by evolution, forming related genes A1 and A2. Therefore, these two genes are called paralogous genes. (iv) Two species x and y evolve from species w, called last common ancestor of x and y, due to speciation event. The descendants of the gene A1 are denoted by A1x and A1y, and the
5’ DNA sequence
C D
A B
descendants of the A2 by A2x and A2y. Therefore, genes A1x and A1y are orthologous genes and genes A2x and A2y are also orthologous genes.
Figure 2.2: Genes A1 and A2 are said to be paralogous genes if they are
derived from a duplication event. Genes A1x and A1y are called orthologous genes if they are derived from a speciation event.
With a rapid enrichment of genome sequences, how to identify orthologous genes between different genomes becomes an important task. The simple assumption is that the sequences of orthologous genes should be more similar to each other than with any other genes in compared genomes. In the following sections, we shall introduce two methods bidirectional best hit and INPARANOID, for identifying the orthologous genes with inparalogs between two give genomes.
Horizontal gene transfer (HGT), the transfer of genes between different species, is recognized as one of the major forces in prokaryotic genome evolution [23]. It was reported that HGT might cause a problem
in the determination of orthologous and paralogous relationships [22]. For example, as shown in Figure 2.3, species A and B may have homologous genes XA and XB, where in fact gene XA is vertically derived from the ancestor, but gene XB has been acquired via HGT from an outside species C. In a careless analysis (e.g., using BBH method, which will be introduced later), XA and XB would be considered as orthologs. However, these two genes are not orthologs by definition, because they do not come from an ancestral gene in the last common ancestor of the compared species. In prokaryotic genomes, such confusion caused by HGT is very common.
Figure 2.3: Effect of HGT on orthology. Gene XB in species B is
acquired by HGT from gene XC in species C.
2.3
Bidirectional Best Hit
A simple method, called the bidirectional best hit (BBH), for prediction of orthologous genes in two organisms is to search for a pair of sequences by performing a BLAST. BBH is defined to be a pair of genes
a and b from two genomes Gi and Gj such that b is the best hit (i.e., most
similar gene) when a is compared against all genes of Gj , and vice versa
(see Figure 2.4 for illustration). It has been evidenced that such a BBH approach of identifying putative orthologs works reasonably well for bacterial genomes [15].
Figure 2.4: Gene a in genome Gi and gene b in genome Gj form a BBH,
if gene a is the most similar to gene b than any other gene in genome Gj,
and vice versa.
2.4
INPARANOID
Remm et al. [18] have developed a program, called INPARANOID (http://www.cbg.ki.se/inparanoid/) , for finding orthologs with inparalogs from two species genomes, based on the following steps.
Given two species genomes, the first step of INPARANOID is to run BLAST search between all pairs of gene sequences. Consequently, the pairs with similarity scores above the predefined threshold are reserved for further analyses on the next step.
Next, INPARANOID continues to find two-way best hits (i.e., BBH) as potential orthologs and further include inparalogs to form putative orthologous groups, based on the idea that the main ortholog has more similarity to inparalogs from the same species than to any sequence from
G
iG
jBBH
gene a
another species.
Third, INPARANOID applies a clustering algorithm to all the putative orthologous groups as follows:
(1) Merge two orthologous groups if the symmetric best orthologous genes are already clustered in the same group.
(2) Merge two orthologous groups if a main orthologous gene in one genome has equally best hit to two orthologous genes in the other genome.
(3) Delete a new group if one of the orthologous genes already belongs to a much stronger (i.e., high similarity) group.
(4) Merge two groups if one gene of the orthologous gene pair has a high similarity in another group.
(5) All other overlapping groups of inparalogs are separated based on their similarity to the orthologous gene.
Finally, the confidence values of a set of orthologous groups are calculated to estimate the reliability of each group (for details, we refer the reader to [18]).
2.5 Rearrangement distance
Genome rearrangement studies based on genome analysis of gene orders play an important role in the phylogenetic tree reconstruction. In the studies of genome rearrangements, a gene is usually represented by a signed integer, where the associated sign indicates its transcriptional orientation. Given two genomes of the same (orthologous) genes, the
genome rearrangement problem aims to compute a minimum sequence of
rearrangement operations required to transform one genome into another. The rearrangement events within genomes with single chromosomes include reversals, transpositions and block-interchanges, where reversals, also called inversions, affect a block of consecutive integers in the
chromosome by reversing the order and flipping the signs of the integers;
transpositions affect two adjacent blocks in the chromosome by
exchanging their positions; block-interchanges are generalized transpositions by allowing the exchanged blocks not being adjacent in the
chromosome. In genomes with multiple chromosomes, the rearrangement operations include translocations, fusions and fissions, where
translocations exchange the end segments between two chromosomes; fusions join two chromosomes into a bigger one; fissions break a
Chapter 3
Methods
In this chapter, we shall first introduce overlapping-gene distance, and then present our algorithm for construction of genome trees based on the overlapping-gene distance between species whole genomes.
3.1
Overlapping-Gene Distance
As used in the studies of genome rearrangements, we utilize a signed integer to represent a gene encoded in a chromosome, with its sign indicating the transcriptional orientation of the corresponding gene (e.g.,”+” stands for 5’ → 3’ and”−” stands for 3’ ← 5’). Moreover, we use a pair of signed integers ( x, y ) to represent an OG of x and y. Basically, there are three possible overlapping types (or structures / directions) of OGs [11, 13]: (1) unidirectional OGs with sign (+, +) or (−,−), that is, the 3’ end of one gene overlaps with the 5’ end of the other, (2) convergent OGs with sign (+,−), that is, the 3’ ends of the two genes overlap, and (3) divergent OGs with sign (−, +), that is, the 5’ ends of the two genes overlap. It has been reported that in prokaryotic genomes unidirectional OGs are most widespread, convergent OGs are less
common, and divergent OGs are rare [8, 9, 13].
For our purpose, the orthologous OG pairs we considered here are further restricted to those orthologous OG pairs with the same (i.e., conserved) overlapping structures. Suppose that there are totally n orthologous OG pairs between Gi and Gj. Then we define the
overlapping-gene distance Di,j between Gi and Gj as follows.
In the above formula, ri,j denotes the genome rearrangement distance
between Gi and Gi using reversals, block-interchanges (i.e., generalized
transpositions) and translocations (including fusions and fissions), which can be computed in polynomial time when block-interchanges are weighted 2 and the others are weighted 1 [17], and xi and xj denote the numbers of total OGs in Gi and Gj , respectively. Basically, Di,j
evaluates the distance between Gi and Gj by considering the orthologous
OG order measure as defined in the first term and the OG content measure as defined in the second term. Then wo and wc can be considered
as the weight of orthologous OG order and the weight of OG content, respectively, where both of their defaults are 1’s in our OGtree2.
3.2
Algorithm
Figure 3.1 shows the flowchart of our algorithm for constructing the genome tree of prokaryotes based on overlapping-gene distance.
Given the accession numbers of several species, the first step is to download complete genomes from the National Centre for Biotechnology
ln
1
ln(
/
2
)
j i c ij ox
n
x
n
w
n
r
w
Dis
Information (NCBI) according to the accession numbers specified by the user. The putative genes are then extracted from each of these genomes on the basis of the coding sequence (CDS) annotation. However, it is inevitable that some of these putative genes may be misannotated in each genome downloaded from the NCBI. We may therefore exclude those genes that were annotated as being unknown, hypothetical or putative for a stringent analysis. In addition, horizontal gene transfer (HGT), the transfer of genes between different species, has been reported to be very common in prokaryotes [19]. It may obscure the OG pairs with which we hope to reconstruct the genome tree of prokaryotes. Hence, we offer an additional option in our OGtree2.0 to remove those genes that were annotated as horizontally transferred genes at the HGT-DB database [19], where HGT-DB currently provides the lists of putative horizontally transferred genes for a large number of prokaryotic complete genomes.
Next, we use BLASTP program to determine putative orthologous genes between two genomes by using bidirectional best hit (BBH) approach. A BBH is defined to be a pair of genes a and b from two genomes
Gi and Gj such that b is the best hit (i.e., most similar gene) when a is
compared against all genes of Gj , and vice versa. It has been evidenced
that such a BBH approach of identifying putative orthologs works reasonably well for bacterial genomes [25]. In addition, we use Inparanoid [19] as an alternative to identify putative orthologous genes between any two genomes. It has been demonstrated that Inparanoid is the best among five currently existing methods of automatically detecting orthologous genes [26]. Recall that the term ”gene” defined in this study can be expanded to include not only its coding region but also regulatory regions, such as promoters and transcription terminators. Basically, the promoters of prokaryotes are always located immediately upstream of the transcription start site (TSS), the TSSs are located upstream of the start
codon, and the transcription terminators are located downstream of the stop codon. In this case, the CDSs of genes are further extended at their 5’ and 3’ ends to their regulatory promoter and terminator regions. Then two adjacent genes in each genome are identified as overlapping genes (OGs), or an OG pair, if their CDSs (or extended CDSs) overlap partially or completely. Two OGs, say (a, c) and (b, d), from different genomes are then considered as an orthologous OG pair if a and b, as well as c and d, are orthologous to each other, and (a, c) and (b, d) have the same directional pattern.
Finally, for any two genomes Gi and Gj , we compute their OG
distance Di,j on basis of their OG pairs. Then we apply distance-based
approaches of building trees, such as UPGMA, NJ and FM, to the matrix of overlapping-gene distance between genomes for constructing genome trees of the input prokaryotic genomes.
Based on the algorithm described above, we have implemented a web server named OGtree2.0 (http://bioalgorithm.life.nctu.edu.tw/OGtree2.0/) that allows the user to reconstruct prokaryotic genome trees with overlapping genes retrieved from the prokaryotic genomes.
Figure 3.1:The flowchart of our method for OGtree2.0 Step 1: Download
specified genomes from the NCBI
Step 2: Extract CDSs from
each specified genome
Step 3 (optinal): Remove
those CDSs annotated as unknown, hypothetical or putative genes
Step 4 (optinal): Remove
those CDSs annotated as horizontally transferred genes
Step 5: Identify
orthologous genes between any two specified genomes using BBI or Inparanoid
Step 6 (optinal): Extend
CDSs to include their promoters and
transcription terminators
Step 7: Identify OGs in
each specified genome and orthologous OG pairs between any two
genomes
Step 8: Calculate the OG
distance between any two specified genomes
Step 9: Reconstruct
genome tree using the UPGMA, NJ or FM method
Chapter 4
Implementation
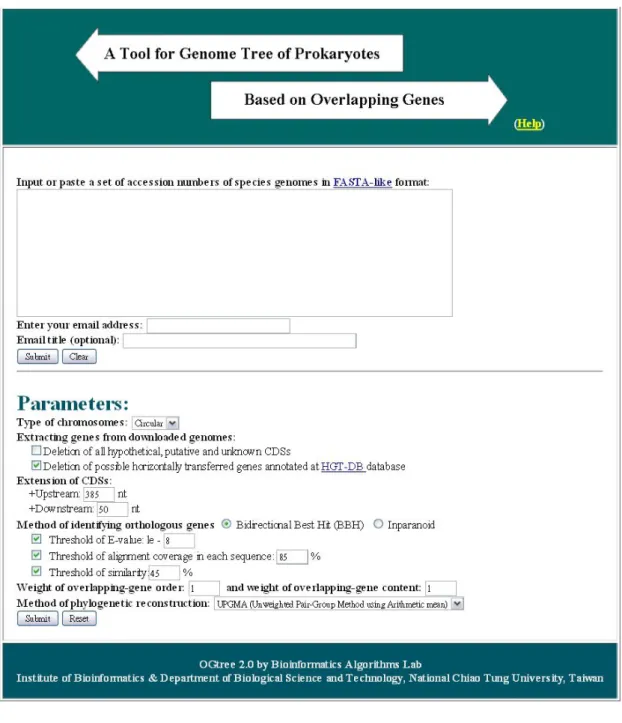
Based on the algorithm we described in the previous chapter, we have implemented a web server called OGtree2.0 (short for genome tree using Overlapping Genes). The kernel programs of OGtree2.0 were written in C and Perl. Its web interface was implemented in PHP. It is available at http://bioalgorithm.life.nctu.edu.tw/OGtree2.0/ for online analysis and can be easily accessed via a simple web interface, as shown in Figure 4.1.
4.1
Input of OGtree2.0
Enter or paste a set of accession numbers of prokaryotic genomes in
FASTA-like format. The so-called FASTA-like format starts with a single-line description beginning with a right angle bracket (">"), followed by a line of accession number of a prokaryotic species. The following is an input example with 3 γ-proteobacterial genomes.
>Ba NC_002528 >Ec NC_000913 >Hi NC_000907
Figure 4.1: OGtree2.0 web interface
Then OGtree2.0 will automatically download the whole genomes of all the specified prokaryotes from the NCBI.
1. Enter an email address in the email box, via which the user will be notified of the result obtained by OGtree2.0 when the submitted job is finished. If necessary, the user can enter a text into the box of email title that will be served as the subject of the returned email later.
2. Just click "Submit" button, if the user would like to run OGtree2.0 with default parameters; otherwise, the user continues with the following parameter settings.
3. Choose the chromosomal type of the input prokaryotic genomes, which currently can be either circular (default) or linear.
4. Check the box that deletion of all hypothetical genes, if the user would like OGtree2.0 to delete all the CDSs whose translated products were annotated as hypothetical, putative and unknown proteins in the NCBI.
5. Check the box that deletion of all horizontally transferred genes, if the user would like OGtree2.0 to delete all the CDS that were annotated as horizontally transferred genes at the
HGT-DB database.
6. Extend the region of CDS by specifying the upstream length at its 5’ end and the downstream length at its 3’end.
7. Choose the method used by OGtree2.0 to identify the orthologous genes between any pair of input genomes. This method can be either bidirectional best hit (BBH) or Inparanoid. In addition, the user can further change the default parameters, if necessary, to control the results of BLASTP for determining the putative orthologous genes. They include threshold of E-value (whose default is 1e-8) and threshold of
alignment coverage in each sequence (whose default is 85%), and threshold of similarity (whose default is 45%).
8. Choose the distance measure of OG distance, which currently can be rearrangement.
9. Specify the method used by OGtree2.0 to reconstruct the genome tree. Currently, it can be either UPGMA (default), NJ or FM.
10. Specify the weight of overlapping gene order (whose default is 1) or specify the weight of overlapping gene content (whose default is 1). Note that both of them can be any real numbers. 11. Click "Submit" button to run OGtree2.0.
4.2
Output of OGtree2.0
In the output page, OGtree2.0 will first show the input genome data and user-defined parameters. Next, it will show the overlapping-gene distance matrix computed according to the downloaded genomes from the NCBI, as was shown in Figure 4.2.
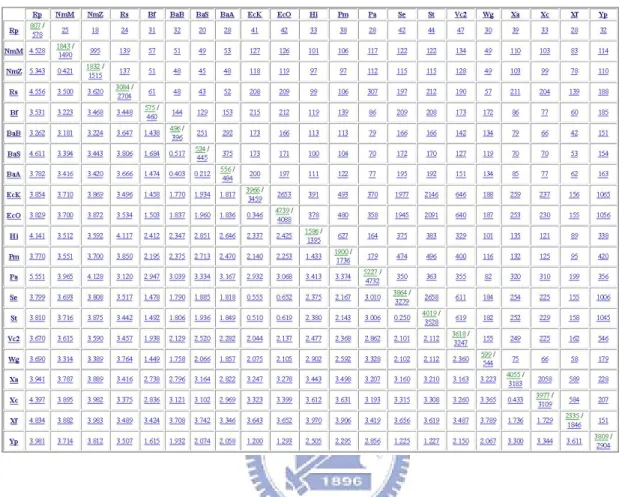
Figure 4.2: An example of OG distance matrix for 21 γ-Proteobacteria.
In each entry of the diagonal, the number of the numerator denotes the number of genes that are extracted from the corresponding genome, or remain in the genome after deleting those genes that were annotated as horizontally transferred genes and/or hypothetical, putative and unknown genes; the number of the denominator denotes the number of OG pairs identified by OGtree2.0 in the corresponding genome. Note that both of numerator and denominator are associated with a link, via which the user can further view the details about all the extracted genes or all the identified OG pairs from each corresponding genome. For example, the numerator link will show the gene ID, protein ID, gene name, locus-tag, start and end positions, and strand for each extracted gene, and the denominator link will display the gene IDs of each GO pair, as well as their overlapping direction.
In the upper triangle, each entry contains an integer denoting the number of identified orthologous OG pairs between the two corresponding genomes. Note that the entry link will show the details of each orthologous OG pair, including its overlapping direction and length, the number of its orthologous OG pairs found in other genomes, as well as the details of its component genes, including gene ID, gene name, location, strand, locus-tag, protein ID and product, COG ID (if have), and translated protein.
In the lower left triangle, each entry denotes the computed overlapping-gene distance between the two corresponding genomes. Note that the user can click the entry link to view the details about the orthologous OG orders in the two corresponding genomes, their rearrangement distnaces, and their overlapping-gene distance.
Finally, OGtree2.0 will show a genome tree according to estimated OG distance between any pair of genomes using UPGMA, NJ or FM method. Note that our OGtree2.0 also provides in the output page with a text file of computed OG distance matrix in the PYLIP format and a text file of constructed genome tree in the Newick format, so that the user can download them for post-processing analysis.
Chapter 5
Result and Discussion
In this study, we have selected 21 genomes of Proteobacteria retrieved from the NCBI as the testing dataset, including R.
prowazekii (abbreviated as Rp, NC_000963), R. solanacearum
(Rs, NC_003295), N. meningitidis MC58(NmM, NC_003112),
N. meningitidis Z2491 (NmZ, NC_003112), E.coli K12 (EcK,
NC_000913), E.coli O157:H7 EDL933 (EcO, NC_002655), S.
enterica subsp. enterica serovar Typhi Ty2 (Se, NC_003198), S.typhimuriu LT2 (St, NC_003197), Y. pestis KIM (Yp,
NC_004088), B. floridanus (Bf, NC_005061), B. aphidicola str.
Bp (BaB, NC_004545), B. aphidicolastr. Sg (BaS, NC_004061),
B. aphidicola str. APS (BaA, NC_002528), W. glossinidia
brevipalpis (Wg, NC_004344), V. cholerae El Tor N1696 (I)(Vc,
NC_002505), V. cholerae El Tor N1696 (II) (Vc, NC_002506),
H. influenzae (Hi, NC_000907), P.aeruginosa (Pa, NC_002516), P. multocida (Pm, NC_002663), X. axonopodis (Xa,
NC_003919), X.campestris (Xc, NC_003902) and X. fastidiosa (Xf, NC_002488). In addition, we used the phylogenetic trees constructed based on concatenated sequences for 60 homologous proteins [18] and 16S rRNAs as reference trees (Figures 5.1 and 5.2) and compared the genome trees obtained by our OGtree2.0 to those
phylogenetic trees predicted by our previous OGtree (Figure 5 . 3) [13] and BPhyOG (Figure 5.4) [6]. Basically, the phylogenetic tree in Figure 1 can be considered as a good reference tree, because it coincides with the taxonomy accepted by biologists for these Proteobacteria. Particularly, the three Buchnera species in this reference tree form a monophyletic group with the other insect endosymbionts of B. floridanus and W. glossinidia. In addition, this group of endosymbionts is a sister clade to the cluster of the other four enterobacteria of Yersinia, Esherichia, Shigella and
Salmonella. However, the phylogenetic tree shown in Figure 5.2 is
slightly differ from that in Figure 5.1 mainly with respect to the positions of the Xanthomonadales group (X. axonopodis, X.
campestris and X. fastidiosa ) and V. cholerae. In this reference
tree of 16S rRNAs, the γ-Proteobacteria of X. axonopodis, X.
campestris and X. fastidiosa were placed in the β-Proteobacteria
branch and the species of V. cholerae was placed a little away from P. aeruginosa.
Figure 5.1: Phylogenetic tree obtained from a trimmed
alignment of 60 concatenated homologous proteins using maximum likelihood method, which was adapted from [18].
Figure 5.2: Phylogenetic tree obtained from 16s rRNAs using
Figure 5.3: Genome tree obtained using OGtree with UPGMA
Figure 5.4: Phylogenetic tree constructed using BPhyOG [6,
7].
Inevitably, some misannotated genes may be included in the genomes of public databases. Therefore, we may exclude those CDSs annotated as being unknown, hypothetical or putative from each downloaded genome in our analysis, as was done in [6]. However, we found that most of the CDSs in W. brevipalpisa are currently annotated as unknown, hypothetical or putative, leading
us to find no orthologous OG pair between W. brevipalpisa and other Proteobacteria, if all these CDSs in W. brevipalpisa are removed from our analysis. Here, instead of this method, we first removed those genes currently annotated as horizontally transferred genes at the HGT-DB database [19] and then applied more stringent criteria of identifying putative orthologous genes by using BBH and setting the parameters with a minimum E-value of 10-8, at least 85% of each authentic CDS sequence involved in the alignment, and a minimum similarity of 45%. In addition, we observed that the amount of the orthologous OG pairs between non-γ-Proteobacteria genomes and other Proteobacteria genomes is few, resulting in difficulty measuring the accurate OG distances between them. Recall that the term ”gene” can be expanded to include both of its coding and regulatory regions, such as promoters and transcription terminators. In prokaryotic genomes, a promoter region, which basically contains the so-called −10 hexamer, extended −10 element, −35 hexamer and UP element, usually occupies about 60 base pairs (bp) upstream of the transcription start site (TSS) [20,21] and a terminator region usually occupies about 50 bp downstream of the stop codon [22]. In addition, as exemplified in
E. coli genome, 95% of TSSs occur 325 bp upstream from the
translation start sites (TLS) of their corresponding genes [23]. According to these information, therefore, we extended the region of each CDS by 385 bp at its 5’ end and by 50 bp at its 3’ end, so that any two adjacent genes in a genome were considered as an OG pair if their extended CDSs partially or completely overlap with each other. With default values for all the other parameters (e.g., the distance of OG order was measured using
rearrangements, instead of breakpoints, wc = 1 and wc = 1), we
used OGtree2 to calculate the OG distance between every pair of Proteobacteria for constructing the genome trees for all the Proteobacteria used in this study with the UPGMA, NJ and FM methods.
Consequently, both the NJ and FM trees (see Figures 5.5 and 5.6, respectively) we obtained using OGtree2 have almost the same tree topology, which differs from the one in the UPGMA tree (see Figure 5.7) with respect to the positions of R.
prowazekii and V. cholerae. In both the NJ and FM trees, the α-Proteobacterium R. prowazekii was placed in the branch of γ-Proteobacteria and V. cholerae was placed as a neighbor (or
sister) of the Pasteurellaceae cluster. As to the UPGMA tree, its topology was greatly congruent with that of the reference tree as shown in Figure 5.1. Particularly, the UPGMA tree clearly and correctly divided the 21 Proteobacteria into three monophyletic clades and it also reflected monophyly not only for the three
Buchnera species but also for a wider group including the other
insect endosymbionts of B. floridanus and W. glossinidia. However, V. cholerae in the UPGMA tree was placed a little away from P. aeruginosa, which is the same as the reference tree of 16S rRNAs in Figure 2. Among the three tree-building methods in this experiment, the UPGMA method produced a genome tree that is much more congruent with the reference tree constructed using a trimmed alignment of 60 concatenated protein sequences, when compared to both the NJ and FM methods. This characteristic may be due to that, as reported in [8, 9], evolution of OGs occurs at a universal mutation rate across bacterial genomes.
Figure 5.5
Genome tree obtained using OGtree2.0 with NJ method.
Figure 5.6
Figure 5.7
Genome tree obtained using OGtree2.0 with UPGMA method.
In the comparison of the phylogenetic tree inferred by BPhyOG (Figure 5.4), our genome tree produced using OGtree2 with the UPGMA method shows more precise and robust phylogenies for the 21 Proteobacteria genomes. In the BPhyOG tree, the relationship of endosymbionts was paraphyletic and particularly the two insect endosymbionts, W. brevipalpis and B.
aphidicola, were separated far away from each other. In addition,
the three β-Proteobacteria were placed just as neighbor taxa rather than a sister cluster. In contrast, W. brevipalpis, B.
aphidicola and other three Buchnera species in our UPGMA tree
(Figure 5.7), as well as in both reference trees (Figures 5.1 and 5.2), were placed as a sister group, suggesting that there should be a common origin for these five endosymbionts. Moreover, our current OGtree2.0 indeed outperformed over its previous version OGtree in phylogeny reconstruction for prokaryotes, because in the genome tree predicted by OGtree (Figure 5.3), the
α-Proteobacteria of R. prowazekii and the β-Proteobacteria of R. solanacearum were placed together as a sister group and the
insect endosymbiont of B. floridanus was placed in the branch of enterobacteria.
Chapter 6
Conclusion
Previously, we have implemented a web server named OGtree2.0 to demonstrate that overlapping genes can be served as a useful genomic marker for reconstructing genome trees of some prokaryotes. In contrast to BPhyOG, the OG distance we defined to measure the difference between two prokaryotic genomes in our OGtree2.0 was based on a combination of their OG content and orthologous OG order.
In this study, we have improved the accuracy of our OGtree2.0 in reconstruction of prokaryotic genome trees by extending the regions of genes to include their regulatory regions and redefining the distance measure between two orthologous OG orders using genome rearrangements rather than breakpoints.
According to our experiments, the genome trees constructed by our OGtree2.0 are quite consistent with those reference trees that were reconstructed based on 16S rRNAs as well as concatenated sequences of 60 homologous proteins, compared with the phylogenetic trees produced by Luo et al. [6, 7] and OGtree2.0. Furthermore, among the tree-building methods in our experiments, the UPGMA method produced much more congruent genome trees compared to both the NJ
and FM methods, if they were based on the OG distance we defined in this study. This characteristic was also pointed out by Luo et al. in their studies [6, 7] only on the basis of the content of OG pairs. It has been reported that evolution of OGs occurs at a universal mutation rate across bacterial genomes [8, 9]. Perhaps due to this property, the UPGMA method is more suitable for the reconstruction of phylogenies particularly based on OG pairs, when compared to the NJ and FM methods. Our experimental results on a set of 21 Proteobacteria have shown that the above modifications indeed helped us to reconstruct a more precise and robust genome tree that coincides with the taxonomy accepted by biologists for these Proteobacteria. This suggests that our current OGtree2.0 can provide interesting insights into the study of evolutionary relationships of completely sequenced prokaryotic genomes.
References
[1] Snel B, Bork P, Huynen MA: Genome phylogeny based on gene content. Nature Genetics 1999, 21:108–110.
[2] Snel B, Huynen MA, Dutilh BE: Genome trees and the nature of genome evolution. Annual Review of Microbiology 2005,
59:191–209.
[3] Blanchette M, Kunisawa T, Sankoff D: Gene order breakpoint evidence in animal mitochondrial phylogeny. Journal of Molecular Evolution 1999, 49:193–203.
[4] Sankoff D: Genome rearrangement with gene families. Bioinformatics 1999, 15:909–917.
[5] Belda E, Moya A, Silva FJ: Genome rearrangement distances and gene order phylogeny in γ-Proteobacteria. Molecular Biology and Evolution 2005, 22:1456–1467.
[6] Luo Y, Fu C, Zhang DY, Lin K: Overlapping genes as rare genomic markers: the phylogeny of γ-Proteobacteria as a case study. Trends in Genetics 2006, 22:593–596.
[7] Luo Y, Fu C, Zhang DY, Lin K: BPhyOG: an interactive server for genome-wide inference of bacterial phylogenies based on overlapping genes. BMC Bioinformatics 2007, 8:266.
[8] Fukuda Y, Nakayama Y, Tomita M: On dynamics of overlapping genes in bacterial genomes. Gene 2003, 323:181–187.
[9] Johnson ZI, Chisholm SW: Properties of overlapping genes are conserved across microbial genomes. Genome Research 2004, 14:2268–2272.
[10] Fukuda Y, Washio T, Tomita M: Comparative study of overlapping genes in the genomes of Mycoplasma genitalium and Mycoplasma pneumoniae. Nucleic Acids Research 1999, 27:1847–1853.
[11] Krakauer DC: Stability and evolution of overlapping genes. Evolution: International Journal of Organic Evolution 2000, 54:731–739.
[12] Sakharkar KR, Sakharkar MK, Verma C, Chow VT: Comparative study of overlapping genes in bacteria, with special reference to Rickettsia prowazekii and Rickettsia conorii . International Journal of Systematic and Evolutionary Microbiology 2005, 55:1205–1209. [13] Jiang LW, Lin KL, Lu CL: OGtree: a tool for creating genome trees
of prokaryotes based on overlapping genes. Nucleic Acids Research 2008, 36:W475–480.
[14] Snyder M, Gerstein M: Defining genes in the genomics era. Science 2003, 300(5617):258–560.
[15] Scherbakov DV, Garber MB: Overlapping genes in bacterial and phage genomes. Molecular Biology 2000, 34:485–495.
[16] Bourque G, Pevzner PA: Genome-scale evolution: reconstructing gene orders in the ancestral species. Genome Research 2002, 12:26–36.
[17] Yancopoulos S, Attie O, Friedberg R: Efficient sorting of genomic permutations by translocation, inversion and block interchange. Bioinformatics 2005, 21:3340–3346.
[18] Koonin E.V. (2005): Orthologs, Paralogs, and Evolutionary Genomics. Annual review of genetics, 39, 309-338.
[19] Koonin E.V., Makarova, K.S. and Aravind, L. (2001): Horizontal gene transfer in prokaryotes: Quantification and Classification. Annual review of microbiology, 55, 709-742.
[20] Remm, M., Storm, C.E. and Sonnhammer, E.L. (2001): Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. Journal of molecular biology, 314, 1041-1052. [21] Comas I, Moya A, Gonzalez-Candelas F: From phylogenetics to
phylogenomics: the evolutionary relationships of insect
endosymbiotic γ-Proteobacteria as a test case. Systematic biology 2007, 56:1–16.
[22] Garcia-Vallve S, Guzman E, Montero MA, Romeu A: HGT-DB: a database of putative horizontally transferred genes in prokaryotic complete genomes. Nucleic Acids Research 2003, 31:187–189. [23] Browning DF, Busby SJW: The regulation of bacterial transcription
initiation. Nature Reviews Microbiology 2004, 2:57–65. [24] Janga SC, Collado-Vides J: Structure and evolution of gene
regulatory networks in microbial genomes. Research in Microbiology 2007, 158:787–794.
[25] Unniraman S, Prakash R, Nagaraja V: Conserved economics of
transcription termination in eubacteria. Nucleic Acids Research 2002, 30:675–684.
[26] Burden S, Lin YX, Zhang R: Improving promoter prediction Improving promoter prediction for the NNPP2.2 algorithm: a case study using Escherichia coli DNA sequences. Bioinformatics 2005,
21:601–607.
[27] Rogozin IB, Spiridonov AN, Sorokin AV, Wolf YI, Jordan IK, Tatusov RL, Koonin EV: Purifying and directional selection in overlapping prokaryotic genes. Trends in Genetics 2002, 18:228–232.
[28] Tatusov RL, Koonin EV, Lipman DJ: A genomic perspective on protein families. Science 1997, 278:631–637.
[29] Hulsen T, Huynen MA, de Vlieg J, Groenen PM: Benchmarking ortholog identification methods using functional genomics data. Genome Biology 2006, 7:4.




![Figure 5.1: Phylogenetic tree obtained from a trimmed alignment of 60 concatenated homologous proteins using maximum likelihood method, which was adapted from [18]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8152115.167133/35.892.199.699.109.780/figure-phylogenetic-obtained-alignment-concatenated-homologous-proteins-likelihood.webp)
![Figure 5.3: Genome tree obtained using OGtree with UPGMA method [13].](https://thumb-ap.123doks.com/thumbv2/9libinfo/8152115.167133/37.892.207.716.115.847/figure-genome-tree-obtained-using-ogtree-upgma-method.webp)
![Figure 5.4: Phylogenetic tree constructed using BPhyOG [6, 7].](https://thumb-ap.123doks.com/thumbv2/9libinfo/8152115.167133/38.892.203.711.114.822/figure-phylogenetic-tree-constructed-using-bphyog.webp)
