國立交通大學
生物科技學系
博士論文
同源蛋白質-蛋白質交互作用之研究
A Study of Homologous Protein-protein
Interactions
研究生:陳俊辰
Chen Chun-Chen
指導教授:楊進木博士
Advisor: Yang Jinn-Moon, Ph. D.
誌謝
此論文能夠完成,俊辰得到了太多的幫助和愛護──師長、父母、朋友──謹藉著 這小小的篇幅,致上我最深切的謝意。 首先最應當深深感謝的,是恩師楊教授進木。是老師將俊辰引領進科學的領域,在 我遇上困難時,您竭心盡力給予支持和指導;您嚴謹的治學態度和寬廣的視野,更令俊 辰獲益良多。這樣的成長與變化,不但是在治學上,也更是變化了俊辰的人生態度。您 與臺大流病所金教授傳春給予的這項寶貴禮物,俊辰將會受益許久,回思此情,感激實 難言喻。 我的父母親陳天華先生、黃鳳娟女士一直給予俊辰溫暖的包容、無聲的支援。完成 這本論文的過程是順遂和低潮相互倚伏的組合,歡喜的時候,我能與您分享,鬱滯的時 刻,您們鼓勵我,是我最大的心靈支柱。感謝妹妹怡安,聽我訴苦,分擔我的情緒,在 忙碌的工作中抽空關懷問候,謝謝你們,我卻愧無回報,彌增慚怍而已。 感謝實驗室的好朋友們:宇書、峻宇,如果沒有你們,這本論文無法完成。許多個 一起熬夜的夜晚,拉著你們一起討論的 meeting,我深深感激。彥甫、其樺、章維、怡 馨、凱程、志達、彥修、敬立、彥超、力仁、韋帆,謝謝你們給這論文的許多建議、協 助、還有關心,共同走過來的這一段日子,真的是非常美好的回憶。 俊辰何其幸運,竟然能得到這樣多的幫助、愛護。最後請容我再一次說:謝謝,感 恩您們。Abstract
The discovery of sequence and structural homologs to a known protein oftenprovides clues, such as biochemical function and domain, for understanding a newly determinedprotein. In this thesis, we have proposed a new concept of "homologous protein-protein interaction (PPI)", which is helpful for understanding a newly determined PPI. We proposed evidence of the existence of homologous PPIs, developed a methodology to rapidly identify homologous PPIs, and infer thetransferability of interacting domains and functions of a queryprotein pair. We found that homologous PPIs can be distinguished when homolog pairswere in the annotated database and have significant joint sequencesimilarity (E-value ≤10-40) with the query protein pair. As an increasing number of reliable PPIs become available and high-throughput experimental methodsprovide systematic identification of PPIs, there is a growingneed for fast and accurate methodologies for discovering homologousPPIs of a PPI. For this demand, we combined the methodology with an annotated PPI database (290,137 PPIs in 576 species) to construct a PPISearch server (http://gemdock.life.nctu.edu.tw/ppisearch/) to supply service for global researchers. In addition, we have utilized the concept of homologous PPIs to cross-species PPI prediction and cross-cross-species network comparison. Currently, large-scale interaction networks are available for only several model organisms. Our preliminary results suggested that the concept of homologous PPI are useful for a systematic transfer of PPI networks between multiple species.
中文摘要
對 新 發 現 的 蛋 白 質 而 言 , 其 同 源 序 列(sequence homolog) 及 同 源 結 構 (structural homolog)通常能提供研究者關於該蛋白質生物功能(function)、功能區塊(domain)的若干 線索。更進一步,我們提出一個新的概念「同源蛋白質-蛋白質交互作用(homologous protein-protein interaction, 簡稱homologous PPI)」,此概念對於了解新被發現的蛋白質交 互作用是很有幫助的。在此研究中,我們提出一套合理定義、鑑別homologous PPIs的方 法論,並提供了自然界中homologous PPIs確實存在的證據。而在現今PPI資料迅速增加 的情況下,研究者迫切地需要能夠快速搜尋並提供合理homologous PPIs的方法,以對未 知功能的PPI進行深入的研究。為達成此目的,我們將本研究中發展出的方法論與一個 大型PPI資料庫(包含來自 576 個物種的 290,137 筆PPIs)結合,建立了一個網路服務工具 PPISearrh (http://gemdock.life.nctu.edu.tw/ppisearch/),使用者可輸入任一提問蛋白質對 (query protein pair),它具有快速搜尋該提問蛋白質對之homologous PPIs (亦稱PPI family) 的功能,並已開放給全球研究者使用。
本研究亦提供統計基礎,將homologous PPIs所具有的生物功能及功能區塊用來合理 地註解未知性質的蛋白質間交互作用,其觀念與用同源序列(或結構)來了解某個新發現 蛋白質相類。本研究中我們提出,homologous PPIs須具備三個條件:是提問蛋白質對中
兩個蛋白質的同源序列之配對;已知紀錄在資料庫中;以及足夠顯著的(E-value ≤ 10-40)
合成序列相似度(joint sequence similarity)。
在本研究中,我們也初步將homologous PPI 的新概念運用於兩個研究課題,第一是
跨物種的蛋白質間交互作用之預測(cross-species PPI prediction),另一個是跨物種間蛋白 質交互作用網絡之比對(cross-species network comparison)。目前只有數種模式生物(model organisms)已累積大量的 PPI 資料,其蛋白質交互作用網絡可被較完整地了解,我們的 初步成果顯示,homologous PPI 之概念可以對研究上述兩個課題有所助益。
Contents
Contents ... 1 Chapter 1. Introduction ... 3 1.1 Motivation ... 3 1.2 Background ... 3 1.3 Thesis overview... 5Chapter 2. Methods and Materials for Finding Homologous Protein-protein Interactions ... 7
2.1 Overview of homologous protein-protein interaction search... 8
2.2 Homologous protein-protein interaction ... 10
2.3 Non-redundant data set for searching homologous PPIs ... 10
2.5 Data sets for evaluating the approach of searching homologous protein-protein interactions ... 14
2.5.1 HOM data set ... 14
2.5.2 ORT data set ... 14
Chapter 3. Evidence Supplying the Existence of Homologous Protein-protein Interactions ... 16
3.1 Evidence of the existence of homologous PPIs ... 16
3.1.1 Conservation of molecular function in PPI families ... 17
3.1.2 Conservation of domain pairs in PPI families... 20
3.1.3 Conservation of interacting domains in PPI families... 21
3.1.4 Conservation of binding model in PPI families ... 23
3.2 Input, output, and options of the PPISearch server... 26
3.3 Example analysis of homologous PPI search... 30
3.3.1 σ1A-adaptin and γ1-adaptin ... 30
3.3.2 MIX-1 and SMC-4 ... 31
3.4 Discussion ... 32
3.4.1 Example analysis for giving more insights into PPI family... 32
3.4.2 A question caused by local sequence alignment ... 35
Chapter 4. Applications of Homologous Protein-protein Interactions ... 38
4.1 Cross-species prediction of protein-protein interactions... 39
4.1.1 Background ... 39
4.1.2 Results and discussion... 40
4.1.3 Discussion ... 45
4.1.4 Summary ... 48
Chapter 5. Conclusion... 56
5.1 Summary ... 56
5.2 Future works... 57
5.2.1 Directions for future research... 57
5.2.2 Combination of sequence-based and structure-based interolog mapping.. 58
References ... 62
Appendix A ... 66
List of publications... 66
Appendix B ... 67
Chapter 1.
Introduction
1.1
Motivation
Many sequence and structure alignment methods have been developed to identify homologs of newly determined sequences and structures1, 2. The discovery of sequence and structural homologs to a known protein often provides clues, such as function and domain, for understanding a newly determinedprotein. Likewise, we proposed a new concept "homologous protein-protein interaction (PPI)". As an increasing number of reliable PPIs become available, there is a growing need for fast and accurate methodolgies for discovering homologous PPIs to understand a newly determined PPI.
In this thesis, we provided evidence to supply the existence of homologous PPIs, developed a methodology tosearch homologous PPIs across multiple species, and annotatethe query protein pair. Public PPI databases, such as IntAct, BioGRID and MINT, offer a good basis for researchers to investigate this issue. In addition, we supplied the new concept of homologous PPIs, and combined our method identifying homologous PPIs with an annotated PPI data set (was consisted of five public PPI database) to construct a web server, PPISearch, to supply service for global researchers.
1.2
Background
interactions of the signaling molecules. This process, called signal transduction, plays a fundamental role in many cellular processes and in multiple diseases (e.g. cancers). Those cellular behaviors and phenotypes, for example, growth, cell division, cellular differentiation, and apoptosis, mainly are mediated by protein-protein interactions.
The interactions between proteins are critical to most of cellularprocesses. To identify and characterize protein-protein interactions and their networks, many high-throughput experimental approaches, such as yeast two-hybrid screening,mass spectroscopy and tandem affinity purification, and computational methods [phylogenetic profiles3, known 3D complexes4 andinterologs5] have been proposed6. Some PPI databases,such as IntAct7, MIPS8, DIP9, MINT10, and BioGRID11 have accumulated PPIs submitted by biologists, and thosefrom mining literature, high-throughput experiments and otherdata sources. As these interaction databases continue growingin size, they become increasingly useful for analysis of newly identified interactions.
Sequence homologs of a known protein oftenprovide clues to understand the function of a newly sequencedgene. As an increasing number of reliable PPIs become available,identifying homologous PPIs should be useful to understand anewly determined PPI. Recently, several PPI databases (e.g.IntAct and BioGRID) allow users to input one or a pair of proteinsor gene names to acquire the PPIs associated with the queryprotein(s). Few computational methods12,
13 applied homologous interactions to assess the reliability of PPIs. Otherwise, several
computational approaches, such as Yu et al. (2004)5, Scott et al. (2007)14, and InteroPORC15, combined known PPIs from one or more species and orthology or homology relationships between the source and targets species. These data sets of predicted PPIs, which are constructed by these methods, are available online. However, only a few PPIs in these data sets have been identified by experiments and these sets have not supplied service of searching PPIs
and provides their homologous interactions from known PPIs for investigating the query protein pair.
According to our knowledge, this study first showed the existence of homologous PPIs, querying a limited number of databases. Additionally, PPISearchis also the first server that identifies homologous PPIs from annotated PPI databases and infers the transferability of interactingdomains and functions between homologous PPIs and the query.PPISearch is an easy-to-use web server that allows users toinput a pair of protein sequences. Then, this server finds homologousPPIs in multiple species from five public databases (IntAct,MIPS, DIP, MINT and BioGRID) and annotates the query. We supplied a threshold of conservation ratio of biological characteristics (e.g. domains and function). These characteristics have higher ratio than the threshold would be reliably assigned to the query protein pair. Our results demonstrated that this server achieves high agreements on interactingdomain-domain pairs and function pairs between query proteinpairs and their respective homologous PPIs.
1.3
Thesis overview
The thesis is organized as follows. In Chapter 2, we proposed the PPI concept and the methodology to find the homologous PPIs for a given query PPI. Then, we combined the methodology with a non-redundant PPI data set to construct a web server, PPISearch. The PPI data set consists of 290,137 PPIs derived from 576 species.
In Chapter 3, we proposed evidence supplying the existence of PPI families, the results of homologous PPIs search, and discussed the observations of PPI families. We examined the new concept of homologous PPI based on four insights. Moreover, we used case studies to describe the insights into the concept of homologous PPI, and the statistical analyses of PPI families. Our results demonstrated the utility and feasibility of the PPISearch server in
identifying homologous PPIs and inferring conserveddomains and functions from PPI families. By allowing users to inputa pair of protein sequences, PPISearch is the first server thatcan identify homologous PPIs from annotated PPI databases andinfer transferability of interacting domains and functions betweenhomologous PPIs and a query. We showed that PPISearchis a fast homologous PPIs search server and is able to providevaluable annotations for a newly determined PPI.
In Chapter 4, we applied the concept of homologous PPI (i.e. PPI family) to cross-species prediction of PPIs and cross-cross-species network comparisons. In recent years, for complementing experimental techniques (e.g. yeast two-hybrid system and mass spectroscopy), a number of computational methods, such as PathBLAST16, 17 and interologs5, 18, have been developed to predict PPIs19. The concept of interologs has been extended to be a “generalized interolog mapping” method5. Our results showed that our discovery can be used to advance the generalized interolog mapping method. In addition, we used case studies to present that the concept of homologous PPI are useful for a systematic transfer of PPI networks between multiple species.
Chapter 2.
Methods and Materials for Finding Homologous
Protein-protein Interactions
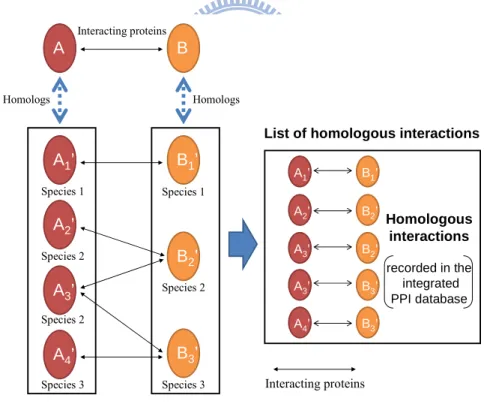
In this chapter, we presented the concept of PPI family and the method to find the homologous PPIs for a given query PPI. Figure 1 illustrates the concept of searching homologous PPIs. For this purpose, we constructed a non-redundant PPI data set for searching homologous PPIs. The data set consists of PPIs derived from five public databases, IntAct,MIPS, DIP, MINT and BioGRID. Total number of PPIs in this data set is 290,137 in 576 species.
Interacting proteins A B A1’ A2’ B1’ A3’ B2’ A4’ B3’ Homologs Homologs Interacting proteins A1’ B1’ A2’ A3’ B2’ B2’ A3’ B3’ A4’ B3’ Homologous interactions recorded in the integrated PPI database Species 1 Species 2 Species 2 Species 3 Species 1 Species 2 Species 3
List of homologous interactions
Figure 1. Illustration of searching homologous interactions. Interacting proteins A and B is the query protein pair given by users. A1'-A4' and B1'-B3' are the homologs (defined by BLASTP
Additionally, we presented how we evaluate the reliability of homologous PPIs, which are defined by sequence similarity (BLASTP E-values) and joint sequence similarity (joint E-value) between the query protein pair and these homologous PPIs.
2.1
Overview of homologous protein-protein interaction search
In this study, we developed a methodology for searching homologous PPIs and used it to constructing a web server, PPISearch. Figure 2 shows the details of the PPISearch server to searchhomologous PPIs of a query protein pair (A and B) by the followingsteps (Figure 2A). This server first identifies the homologousfamilies (A' and B') of A and B, respectively, with E-value ≤10-10 by using BLASTP to scan the annotated PPI databases (Figure 2B and C). All protein pairs of A' and B' are considered candidates of homologous PPIs. We selected homologous PPIs from these candidates,which are recorded in the annotated databases, and have significantjoint sequence similarity (E-value ≤ 10-40) between candidatesand the query (Figure 2D). Then, we measure the conservationratios of domain-domain pairs (DDPs; Pfam20 and InterPro21 domains) and protein functions (Gene Ontology annotations22) derived from these homologous PPIs of the query (Figure 2E).
Annotated databases (290,137 protein-protein interactions)
BLASTP E-value ≤ 10-10 BLASTP E-value ≤ 10-10
Homologous PPIs σ1A-adaptin of mouse (P61967) γ1-adaptin of mouse (P22892) A B C P61966 P61967 P56377 Q9DB50 Q9VCF4 P35181 P62743 P53680 O75843 O43747 Q86B59 Q9W388 Q9UPM8 Q12028 P17427 O95782 O75843 P61966 P61967 O43747 P56377 O75843 O43747 O43747 Q9DB50 O75843 Q9VCF4 P61966 Mouse Human Human Human Human Yeast Human Human Human Fruit fly Human Human Fruit fly Human Human Human Mouse Mouse Human Human Mouse Mouse Fruit fly Fruit fly Human Yeast Mouse Human
Domain-domain pairs Conservation ratio 14/14 = 1.0 13/14 = 0.93 2/14 = 0.14 Step 1: Query a pair of protein sequences
(A and B)
Step 2: Identify homologous protein
families (A' and B') of A and B,
respectively, with E-values ≤ 10-10using
BLASTP from annotated PPI databases
0.6 E 1/14 = 0.07 PF01602 PF02883 PF02296 PF07718 PF01217 PF01217 PF01217 PF01217 interacting domains
Step 4: Measure the conservation ratios
of all of domain-domain pairs (DDPs) and protein functions derived from these homologous PPIs of a query. The DDPs and function terms are considered as conservation if their ratios ≥ 0.6.
Step 5: Output homologous PPIs,
conserved DDPs and functions, and multiple sequence alignments across multiple species for the query
Step 3: Identify homologous PPIs which
are protein pairs of A' and B' and
recorded in annotated databases with joint
E-values (JE) ≤ 10-40. D P61967 Human Mouse P61966 O43747 Human Human P56377 O75843 Q86B59 Fruit fly Human Q9W388 Fruit fly Mouse Q9UPM8 Q9DB50 Human Fruit fly Q12028 Human Human P53680 O95782 Q9VCF4 Yeast P35181 Yeast P62743 P17427 Mouse Mouse … …
σ1A-adaptin family γ1-adaptin family
1.7e-134 1.7e-134 1.7e-134 1.7e-134 1.7e-129 1.7e-129 1.0e-128 1.0e-128 2.0e-124 2.0e-124 5.5e-73 6.5e-63 9.5e-55 6.0e-54 JE
Figure 2. Overview of the PPISearch server for homologous protein-protein interaction search and conservation analysis using proteinsσ1A-adaptin and γ1-adaptin as the query. (A) The main procedure. (B) Identify homologs ofσ1A-adaptin and γ1-adaptin using BLASTP to scan the annotated PPI databases. (C) The homologous families of σ1A-adaptin and γ1-adaptin with E-values ≤10-10. (D) Homologous PPIs of the query. (E) Conservation ratios of domain-domain pairs derived from homologous PPIs.
2.2
Homologous protein-protein interaction
The concept of homologous PPI is the core of the this studyto identify the PPI family and measure DDPs and functional conservationsof a query protein pair (A and B). We defined a homologous PPIas follows: (1) homologs of A and B are proteins with significantsequence similarity BLASTP E-values ≤ 10-10;5, 18 (2)significant joint sequence similarity (joint E-value JE ≤ 10-40)between two pairs, i.e. (A, A1') and (B, B1'), of the queryprotein pair (A and B) and
their respective homologs (A1' and BB1') recorded in annotated PPI databases. This work
followed previous studies5, 18 to define joint sequence similarity as
B A
E
E
E
J
=
×
(1)where EA is theE-value of proteins A and A1'; and EB is the E-value of proteins B and BB 1'. Here,
JE ≤10 is considered a significant similarity according to statistical analysis of 290,137
annotated PPIs and 6,597 orthologous PPI families collected from the PORC database (see Chapter 3).
-40
23
2.3
Non-redundant data set for searching homologous PPIs
Table 1 lists the dates and numbers of PPIs of the five public databases.
Table 1. Five source data sets of PPIs
Database Number of PPIs Date
IntAct 147,634 Dec. 14, 2008 MIPS 18,529 Oct. 1, 2008 DIP 52,445 Oct. 14, 2008 MINT 77,846 Oct. 28, 2008 BioGRID 150,827 Dec. 17, 2008 Total 447,281
After removing redundant PPIs, the annotated data set used in this study has 290,137 PPIs. These PPIs were identified experimentally from 576 species.
We describe briefly these public databases as follows: (1) IntAct: All interactions are derived from literature curation or direct user submissions and are freely available7. IntAct is a freely available and open source database system of protein interaction data; (2) MIPS: The Munich Information Center for Protein Sequences (MIPS) combines automatic processing of large amounts of sequences with manual annotation of selected model genomes. PPIs of MIPS are annotated by the compilation of manually curated databases for protein interactions based on literature to serve as an accepted set of reliable annotated interaction data8; (3) DIP: The DIP database catalogs experimentally determined PPIs. It combines information from a variety of sources to create a single, consistent set of PPIs. The data stored within the DIP database were curated manually by both expert curators and automatically using computational approaches that utilize the knowledge about the PPI networks extracted from the most reliable, core subset of the DIP data9; (4) MINT: The Molecular INTeraction database focuses on experimentally verified protein-protein interactions mined from the scientific literature by expert curators10; (5) BioGRID: The BioGRID (Biological General Repository for Interaction Datasets) database was developed to house and distributes collections of protein and genetic interactions from major model organism species. BioGRID currently contains ~150,000 interactions from six different species, as derived from both high-throughput studies and conventional focused studies11.
The tabular data files of PPIs from the five public databases were downloaded. We merged all of these PPIs and removed duplications by using UniProt accession numbers. A total of 290,137 PPIs in 576 species were included in our investigation.
2.4
Annotations of homologous protein-protein interactions
A query protein pair and its homologous PPIs, significant bothin sequence and joint sequence similarity, can be considereda PPI family. The concept of PPI families is similar to thatof protein sequence family20, 21 and protein structure family24. We believe that PPI families can be applied widely inbiological investigations. Here, we assume that the membersof a PPI family are conserved on specific functions and in interactingdomain(s). Using these conservations of a PPI family, our servercan be used to annotate the protein functions and DDPs of aquery protein pair.
2.4.1 Transferability of molecular function
These members of a PPI family often have similar molecular functions.We used the molecular function (MF) terms of Gene Ontology22 to annotate the functions of a query protein pair. The conservation ratio (CRFm) of an MF term pair (MFP) m in homologousPPIs of a query i is
utilized to measure the agreement and isdefined as
i m CRFm query of PPIs homologous of Number pair term MF GO a with PPIs homologous of Number = (2)
Additionally,the shared ratio of MFPs (SRF), which is statistically derived from 106,997 annotated queries, is utilized to estimate thetransferability of conserved function pairs shared by the queryand its homologous PPIs. The SRF against different ratio k isdefined as
∑
∑
∈ ∈ ≥ ≥ = Q i m i Q i m i k CRF F k CRF f SRF ) ( ) ( (3)whereQ is a set of annotated PPIs in databases; i is a query proteinpair; fi(CRFm ≥k) is the
number of MFPs with CRFm values exceedingk and these MFPs are shared by the query i and
its homologousPPIs; and Fi(CRFm ≥k) is the total number of MFPs with CRFm≥k, where
MFPs are derived from homologous PPIs of the queryi. Here, k is set to 0.6.
2.4.2 Transferability of domain-domain pairs
A query protein pair and its homologous PPIs often show conserveinteracting DDPs. To measure the occurrence of each DDP in aPPI family, we define the conservation ratio (CRDp)
of a DDPpin homologous PPIs of a query protein pair i as
i p CRDp query of PPIs homologous of Number pair domain a with PPIs homologous of Number = (4)
Figure 2D and E show an example to calculatethe CRD values of four DDPs. In addition, to evaluate the transferabilityof DDPs between a query and its homologous PPIs statistically,this study defines the shared ratio (SRD) of DDPs using CRDpand 103,762 annotated PPIs as
query protein pairs. The SRD ofDDPs against different ratio c is given as
∑
∑
∈ ∈ ≥ ≥ = Q i p i Q i p i c CRD D c CRD d SRD ) ( ) ( (5)where Q is a set of annotated PPIs in databases(here, the total number of PPIs in Q is 103 762); i is a queryprotein pair; di(CRDp ≥c) is the number of DDPs with CRDp valuesexceeding c;
and these DDPs are shared by the query i and itshomologous PPIs. Di(CRDp ≥c) is the total
number of the DDPswith CRDp ≥c, where DDPs are derived from homologous PPIs ofthe
of CRDp to yield reliableDDP annotations with an acceptable level of Di. Please notethat
CRDp and SRD are computed from a query protein pair anda set of queries, respectively.
2.5
Data sets for evaluating the approach of searching homologous
protein-protein interactions
For evaluating the usefulness of the PPISearch server for the discoveryof homologous PPIs and for the annotations of a query proteinpair, we constructed two query protein sets, termed HOM and ORT.For searching homologous PPIs, HOM and ORT data sets are used to assess performance of PPISearch and to determine the threshold of joint E-valueJE [Equation (1)]
(Figure 3A).
2.5.1 HOM data set
The HOM set includesall of 290,137 PPIs. The HOM set wasapplied to infer the relations between conservation ratios [CRF and CRD defined in Equations (2) and (4)] and the transferabilityof DDPs and MFPs, respectively, between a query and its homologousPPIs.
2.5.2 ORT data set
The ORT set has 6,597 orthologous PPI families(14,571 PPIs) derived from the annotated PPI database and PORC orthology database. PORC data (putative orthologous clusters) were defined as orthologous families from Integr823 and CluSTr25 databases. These clusters contain all sequenced organisms (1125 bacteria, 125 eukaryota and 50 archaea in the release 94). Each entry in PORC represents a cluster of genes grouped by the similarity of their longest protein
product. According to the construction process of PORC, a gene cluster contains at most a single protein from a given species and a protein can be assigned to only a single cluster.
Chapter 3.
Evidence Supplying the Existence of Homologous
Protein-protein Interactions
In this chapter, we presented the evidence of existence of homologous PPIs (Section 3.1), the results of homologous PPIs search (i.e. PPISearch) (Sections 3.2-3.3) and discussed the observations of PPI family. We used case studies to describe the insights used to examine the concept of homologous PPI and statistically analyze PPI families (Section 3.4).
3.1
Evidence of the existence of homologous PPIs
We analyzed the results of homologous PPIs by four views. Firstly, we observed the conservation of biological function in PPI families. Secondly, we observed the conservation of domain pairs in PPI families. Thirdly, because of the sharing of conserved domain pairs in PPI families, we observed the conservation of interacting domains in PPI families (based on protein 3D structures). Finally, because of the sharing of conserved interacting domains in PPI families, we observed the conservation of binding interface between two proteins of each PPI in PPI families. These evidences from the four views showed the existence of homologous PPIs (i.e. PPI family).
3.1.1 Conservation of molecular function in PPI families
To verify the discoveryof homologous PPIs, we selected two query protein sets, termed HOM and ORT.To search homologous PPIs, HOM and ORT are used to assess PPI families and to evaluate the threshold of joint E-valueJE (Figure 3A). In addition, the HOM set wasapplied to
infer the relations between conservation ratios [CRF defined in Chapter 2] and the transferabilityof MFPs, respectively, between a query and its homologousPPIs (Figure 3B). The HOM set includesall 290,137 PPIs and the ORT set has 6,597 orthologous PPI families (14,571 PPIs) derived from the annotated PPI database and PORCorthology database.
HOM and ORT were used to assess the PPISearch server in identifyinghomologous PPIs and orthologous PPIs, respectively, by searchingthe annotated PPI database (290,137 PPIs with 54,422 proteins).Figure 3A shows the relationships between joint E-value JE andnumber
of orthologous PPIs (black) and homologous PPIs (red).The orthologous PPIs often have the same functions and domains. When JE ≤10-40, the number of orthologous PPIs decreases
significantly; conversely, the number of homologous PPIs decreasesmore gradually than that at JE ≥ 10-40. This result showsthat the proposed method is able to identify 98.2% orthologous
0 5000 10000 15000 20000 25000 10 20 30 40 50 60 70 80 90 100 110 120 -log(Joint E-value) N um be r of or thol ogo us P P Is
A
0 50000 100000 150000 200000 250000 300000 350000 400000 450000 500000 N um be r of ho m ol ogo us P P Is 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 Conservation ratio of MF pairs in homologous PPIsS har ed r at io o f M F p ai rs ( S R F ) 0 100000 200000 300000 400000 500000 600000 700000 Nu m be r of M F p ai rs (N M F P ) SRF (logJE<-10) SRF (logJE<-40) SRF (logJE<-100) NMFP (logJE<-10) NMFP (logJE<-40) NMFP (logJE<-100)
B
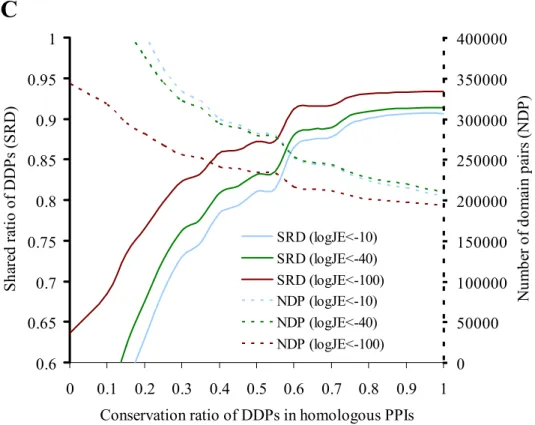
0.6 0.65 0.7 0.75 0.8 0.85 0.9 0.95 1 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 Conservation ratio of DDPs in homologous PPIs
S har ed r at io o f D D P s ( S R D ) 0 50000 100000 150000 200000 250000 300000 350000 400000 N um be r of dom ai n pa ir s ( N D P ) SRD (logJE<-10) SRD (logJE<-40) SRD (logJE<-100) NDP (logJE<-10) NDP (logJE<-40) NDP (logJE<-100)
B
C
Figure 3. Conservations of biological functions and domain pairs in PPI families. (A) The relationships between joint E-value JE and the numbers of orthologous PPIs (black) and
homologous PPIs (red) derived from 290,137 annotated PPIs. (B) The relationships between the conservation ratios of molecular function pairs (MFPs) with the shared ratios of MFPs and with the number (dotted lines) of MFPs derived from 106,997 PPI families. The shared ratio of MFPs is 0.69 and the number of MFPs is 454,251 if the conservation ratio is 0.6 and the joint E-value is 10-40 (green lines). (C) The relationships between conservation ratios of DDPs with shared ratios of DDPs and with the number (dotted lines) of DDPs derived from 103,762 PPI families. The shared ratio of DDPs is 0.88 and the number of DDPs is 252,728 when the conservation ratio is 0.6 and joint E-value is 10-40 (green lines).
To evaluate the transferability of MFPs between a queryand its homologous PPIs, we used the SRF [Equation (3)]. The HOM set is also used to evaluate the utilityof the PPISearch server in annotating the query protein pair.By excluding proteins without molecular function annotations of GO from the queryset, 106,997 PPIs are used to evaluate the transferability (SRF)of conserved MFPs between these query PPIs and their respectivehomologous PPIs (Figure 3B). Themembers of a PPI family have similar molecular functions, andSRF ratios are highly correlated with conservation ratios (CRF)of MFPs. When the CRF is 0.6 and the joint E-value is 10-40 (green lines), the SRD is 0.69 and the number of MFPs is 454,251.
3.1.2 Conservation of domain pairs in PPI families
In addition, the HOM set wasapplied to infer the relations between conservation ratios [CRD defined in Chapter 2] and the transferabilityof DDPs, respectively, between a query and its homologousPPIs (Figure 3C). To evaluate the transferability of DDPs between a queryand its homologous PPIs, we used the SRD [Equation (5)]. By excluding proteins without domain annotations from the queryset, 103,762 PPIs are used to evaluate the transferability (SRD)of conserved DDPs between these query PPIs and their respectivehomologous PPIs (Figure 3C).
Figure 3C shows the relationship between conservation ratios(CRD) of DDPs and the SRD ratios. The SRD ratio increases significantly(solid lines) when the CRD increases and CRD ≤ 0.6. Conversely,the number of DDPs derived from 103,762 PPI families decreases (dotted lines) as CRD increases. If the CRD is set to 0.6 andthe joint E-value is set to 10-40 (green lines), the SRDis 0.88 and the number of DDPs is 252,728. This result demonstrates that members of a PPI family reliably share DDPs (or interacting domains). Additionally, similar resultswere obtained for the transferability of conserved functions betweenhomologous PPIs and the query (Figure 3B).
These results reveal that PPI families achieve a highSRD with a reasonable number of DDPs when the joint E-value is set to 10-40. In summary, these experimental results demonstrate that this server achieves high agreement on MFPs and DDPsbetween the query and their respective homologous PPIs.
3.1.3 Conservation of interacting domains in PPI families
The two above evidence were acquired by sequence-based searching. As an increasing number of structural data was available (e.g. protein complexes in PDB), we used structure-based views to examine the concept of homologous PPIs. In this section 3.1.3, we observed the conservation of interacting domains in PPI families because of the assumption of "the members of a PPI family have similar interacting domains".
Firstly, we collected a data set of protein complexes. Each complex was composed of two protein chains (i.e. heterodimer or homodimer) and was (1) recorded in the annotated PPI database (290,137 PPIs) and (2) recorded in iPfam database. iPfam is a database []. After selecting protein complexes from PDB, a data set of 1,014 complexes (in other words, 1,014 PPIs) was constructed.
Figure 4 shows the method we calculated the conservation of interacting domain in PPI families. Proteins A-B is an interacting protein pair, in which there are two physical domain-domain interactions (DDIs). If a protein pair between homologs (E-value ≤10-10) A' and B' kept ≥ 1 DDI, we considered the protein pair has similar interacting domains to the query pair A-B. We compared the two partitions of "PPI families" and "Non PPI families". The "PPI families" consisted of PPIs with JE ≥10-40, for example, the PPIs circled by blue. Conversely, the "Non
Conservation of interacting domain
Homologs Homologs A B B1’ 0.0 5e-45 1e-100 5e-30 B2’ B4’ B5’ A1’ A2’ A4’ A5’ 1e-80 B3’ A3’ 5e-20 B6’ A6’ JE 5e-10 B7’ A7’ 1. iPfam 2. Database (290,137 PPIs) Keeping 1 DDI Yes Yes Yes Yes Yes No No 1 2 ≥PPI family
Non PPI family
Figure 4. Illustration of the method we calculated the conservation of interacting domain in PPI families. The rectangles colored by green, blue, light blue, yellow, and red mean domains. The query interacting protein pair A-B has two DDIs. All of the pairs of A' and B' homologs are marked "Yes" (keeping ≥ 1 DDI with the query PPI A-B) or "No" (keeping no DDI with the query PPI A-B).
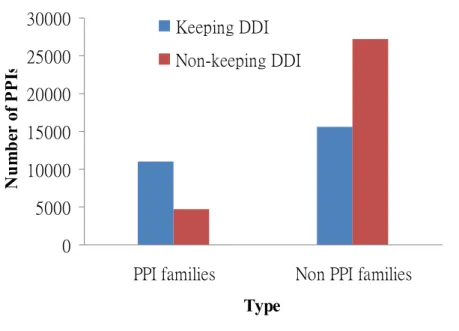
Figure 5 indicates the results of observing the conservation of interacting domains in PPI families. We found that the number of PPIs keeping ≥ 1 DDI (11,060 PPIs) were 2.35-fold more than that of PPIs not keeping DDI (4,699 PPIs) in the set "PPI families". In comparison, the number of PPIs not keeping (27,264 PPIs) were 1.74-fold that of PPIs keeping DDI ≥ 1 DDI (15,653 PPIs) in the set "Non PPI families".
0 5000 10000 15000 20000 25000 30000
PPI families Non PPI families
Type N u m b er o f PPI s Keeping DDI Non-keeping DDI
Figure 5. Conservation of interacting domains in PPI families. The number of PPIs keeping ≥ 1 DDI is 11,060 PPIs (blue) and that of PPIs not keeping DDI is 4,699 PPIs (red) in the set "PPI families". In comparison, the number of PPIs not keeping is 27,264 PPIs (red) and that of PPIs keeping DDI ≥ 1 DDI is 15,653 PPIs (blue) in the set "Non PPI families".
These results indicated that there was higher conservation of interacting domains in PPI families than in non-PPI families. In the Section 3.4 Discussion, we supplied a possible reason of why there were 4,699 PPIs which not keeping DDI in the set "PPI families".
3.1.4 Conservation of binding model in PPI families
After we acquired the evidence of conservation of interacting domain, we were interesting to observed the similarity of structural binding interfaces within PPI families. This idea was derived from the assumption that the PPI members of a PPI family have similar structural binding interfaces between two protein partners of each PPI. We have developed 3D-partner,
which is a web tool to predict interacting partners and binding models of a query protein sequence through structure complexes and a new scoring function.
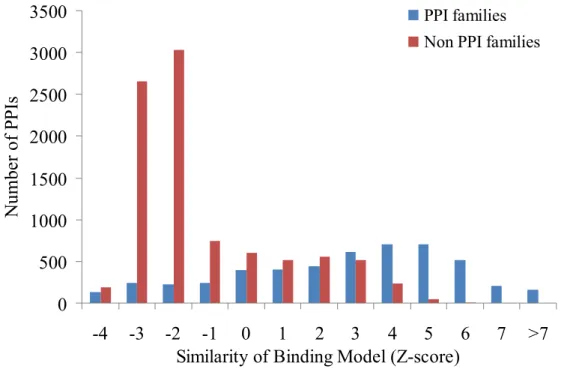
For above purpose, we collected a data set of protein complexes from PDB, which was composed of 517 heterodimers because 3D-partner was developed based on protein structures of heterodimers. Similar to the description in Section 3.1.3, we compared the two subsets of "PPI families" (4,998 PPIs) and "Non PPI families" (9,102 PPIs). The results of comparison between the two data subsets are showed in Figure 6. We used a threshold Z-score to measure the similarity of binding model and identify interacting partners with the query. The Z-score reveals that the proportion of true positives rises when a higher Z-score is utilized. The P value of T test between the Z-scores of the two subsets "PPI families" and "Non PPI families" is less than 10-30. The results indicated that there was significant difference between the two subsets.
-log(JE) > 40 0 500 1000 1500 2000 2500 3000 3500 -4 -3 -2 -1 0 1 2 3 4 5 6 7 >7
Similarity of Binding Model (Z-score)
Nu m be r o f P P Is PPI families Non PPI families
Figure 6. Distribution of Z-score (i.e. similarity of binding models) in two subsets of "PPI families" (blue) and "Non PPI families" (red). There are 4,998 and 9,102 PPIs in the two subsets, respectively.
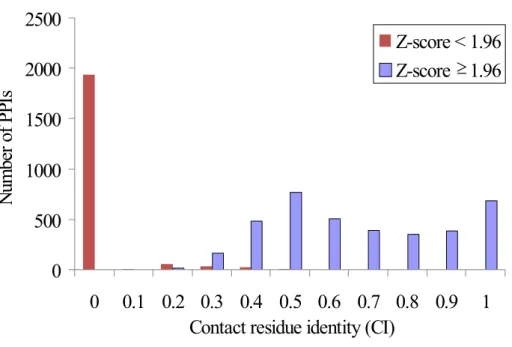
In addition, we were interesting that why many PPIs in “PPI families” subset have low similarity with the query PPIs. For this purpose, we used a descriptor, aligned contact residue identity (CI), to observe the similarity of binding interface in a PPI family. Figure 7 shows an illustration of how we calculated CI values.
We selected the PPIs in "PPI families" subset for observation (Figure 8) and example analysis. Figure 8 indicates the distributions of CI values in PPIs with Z-score ≥ 1.96 (i.e. 95% confidence interval) and that with Z-score < 1.96. We found that 93.5% of PPIs with Z-score < 1.96 have CI = 0. In other words, these results suggested that we would get PPIs with different binding model from the query PPIs through the search method we currently used. In the Section 3.4 Discussion, we supplied a possible reason of causing this observation.
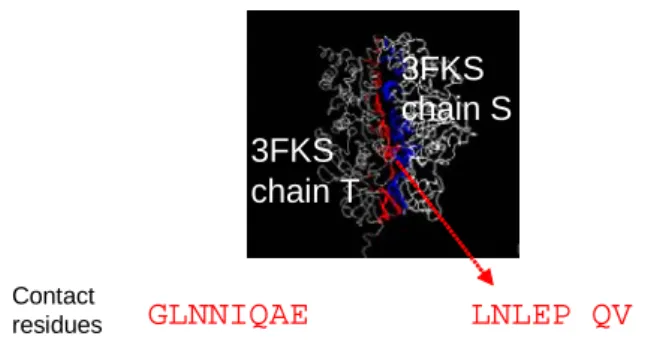
3FKS chain S 3FKS chain T GLNNIQAE LNLEP QV VFGLNNIQAEESGVKGMALNLEPGQVG VFGLNNLQAEELVEFGMALNLEPGQVG AHGLDNVMSGENAVMGMALNLEENNVG CI = 14/15 = 0.93 Contact residues CI = 8/15 = 0.53 P0ABB0 P09219 P09219 P0ABB0 3FKS:T
Figure 7. An example of how to calculate CI values. The 15 residues colored by red are part of contact residues (on 3FKS chain T) in the interacting interface between 3FKS chains T and chain S. The underlined residues in the aligned sequences P09219 and P0ABB0 are the residues which are identical to the contact residues on 3FKS chain T. In this case, CI of
0 500 1000 1500 2000 2500 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Contact residue identity (CI)
Nu m be r o f P PI s Z-score < 1.96 Z-score > 1.96≥
Figure 8. Distribution of CI values in PPIs with Z-score ≥ 1.96 (blue) and Z-score < 1.96 (red).
3.2
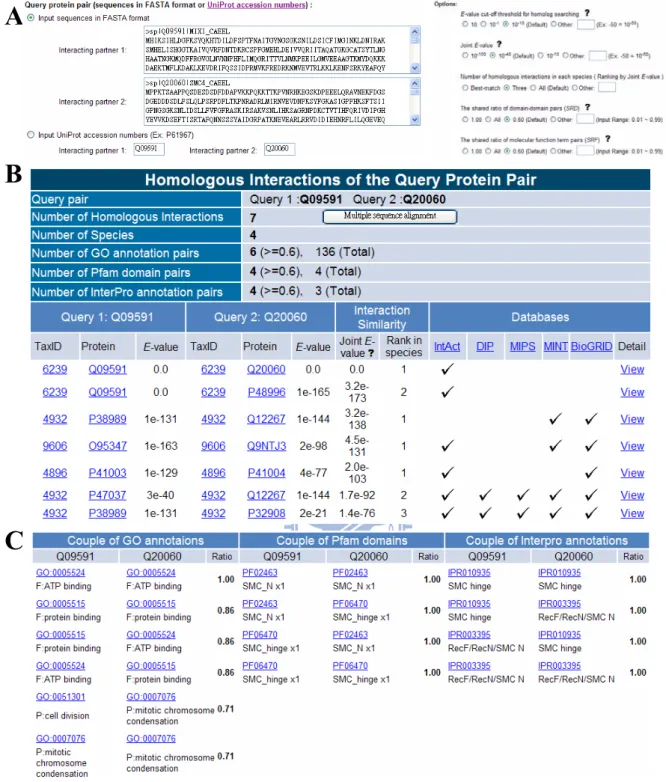
Input, output, and options of the PPISearch server
The PPISearch is an easy-to-use web server (Figure 9). Usersinput a pair of protein sequences in FASTA format or UniProt ID, and choose E-value thresholds for homologs and for homologousPPIs (Figure 9A). In addition, users can assign the CRD andCRF thresholds, specific species and the number of homologousPPIs in a species.
To evaluate the usefulness of the PPISearch server for the discoveryof homologous PPIs and for the annotations of a query proteinpair, we selected two query protein sets, termed HOM and ORT.To search homologous PPIs, HOM and ORT are used to assess PPISearch performance and to determine the threshold of joint E-valueJE (Figure 3A). In addition, the
HOM set wasapplied to infer the relations between conservation ratios [CRDand CRF defined in Chapter 2] and the transferabilityof DDPs and MFPs, respectively, between a query and its homologousPPIs (Figure 3B and C). The HOM set includesall 290,137 PPIs and the ORT set
has 6,597 orthologous PPI families(14,571 PPIs) derived from the annotated PPI database and PORCorthology database.
HOM and ORT were used to assess the PPISearch server in identifyinghomologous PPIs and orthologous PPIs, respectively, by searchingthe annotated PPI database (290,137 PPIs with 54,422 proteins).Figure 3A shows the relationships between joint E-value JE andnumber of
orthologous PPIs (black) and homologous PPIs (red).The orthologous PPIs often have the same functions and domains. When JE ≤10-40, the number of orthologous PPIs decreases
significantly; conversely, the number of homologous PPIs decreasesmore gradually than that at JE ≥10-40. This result showsthat the proposed method is able to identify 98.2% orthologous
PPIs with a reasonable number of homologous PPIs when JE ≤10-40.
To evaluate the transferability of DDPs and MFPs between a queryand its homologous PPIs, we used the SRD [Equation (3)] andSRF [Equation (5)]. The HOM set is used to evaluate the utilityof the PPISearch server in annotating the query protein pair.By excluding proteins without domain annotations from the queryset, 103,762 PPIs are used to evaluate the transferability (SRD) of conserved DDPs between these query PPIs and their respective homologous PPIs (Figure 3B). The transferability (SRF) of conservedfunctions between the 106,997 PPIs and their homologous PPIsis assessed by excluding proteins without molecular functionterms of GO from the original query set (Figure 3C).
Figure 3B shows the relationship between conservation ratios(CRD) of DDPs and the SRD ratios. The SRD ratio increases significantly(solid lines) when the CRD increases and CRD ≤ 0.6. Conversely,the number of DDPs derived from 103,762 PPI families decreases (dotted lines) as CRD increases. If the CRD is set to 0.6 andthe joint E-value is set to 10-40 (green lines), the SRDis 0.88 and the number of DDPs is 252,728. This result demonstrates that members of a PPI family derived by PPISearch reliably share DDPs (or interacting
functions betweenhomologous PPIs and the query (Figure 3C). Themembers of a PPI family have similar molecular functions, andSRF ratios are highly correlated with conservation ratios (CRF)of MFPs. When the CRF is 0.6 and the joint E-value is 10-40 (green lines), the SRF is 0.69 and the number of MFPs is 454,251.
These results reveal that the PPISearch server achieves a highSRD with a reasonable number of DDPs when the joint E-valueis set to 10-40. In summary, these experimental results demonstrate that this server achieves high agreement on DDPsand MFPs between the query and their respective homologous PPIs.
Typically, the PPISearch server yields homologous PPIs within 20 seconds when sequence length is ≤ 350 (Figure 9B). This server identifieshomologous PPIs in multiple species; conservations and GO annotationsof protein functions; conservations and annotations of DDPs; and the best-matched protein pairs of the query (Figure 9C). Additionally, the PPISearch server provides multiple sequencealignments of homologous PPIs and indicates the conserved residuesbased on amino acid types. For each homologous PPI, this servershows the alignments and experimental annotations (e.g. interactiontypes, experimental methods, gene names and GO terms).
A
B
C
Figure 9. The PPISearch server search results using proteins MIX-1 and SMC-4 of
Caenorhabditis elegans as the query. (A) The user interface for assignments of query protein sequences and E-value thresholds of homologs and homologous PPIs. (B) Homologous PPIs of MIX-1–SMC-4 in multiple species and public databases. (C) Conserved protein functions (GO terms) and domain-domain pairs (Pfam and InterPro) of homologous PPIs with a conservation
3.3
Example analysis of homologous PPI search
3.3.1 σ1A-adaptin and γ1-adaptin
Figure 2C and D show search results using σ1A-adaptin (UniProtaccession number: P61967) and γ1-adaptin (P22892) of Mus musculusas the query. These two proteins are components of the heterotetramericadaptor protein complex 1 (AP-1), which medicates clathrin-coatedvesicle transport from the trans-Golgi network to endosome26.According to the crystal structure (PDB code 1W63)27, thisprotein pair is a physical interaction, but it is not recordedin the annotated PPI database. For this query, the PPISearchserver identifies 14 homologous PPIs, a PPI family, from fourspecies (human, mouse, fruit fly and yeast). This PPI familyhas four DDPs (Figure 2E) — PF01217-PF01602 (CRD is 1.0),PF01217-PF02883 (0.93), PF1217-PF02296 (0.14) and PF01217-PF07718(0.07). Two DDPs (PF01217-PF01602 and PF01217-PF02883) with highest CRD ratios are the domain compositions of the queryand PF01217-PF01602 is the interacting domains27.
This server allows users to choose the JE threshold of homologousPPIs. For example,
when JE is set to 10-100 (default valueis 10-40), the number of homologous PPIs decreases from
14 to 10 by filtering out the last four PPIs (Figure 2D). These10 homologous PPIs consistently include the two DDPs PF01217-PF01602and PF01217-PF02883, each with a CRD = 1.0. Furthermore, userscan choose the best match or number of homologous PPIs in aspecies. In this manner, the PPISearch server is able to select the primary homologous PPIs of each species for specific applications,such as evolutionary analysis of essential proteins.
3.3.2 MIX-1 and SMC-4
Mitotic chromosome and X-chromosome-associated protein (MIX-1,Q09591) and structural maintenance of chromosomes protein 4 (SMC-4, Q20060) of Caenorhabditis elegans are members of SMCprotein family, and are required for mitotic chromosome segregation28. Both MIX-1 and SMC-4 are essential components in formingthe condensin complex for interphase chromatin to convert intomitotic-like condense chromosomes28, 29. Using C. elegansMIX-1 and SMC-4 as the query protein pair and JE is set to 10-40,the PPISearch server found seven
homologous interactions fromannotated PPI databases (Figure 9B). These seven homologous PPIs are consistently SMC–SMC protein interactions, including SMC-2–SMC-4, SMC-3– SMC-4 and SMC-2–SMC-1, in four species. Among these homologous PPIs, two PPIs, Q95347-Q9NTJ3 (Homo sapiens) and P38989-Q12267 (Saccharomyces cerevisiae), are orthologous interactions of the query MIX-1–SMC-423.
These seven homologous PPIs of MIX-1 and SMC-4 include 136 GOterm pairs. Among these GO terms, the CRF ratios of four GOMF term pairs and two GO BP term pairs exceed 0.6 (Figure 9C).These six GO term pairs are consistent with the term-pair combinationsof MIX-1 and SMC-4. For example, MIX-1 and SMC-4 have the sametwo GO MF annotations, protein binding (GO:0005515) and ATP-binding (GO:0005524). Additionally, these seven homologous PPIs containfour DDPs with CRD ratios of 1.0. These four DDPs, PF02463-PF02463, PF06470-PF02463, PF02463-PF06470 and PF06470-PF06470, are recorded in iPfam20 and are consistent with the query pair. The hinge-hingeinteraction (PF02463-PF02463) is experimentally proved, andis conserved in the eukaryotic SMC-2–SMC-4 heterodimer30. These analytical results reveal that the PPISearch serveris able to identify homologous PPIs that share conserved DDPsand MFPs with the query.
3.4
Discussion
3.4.1 Example analysis for giving more insights into PPI family
In above content, we brought up the concept of homologous PPIs, and give statistic evidence and biological examples to support it. At next step, we will provide more evidence to verify the homologous PPIs identified by our methodolgy. For this purpose, we will verify this issue based on four views: (1) domain composition of PPIs, (2) biological functions of PPIs, (3) the locations of PPIs in pathways, and (4) PPIs in manually curated complexes. In other words, we assume that if a PPI is “homologous” to another PPI, they have the same specific function, interacting domains, and they are experimentally identified in the same pathway and/or in the same protein complex.
The first two views have been used to evaluate the concept of homologous PPIs through Pfam annotations and GO terms. Currently, we are starting to gain insights into homology of PPIs by the last two views. Preliminarily, we use components of the transforming growth factor β (TGF-B) system as an example to test our assumption.
Nearly 30 members of the TGF-B family have been described in human, and many orthologs are known in mouse and other vertebrates31. The family is divided into two general branches, the BMP/GDF and TGF-B/Activin/Nodal branches, whose members have diverse, while often complementary, effects31. TGF-Bs are potent fibrotic factors responsible for the synthesis of extracellular matrix. TGF-Bs act through the TGF-B type I and type II receptors (TGFBR-1 and TGFBR-2) to activate intracellular mediators, such as Smad proteins, the p38 mitogen-activated protein kinase (MAPK), and the extracellular signal-regulated kinase pathway32. Figure 10A shows two branches of the Smad pathway mediate signaling by the two main groups of TGF-B family agonists. The BMPs and related GDFs, as well as AMH/MIS,
trigger receptors that signal through Smads 1, 5, and 8. The TGF-Bs, Activins, and Nodals (and the Nodal-related Xnr factors from Xenopus) trigger receptors that phosphorylate Smads 2 and 3. Orthologs from fruit fly are presented in red color (Figure 10B). Alternative type I receptor names are: ALK3 (BMPR-IA), ALK4 (ActR-IB), ALK5 (TbR-I) and ALK6 (BMPR-IB). Activins and BMPs share some of their type II receptors, as indicated31.
? dSmad2 Mad cytoplasm cytoplasm BMP2 BMP4 BMP7 TGF-B BMPR-IA BMPR-IB TGFBR-1 Smad2 Smad3 Smad1 Smad5 Smad8 ALK1 ? BMPR-IB AMH/MIS AMHR-II ALK2 BMP7 ActR-II ActR-IIB CDF5 BMP4 BMPR-IA BMPR-IB ActR-II ActR-IIB BMPR-II Activin Nodal ActR-IB ActR-II ActR-IIB TGFBR-2 Saxophone Screw Gbb Punt Dpp Thickveins Punt dActivin Baboon Punt Nucleus Nucleus A B
Figure 10. Ligand, receptor, and Smad relationships in the TGF system. (A) Two branches of the Smad pathway mediate signaling by the two main groups of TGF-B family agonists. The TGF-Bs, Activins, and Nodals (and the Nodal-related Xnr factors from Xenopus) trigger receptors that phosphorylate Smads 2 and 3. The BMPs and related GDFs, as well as AMH/MIS, engage receptors that signal through Smads 1, 5, and 8. (B) Orthologs in fruit fly are presented in red color.
Homologs Homologs
P10600 P36897
Transforming growth factor beta-3 (human)
P27091 P61812 P07200 P27091 1e-129 3e-83 1e-17 1e-17 Q95SI0 P36897 0.0 5e-60 1e-113 1e-111 Q24326 TGF-beta receptor type-1 (human) Q95U21 P27091 1e-17 (412 a.a.) (503 a.a.) 1e-110 P37173 PF00069 PF01064 PF00019 PF00688 TGF beta-2 (human) TGF beta-1 (human) Protein 60A; TGFb-60A (Fruit fly) P10600 0.0 P37023 1e-112 TGF beta-3 (human) PF08515 TGF-B superfamily receptor type I (human) TβR-II (human) Saxophone (Fruit fly) BMP signaling pathway Thickveins (Fruit fly) DTFR (Fruit fly) P10600 P36897 2PJY chain A, C PF08917
Figure 11. Illustration of cross-species PPI family of TGF-B3-TGFBR-1 interaction. Here we present six of 42 homologous PPIs (JE ≤ 10-40) across four species, human, fruit fly, mouse,
and chicken. All of the 42 PPIs are protein pairs between TGF-B family and TGFBR family. Interacting domain pairs are circled by purple lines.
The search result of homologous PPIs is showed in Figure 11. PPIsearch found 42 PPIs from the annotated PPI database. All of the 42 PPIs are protein pairs between members of TGF-B family and TGFBR family. Protein structure of TGF-B3-TGFBR-1 complex (PDB code: 2PJY)33 gives us the information of interacting domains. TGF-B3 engages with TGFBR-1 with domain PF000TGFBR-19 (TGF-B like domain) and PF0TGFBR-1064 (Activin types I and II receptor domain).
In this example, we observed homology of PPIs through the two views: pathway and complex insights. The result shows that the PPIs searched by PPISearch are in the TGF signaling pathway, which is identified by experiments, and in protein complexes, which are evolutionarily homologous to each other. The preliminary observation suggests that the two views may be useful to help us to identify homologous PPIs. In the future, we will collect data sets of pathways (e.g. from KEGG34) and complexes (e.g. EcoCyc35) to verify our concept on large scale.
3.4.2 A question caused by local sequence alignment
In this study, we supplied the concept of homologous PPIs and inferred the transferability of domain and function pairs. Moreover, we applied the concept to construct a web server, PPISearch. In the process that we evaluated the results of searching homologous PPIs, a question was found. We utilized BLASTP as the fast sequence alignment tool to search potential homologs, however, these search results might be biased by local sequence alignments.
We presented an example to describe this question in Figure 12. Q8INB9 is a RAC serine/threonine-protein kinase of fruit fly with three Pfam domains. In this kinase, a protein kinase domain (red) has 258 amino acids, which covers 42% of the whole sequence length, 611 amino acids. The potential homologs with BLASTP E-value ≤ 10-10 have the protein kinase domain, however, they lose the interacting domain PF00169 (deep blue, pleckstrin homology domain). This example indicated the question of searching potential homologs. The question will be reformed in the future works.
E-value ≤10-10 Q8INB9 Q9VPR4 4E-90 Q6C292 P25382 1E-115 … 1E-86 P18961 7E-86 P12688 2E-85 P11792 2E-26 P38622 RAC
serine/threonine-protein kinase Notchless 42%
Interacting domains
PF00169 PF00400
Homologs found by BLASTP alignments may be biased because of the domain(s) with large coverage
Protein kinase
domain E-value ≤10-10
Figure 12. The search result of an interacting protein pair Q8INB9-Q9VPR4. The potential homologs of Q8INB9 keep the protein kinase domain (colore red) but have no PF00169 domain (colored deep blue) to interact with the potential homolog of Q9VPR4.
Additionally, as described in Section 3.1.3 and 3.1.4, we discussed possible reasons of why members of a PPI family have different domains and binding models with the query protein.
Figure 13 shows an example of our observation. Transcription factor IIIB (TFIIIB), consisting of the TATA-binding protein (TBP), TFIIB-related factor (BRF-1) and BDP-1, is a central component in basal and regulated transcription by RNA polymerase III36. In this case, we found that when we searched homologs of yeast BRF-1 by using BLASTP, there were protein sequences with E-values ≤ 10-10, which had no interacting domains (colored by blue), in searching results. This observation suggested that local alignment methods, such as BLASTP,
may get unreliable homologs because of locally similar regions on sequences. The question will be reformed in the future works, too.
Homologs Homologs P29056 P13393 Transcription factor IIIB; BRF-1 (Yeast) P61998 Q92994 Q00403 P29052 P62000 P20226 75.28 48.61 36.87 21.55 P62380 TATA-box-binding protein (Yeast) Q27896 Q00403 (596 a.a.) (240 a.a.) 21.05 P20226 PF07741 PF08271 Transcription factor IIIB (Human) Transcription Initiation factor IIB (Human) Transcription Initiation factor IIB (Fruit fly)
Q9NG52 61.55 Q27896
RNA polymerase III transcription factor BRF (Fruit fly)
PF08515 TATA-box-binding protein(Human) TATA box-binding protein-like protein 1 (Human) PF00382 Transcription Initiation factor
IIB (Pyrococcus furiosus)
TBP-related factor (Fruit fly)
TATA-box-binding protein (Pyrococcus furiosus) -log(JE) PF07741 PF08515 1NGM chain A, B
Figure 13. An example of our method selecting proteins without interacting domain (E-values ≤ 10-10) as homologs. The interacting domains of the complex 1NGM chains A and B are colored by blue (PF07741) and yellow (PF08515), respectively. Q00403, the transcription initiation factor IIB of human, does not has the domain PF07741 but has E-value ≤ 10-10.
Chapter 4.
Applications of Homologous Protein-protein Interactions
In this chapter, we applied homologous PPI to species prediction of PPIs, and cross-species network comparisons. Many experimental approaches, for example, yeast two-hybrid system, mass spectroscopy, and tandem affinity purification, have been used to decipher PPI networks. To complement these experimental techniques, a number of computational methods for predicting PPIs, such as PathBLAST16, 17 and interologs5, 37 (i.e. conservation of inter-actions across species), have been developed19.
The concept of interolog (originally introduced by Walhout et al.37) combines known PPIs from one or more source species and orthology relationships between the source and target species to predict PPIs in the target species. Yu et al. (2004)5 extended and assessed the concept of interologs to provide a “generalized interolog mapping” method (see below). We considered that our discovery (described in Section 3.4 Discussion) can be used to advance the generalized interolog mapping method.
In addition, cross-species network comparison provides insights into the relationships between the proteins of an organism thereby contributing to a better understanding of cellular processes. However, large-scale interaction networks are available for only several model organisms. We considered that the concept of homologous PPI are useful for a systematic transfer of PPI networks between multiple species.
4.1
Cross-species prediction of protein-protein interactions
4.1.1 Background
Protein-protein interactions play an essential role in cellular functions. For rapidly increasing of sequenced genomes, it has been of significant value to provide the approaches of predicting PPIs from one organism (with abundant known interactions) to another organism (with less interaction data). In other words, to reliably transfer PPI annotation from one organism to another5.
The concept of “interologs” means: If interacting proteins A and B in one organism (source) have interacting orthologs A' and B' in another organism (target), the pair of A-B and A'-B' are called interologs. Operationally, the ortholog of a protein is defined as its best-matching homolog in another organism. Matthews et al. (2001)18 proposed a “best-match mapping” method to predict worm (C. elegans) interactions from yeast (S. cerevisiae) inter-actome. This method considered all pairs of best-matching homologs (homologs are defined by BLASTP E-value ≤ 10-10) of interacting yeast proteins as potential interologs.
Additionally, Yu et al. (2004)5 extended and assessed the concept of interologs to provide a “generalized interolog mapping” method. The mapping method regards all pairs of homologs, which have joint similarities (see Section 4.1.5) larger than a certain cutoff, as possible interologs. Their results showed that interaction annotation could be reliably transferred between two organisms if a pair of proteins has a joint E-value (JE) < 10-70.
There are interesting questions in best-match and generalized interolog mapping methods. Firstly, best-match mapping method suffers from low coverage of the total interactome5, because of using only best matches. For this question, Yu et al. (2004)proposed the method of
homologs of a query protein selected at a certain E-value would sometimes be different in subcellular compartment, biological process, or function from the query protein. For example, YLL034C in yeast has a low E-value (< 10-120) with protein Q01853 in mouse, but YLL034C has no CDC48 domain (for protein degradation)38. The protein pairs having these sequences may be not reliable candidates of interologs.
Orthology of different organisms are usually used in predicting interactions39. The third question is that, orthologous protein interactions between two species have various JE. For
example, two protein pairs P47857-P12382 and Q8R317-P40142 in mouse have orthologous interactions YMR205C-YGR240C and YMR276W-YBR117C with JE =10-171 and 10-27 in
yeast, respectively. In other words, a certain cutoff would usually lose part of orthologous interactions.
To improve these three questions, we preliminarily propose a new “ranked-based interolog mapping” method for predicting protein-protein interactions between species. This method uses only part, not all, of homologs of interacting proteins to gather possible interologs.
4.1.2 Results and discussion
4.1.2.1 Accuracy of rank-based interolog mapping
For practicability of approach to predict interactions, we wish to develop a method which has reliable predicting accuracy and acceptable coverage. In this preliminary study, we propose a new “rank-based interolog mapping” method. This method looses the best-match mapping to get a higher coverage of the total interactome. On the other hand, this method selects part, not all, of homologs in the target organism to amend the two questions of generalized interolog mapping.
0 0.05 0.1 0.15 0.2 0.25 0.3 0 100 200 300 400 500 600 700 800 900
Number of true positives
A ccu ra cy All E-values E70+rank E40+rank E10+rank Best-match A 0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 0 500 1000 1500 2000 2500 3000 3500 4000 4500 5000 5500 Number of true positives
A ccu ra cy All E-values E70+rank E40+rank E10+rank Best-match B
Figure 14. The comparison of accuracy of rank-based interolog (yellow, blue, and pink lines), best-match (green line), and generalized interolog mapping (deep blue line) methods in (A) worm-yeast mapping and (B) the four mappings. “E10+rank”, “E40+rank”, and “E70+rank” mean Acc(10-10,R), Acc(10-40,R), and Acc(10-70,R), R∈[1,5,10,15,20,25,30,35,40,45,50,55, 60,65,70,75,80,85,90,95,100, 'All'], respectively.
First, we map only worm interactions onto the yeast genome. We assess the predicting accuracy of our method, best-match and generalized interolog mapping against sets of gold standard positives P and negatives N (see Section 4.1.5). Figure 14A shows the relationship between accuracy and coverage in the worm-yeast mapping. The blue line indicates the accuracy of generalized interolog mapping from JE < 10-190 to 10-10. The green line indicates
the accuracy of best-match mapping at JE < 10-10. There are three clear observations:
1. While selecting only pairs of top R homologs (see Section 4.1.5) as candidate inter-actions (i.e. rank-based interologs), the accuracy would be usually better than the accuracy of generalized interolog mapping. For example, the purple line consists of plots Acc(10-70,R),
R∈[1,5,10,15,20,25,30,35,40, 45,50,55,60,65,70,75,80,85,90,95,100, 'All'], JE < 10-70 was
used as a good threshold of predicting interactions in Yu et al. (2004). The accuracy of R = 1, 5, and 10 are 0.22, 0.26, and 0.21, respectively, are better than Acc(10-70, 'All') = 0.11. 'All' means R has no limit.
2. If JE < 10-110, Acc(JE, 'All') will raise sharply but the number of true positives are < 25. In
other words, a very low coverage of yeast interactions.
3. The max number of true positives in best-match mapping is 50 (at JE < 10-10). Similarly,
there is a low coverage of yeast interactions.
To gather better statistics, we map inter-actions in worm, fruit fly, mouse, and human onto the yeast genome, assessing them against our gold standards. We perform a similar analysis in
Figure 14B. The number of true positives dotted in Figure 14B is sum of the true positives in worm-yeast, fly-yeast, mouse-yeast, and human-yeast mappings. The accuracy is calculated by sum of the true and false positives in the four mapping processes.
In Figure 14B, the comparison among rank-based interolog, best-match, and generalized interolog mapping is similar to that in Figure 14A. The accuracy of R = 1, 5, and 10 are 0.21, 0.17, and 0.12, respectively, are better than Acc(10-70, 'All') = 0.04.
4.1.2.2 Functional similarity between homologous protein pairs
For quantitatively assessing the unreliable homologous pairs in rank-based interologs, we construct sets of P’ and N’ (see Section 4.1.5). Genome Ontology (GO) consortium provides a standardized vocabulary, in which three structured ontologies have been proposed, which allow the description of molecular function (MF), biological process (BP) and cellular component (CC)40. This annotation particularly allows for assessing the functional similarity of genes or
their products. Based on Wu et al. (2006)41, we calculate the functional similarities between query (in the four organisms) and target (in yeast) interactions by using GO annotations. However, not all of protein sequences have GO annotations. Table 2 shows the percentage of TP’(10-10, 'All') and FP’(10-10, 'All') with the terms of CC and BP ontologies in each organism. Here TP’(10-10, 'All') = TP(10-10, 'All') ∩ P’ and FP’(10-10, 'All') = FP(10-10, 'All') ∩ N’.
Table 2. Comparison of true and false positives selected by JE and that evaluated by CC and
BP annotations
Species TP(10-10, 'All') FP(10-10, 'All') TP’(10-10, 'All') FP’(10-10, 'All')
Worm 788 13971 412 (52.3%) 2778 (19.9%)
Fly 780 73235 362 (46.4%) 23148 (31.6%)
Mouse 912 37636 685 (75.1%) 27770 (73.8%)
Human 2790 187149 1661 (59.5%) 128752 (68.8%)
The statistics of recall of TP’(10-10, 'All') and FP’(10-10, 'All') in four mappings are showed in Figure 15. Figure 15A indicates the relationship between recall and rank. For example, in worm-yeast mapping, the recall of TP’(10-10, 'All') at R = 1, 5, 10 are 5.1% (21/412), 59.0% (243/412), and 98.3% (405/412), respectively. At R = 1, 5, 10, the recall of FP’(10-10, 'All') is 0.5% (15/2778), 6.7% (185/2778), and 14.8% (411/2778). There are similar trends in the four mappings from the source organisms to yeast.
Otherwise, there is no given JE could satisfy the demands together: High recall of true
positives and low recall of false positives. For example, the recall of FP’(10-10, 'All') is 16.3% at JE < 10-40, near that at R = 10, but the recall of TP’(10-10, 'All') is only 58.3%. At JE < 10-40,
the recall of true and false positives are 10.4% and 2.6%, respectively. This result suggests that rank-based mapping method could predict more reliable interactions under a given percentage of false positives than best-match and generalized interolog mapping methods.